

Abstract
The transient receptor potential channel C6 (TRPC6) is a slit diaphragm–associated protein in podocytes involved in regulating glomerular filter function. Gain-of-function mutations in TRPC6 cause hereditary focal segmental glomerulosclerosis (FSGS), and several human acquired proteinuric diseases show increased glomerular TRPC6 expression. Angiotensin II (AngII) is a key contributor to glomerular disease and may regulate TRPC6 expression in nonrenal cells. We demonstrate that AngII regulates TRPC6 mRNA and protein levels in cultured podocytes and that AngII infusion enhances glomerular TRPC6 expression in vivo. In animal models for human FSGS (doxorubicin nephropathy) and increased renin-angiotensin system activity (Ren2 transgenic rats), glomerular TRPC6 expression was increased in an AngII-dependent manner. TRPC6 expression correlated with glomerular damage markers and glomerulosclerosis. We show that the regulation of TRPC6 expression by AngII and doxorubicin requires TRPC6-mediated Ca2+ influx and the activation of the Ca2+-dependent protein phosphatase calcineurin and its substrate nuclear factor of activated T cells (NFAT). Accordingly, calcineurin inhibition by cyclosporine decreased TRPC6 expression and reduced proteinuria in doxorubicin nephropathy, whereas podocyte-specific inducible expression of a constitutively active NFAT mutant increased TRPC6 expression and induced severe proteinuria. Our findings demonstrate that the deleterious effects of AngII on podocytes and its pathogenic role in glomerular disease involve enhanced TRPC6 expression via a calcineurin/NFAT positive feedback signaling pathway.
The glomerular capillary filtration barrier consists of endothelial cells, glomerular basement membrane (GBM), and visceral epithelial cells or podocytes linked by the slit diaphragm. The slit diaphragm is a complex of interconnected proteins that connect podocyte foot processes, which provides both physical linkage and a signaling unit that regulates podocyte behavior.1 Damage to the glomerular capillary filter, in particular at the level of the podocyte and the slit diaphragm, is of crucial importance in the pathophysiology of proteinuria.1 Previously, the transient receptor potential channel C6 (TRPC6) has been identified as a novel slit diaphragm–associated protein in podocytes.2 Gain-of-function mutations in TRPC6 have been shown to cause autosomal dominant focal segmental glomerulosclerosis (FSGS), and enhanced podocyte expression of wild-type and mutant TRPC6 leads to glomerular damage.2–5
TRP channels are involved in several renal processes and diseases, ranging from tubular Ca2+ and Mg2+ reabsorption, through osmoregulation, to polycystic kidney disease.6–9 Podocytes express TRPC6, and co-immunoprecipitation studies demonstrate that TRPC6 is associated with the slit diaphragm proteins nephrin and podocin, suggesting that TRPC6 is involved in signaling events at the slit diaphragm.2,10 The slit diaphragm complex is mechanically and functionally linked to the actin cytoskeleton. Cytoskeletal rearrangement has been suggested to underlie foot process effacement, which is a crucial early event in the pathophysiology of proteinuria.4 Several gain-of-function TRPC6 mutations have been identified in the TRPC6 encoding gene.2–4,11,12 In addition, glomerular TRPC6 expression is increased in acquired human proteinuric diseases, including nonfamilial FSGS and membranous glomerulopathy.4 Taken together, it is likely that enhanced Ca2+ influx due to an increased number of functional TRPC6 channels at the cell surface and/or enhanced channel activity compromises the structural integrity of the podocyte, leading to proteinuria.
TRPC6 is a receptor-operated cation channel, which can be activated by angiotensin II (AngII) through stimulation of the angiotensin type 1 receptor (AT1R) and secondary generation of diacylglycerol.3,13,14 AngII is a key contributor to the pathogenesis of glomerular disease, and the antiproteinuric effects of angiotensin-converting enzyme (ACE) inhibition and AT1R blockade are undisputed.15,16 In nonrenal cells, AngII activates TRPC6 currents and enhances TRPC6 transcription.14,17,18 In cardiomyocytes, AngII induces a TRPC6 and Ca2+-dependent calcineurin/nuclear factor of activated T cells (NFAT) positive feedback loop, leading to increased TRPC6 transcription, driving cardiac hypertrophy.14,18 Podocytes also express both AT1R and AT2R, and AngII has detrimental effects in podocytes.15,16,19,20 AngII increases intracellular Ca2+ levels and induces changes in the actin cytoskeleton.21–23 When the AT1R is overexpressed in podocytes, transgenic rats develop podocyte damage and glomerulosclerosis.24 Furthermore, the overexpression of renin in mice induces podocyte damage and proteinuria, pathological effects that can be ameliorated by treating these transgenic animals with angiotensin receptor blockers (ARBs).25 In analogy to cardiomyocytes, AngII-induced Ca2+-calcineurin-NFAT–mediated transcription of TRPC6 could also occur in podocytes; therefore, AngII could cause an up-regulation of TRPC6 expression, which results in elevated intracellular Ca2+ levels in podocytes in acquired proteinuric disease.
The aims of this study were to determine whether AngII regulates TRPC6 expression in podocytes, to gain insight into the downstream effectors of AngII/TRPC6-mediated signaling, and to evaluate its in vivo significance in experimental proteinuric glomerular disease.
Materials and Methods
Animal Studies
Unilateral doxorubicin nephropathy was induced in rats by temporary clipping of the left renal artery and vein, followed by injection of 1.5 mg/kg of doxorubicin (Sigma-Aldrich, Zwijndrecht, the Netherlands) via the tail vein. After 12 minutes, when doxorubicin was cleared from the circulation, the clamp was removed. Bilateral doxorubicin nephropathy was induced by injection of 5 mg/kg of doxorubicin. Animals were treated with the ARB L158,809 (150 mg per liter of drinking water) from week 6 to 12 after induction of doxorubicin nephropathy. Additional animals received the ACE inhibitor (ACEi) lisinopril (75 mg per liter of drinking water) from week 6 to 18 after induction of doxorubicin nephropathy. Cyclosporine (20 mg/kg; dissolved in 0.5 mL of olive oil) or vehicle (0.5 mL of olive oil) was administered by daily oral gavage from week 4 to 6 after doxorubicin injection. For the AngII infusion studies, Wistar rats received a continuous AngII infusion (435 ng/kg/min) by subcutaneous osmotic minipumps during 3 weeks. Before termination, animals were housed in metabolic cages for 24 hours. Male homozygous TGR(mRen2)27 (Ren2 transgenic) rats and age-matched Sprague-Dawley rats were purchased from the Max Delbrück Center for Molecular Medicine (Berlin-Buch, Berlin, Germany). Wild-type and Ren2 transgenic rats were treated with a nonhypotensive dose of the ARB candesartan (0.05 mg/kg/d) with osmotic minipumps (Alzet model 2004) for 4 weeks. The animal ethics committees of the Radboud University Nijmegen and the University Medical Centre Groningen approved all animal studies.
Generation of Inducible Transgenic Mice Overexpressing Constitutive Active NFATc1 in Podocytes
The transgenic TetO-HA-NFATc1nuc mouse line was generated in the laboratory of Dr. Gerald Crabtree and provided by Dr. Seung K. Kim (both from Stanford University, Stanford, California).26 In NFATc1nuc, the serine residues that are dephosphorylated by calcineurin are substituted with alanine residues, rendering it constitutively nuclear, constitutively active, and insensitive to nuclear kinases.27 These single transgenic mice were mated with podocin–reverse tetracycline-controlled transactivator (rtTA) mice to generate double transgenic doxycycline-inducible podocin-rtTA/TetO-HA-NFATc1nuc mice.28 Transgenic mice were genotyped using specific primer sets. Podocin-rtTA/TetO-HA-NFATc1nuc F1 littermates were mated to obtain F2 double transgenic mice for experimental procedures. Transgene expression was induced in podocytes by adding doxycycline (Sigma-Aldrich; 2 mg/mL in 7% sucrose, pH ∼ 5) to the drinking water of 6- to 8-week-old double transgenic mice for 4 days. Simultaneously, the mice were fed a special diet chow containing doxycycline (2000 ppm). Control animals were either single transgenic mice that also received doxycycline or double transgenic mice that received no doxycycline but normal chow and 7% sucrose in the drinking water. Induction of NFATc1nuc expression in isolated glomeruli was monitored by RT-PCR using DNase-treated total RNA and NFATc1nuc specific primers.
Immunohistochemistry
Glomerular expression of TRPC6 and other proteins was determined by semiquantitative scoring of immunofluorescence staining in 2-μm cryosections. We first verified our immunofluorescence method detecting TRPC6 expression in the passive Heymann nephritis rat model, in which enhanced glomerular TRPC6 expression was previously shown. TRPC6 was detected using two polyclonal antibodies directed against different epitopes in TRPC6 (Table 1): a rabbit polyclonal antibody against the C-terminal tail of rat TRPC6 and a rabbit polyclonal antibody directed against a conserved epitope in the N-terminal tail of mouse and rat TRPC6. Alexa-conjugated secondary antibodies were used subsequently. Both TRPC6 antibodies detected low levels of TRPC6 in the glomerulus of control animals, and TRPC6 expression was clearly increased in passive Heymann nephritis. Similar distribution patterns were observed using both anti-TRPC6 antibodies. When the primary antibody was omitted and only the secondary antibody was applied, no immunolabeling could be observed.
Table 1.
Antibodies Used in the Study
| Antigen | Antibody | Description | Dilution | Manufacturer or reference |
|---|---|---|---|---|
| TRPC6 | ab62999 | Rabbit anti-rat TRPC6 | 1:1000 | Abcam Plc, Cambridge, England |
| ab12249 | Rabbit anti-mouse TRPC6 | 1:25–300 | Abcam Plc, Cambridge, England | |
| ACC-017 | Rabbit anti-mouse TRPC6 | 1:200 | Alomone Laboratories, Jerusalem, Israel | |
| Desmin | D33 | Mouse anti-desmin | 1:400 | Dako, Glostrup, Denmark |
| α-SMA | 1A4 | Mouse anti-α-SMA | 1:4000 | Sigma-Aldrich, Zwijndrecht, the Netherlands |
| HS | JM403 | Mouse anti-rat HS: N-unsubstituted | 1:300 | 29 |
| Glucosamine domain | ||||
| Agrin | MI-91 | Guinea pig anti-rat agrin | 1:800 | 30 |
| β-actin | AC-15 | Mouse anti-β-actin | 1:10,000 | Sigma-Aldrich, Zwijndrecht, the Netherlands |
| GAPDH | 6C5 | Mouse anti-GAPDH | 1:10,000 | Calbiochem, EMD4Biosciences, San Diego, CA |
Glomerular TRPC6 expression was scored semiquantitatively from 0 to 5 based on the extent of TRPC6 immunofluorescence staining in the glomerulus (negative = 0, 1% to 20% positive = 1, 21% to 40% positive = 2, 41% to 60% positive = 3, 61% to 80% positive = 4, and 81% to 100% positive = 5). Semiquantification of other proteins was performed in a similar way. Scoring was performed independently by two investigators, who scored 35 to 50 glomeruli per animal on blinded sections. Focal glomerulosclerosis (FGS) was scored semiquantitatively on periodic acid–Schiff–stained paraffin sections in 50 glomeruli per kidney on a scale of 0 to 400. FGS lesions were defined as glomerular areas with mesangial expansion and adhesion formation simultaneously present in one segment.
Cell Culture and Transfection
Conditionally immortalized mouse podocytes (MPC-5) were cultured as described previously.31 Differentiated podocytes were treated with doxorubicin (0.25 μg/mL) or puromycin aminonucleoside (PAN; 100 μg/mL) for 24 hours. Depending on the exact experimental setup, AngII (1 μmol/L), losartan (100 μmol/L), captopril (1 mmol/L), chymostatin (100 μmol/L), LaCl3 (50 μmol/L), 2-aminoethyldiphenylborane (2-APB) (10 μmol/L), and/or cyclosporine (csA) (1 μmol/L) was added. A podocyte cell line stably expressing TRPC6 silencing short hairpin RNA (shRNA) was obtained after transfecting a TRPC6 shRNA construct with Lipofectamine 2000 into undifferentiated MPC-5 podocytes cultured at 33°C and subsequent selection in the presence of G418. Single clones were tested for TRPC6 mRNA and protein expression. Podocyte TRPC6 overexpression was achieved by lentiviral transduction of differentiated podocytes. FLAG-tagged wild-type mouse TRPC6 cDNA was cloned into the VVPW lentiviral expression vector (kind gift of G. Luca Gusella, New York, NY). Then 80% confluent HEK 293T cells were transfected in antibiotic-free Dulbecco's modified Eagle's medium, 10% fetal bovine serum with the VVPW plasmid, and the two helper plasmids psPAX2 and pCMV-VSVG (both from Addgene, Cambridge, MA) in a ratio of 3:2:1 using FuGENE, according to the manufacturer's protocol. Control virus was produced using empty VVPW vector together with the same helper plasmids. After 16 hours, the medium was changed to Dulbecco's modified Eagle's medium and 10% fetal bovine serum, containing penicillin and streptomycin. At 24 and 48 hours thereafter, the virus-containing cell culture supernatant was harvested and stored at 4°C, the 24- and 48-hour collections were pooled and centrifuged (600 × g; 5 minutes), and the supernatant filtered through a 0.5 μm filter, aliquoted, and frozen at −80°C. Podocytes stably transfected with the pGL4.30 reporter plasmid expressing the luc2P firefly luciferase gene under the control of the NFAT response element (see below) were transduced with lentivirus 10 days after induction of differentiation in the presence of 4 μg/mL of hexadimethrine bromide for 16 hours, and NFAT activity was measured 4 days later.
NFAT Reporter Assay
To assess NFAT activity in podocytes, a reporter podocyte cell line was generated that stably expresses the pGL4.30 reporter plasmid (Promega, Madison, WI). pGL4.30 includes the luc2P firefly luciferase gene under the control of the NFAT response element. Stably transfected clones were selected in the presence of hygromycin (300 μg/mL). Cells were differentiated for 10 days before lentiviral transduction and for 14 days before drug treatment. Stable cell lines were probed with Bright-Glo Luciferase Assay System (Promega), and luminescence was measured on a SpectraMax L luminescence microplate reader (Molecular Devices, Sunnyvale, CA).
To assess NFAT activity in TRPC6 knockdown cells, undifferentiated wild-type and TRPC6 stable knockdown podocytes were transiently transfected with the pGL4.30 reporter plasmid-expressing the luc2P firefly luciferase gene under the control of the NFAT response element and the pGL4.74 plasmid-expressing hRluc Renilla luciferase as an internal control to correct for transfection efficiency (Promega). Transiently transfected cells were assayed with Dual-Glo Luciferase Assay System (Promega).
Real-Time Quantitative RT-PCR Analysis
Total RNA was isolated from cultured podocytes and RNA was reverse transcribed (Transcriptor Kit; Roche Diagnostics, Mannheim, Germany). Real-time quantitative PCR was performed using SYBR Green Supermix (Roche Diagnostics) on a MyiQ Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). TRPC6 expression was quantified by the delta-delta CT method using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the housekeeping gene. Sample sizes of 5 to 8 separate mouse podocyte cultures were used per experimental condition per experiment. Results were confirmed in at least two distinct experiments.
Immunocytochemistry
Podocytes grown on collagen A–coated plastic SlideFlasks (NUNC, Roskilde, Denmark) were fixed and incubated with a rabbit polyclonal anti-mouse TRPC6 antibody (Table 1). Alexa-conjugated secondary antibodies were applied, cells were embedded, and images were collected as described above.
Immunoblotting
Podocytes or isolated glomeruli were lysed in a 20 mmol/L Tris pH 8 buffer containing 500 mmol/L NaCl, 0.5% (wt/vol) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 1% (vol/vol) Triton X-100, 2 pg of pepstatin, and the Complete Mini cocktail of protease inhibitors (Roche Diagnostics). Protein concentration was determined and samples containing equal amounts of protein were resolved on a 10% (wt/vol) SDS-PAGE gel and blotted to polyvinylidene difluoride membranes (Bio-Rad Laboratories). Blots were incubated with rabbit polyclonal anti-mouse TRPC6 antibody, mouse anti-β-actin antibody, or mouse anti-GAPDH antibody (Table 1) and subsequently with peroxidase-labeled secondary antibodies. Proteins were visualized using chemiluminescence, and signal intensity was determined digitally.
Ca2+ Imaging Studies
Imaging of intracellular Ca2+ concentration ([Ca2+]i) with Fura-2 was performed in selected populations of cells with an inverted fluorescence microscope setup. In brief, conditionally immortalized mouse podocytes expressing either a control shRNA or TRPC6 shRNA construct were incubated with 5 μmol/L Fura-2-AM (Sigma-Aldrich) for 45 minutes at room temperature. Cells were treated with 100 μmol/L 1-oleoyl-2-acetyl-sn-glycerolin (OAG) in the presence or absence of 2-APB, and [Ca2+]i was measured. After stimulation, the extracellular buffer was exchanged with 2 mmol/L Ca2+ to distinguish membrane-associated channel-dependent changes in [Ca2+]i. Fluorescence data are presented as a 340/380-nm ratio. Data from selected cell populations were averaged, and statistical analysis was performed on multiple experiments.
Statistical Analysis
Data are expressed as mean ± SEM. Statistical comparisons were analyzed by one-way analysis of variance and Fisher's multiple comparison. P < 0.05 was considered significant.
Results
Glomerular TRPC6 Expression Correlates with Podocyte Injury in Doxorubicin Nephropathy
Unilateral doxorubicin nephropathy was induced and resulted in a significant proteinuria (161 ± 52 mg/24 h, 246 ± 14 mg/24 h, and 388 ± 28 mg/24 h at 6, 18, and 30 weeks, respectively, after doxorubicin injection). Glomerular TRPC6 protein expression was increased in doxorubicin-exposed kidneys when compared with control kidneys (Figure 1, A and B). TRPC6 expression and proteinuria increased over time (Figure 1B), and proteinuria tended to increase with higher TRPC6 protein levels (r2 = 0.2029; P = 0.16) (Figure 1C). FGS as a direct measure of glomerular injury was significantly increased in doxorubicin-exposed kidneys (8 ± 3 versus 1 ± 1 at 6 weeks, 47 ± 11 versus 12 ± 4 at 18 weeks, and 89 ± 24 versus 33 ± 7 at 30 weeks). The FGS score correlated with TRPC6 protein levels (r2 = 0.8028; P < 0.0001) (Figure 1D). TRPC6 expression was also enhanced in bilateral doxorubicin nephropathy (Figure 1E), as were cortical TRPC6 mRNA levels (Figure 1F), suggesting that doxorubicin nephropathy alters transcription of TRPC6. In glomeruli from doxorubicin nephropathy rats, TRPC6 co-localized with desmin, which is a marker for injured podocytes, whereas in control kidneys desmin and TRPC6 expression levels were low (Figure 1G). No co-localization was found for TRPC6 and glomerular α-smooth muscle actin (α-SMA), which is a mesangial injury marker (Figure 1H). However, α-SMA and TRPC6 were expressed in similar glomerular segments in doxorubicin nephropathy, whereas α-SMA expression was negative in control kidneys. Because controls show, as expected, no α-SMA expression and low TRPC6 expression, we included co-staining of synaptopodin with α-SMA instead as a control. To further study the relationship between glomerular damage and TRPC6 expression, we performed co-stainings of TRPC6 with the heparan sulfate (HS) antibody JM403, which recognizes N-unsubstituted HS moieties present in the GBM. Immunostaining for HS in the GBM was linear in control animals, but in glomerular areas with high TRPC6 expression in doxorubicin nephropathy, HS staining was reduced and aberrantly distributed (Figure 1I).
Figure 1.

Glomerular TRPC6 expression in the doxorubicin nephropathy (DN) rat model. Representative images of glomerular immunolabeling for TRPC6 in control rats (CTR) and rats with DN (A). TRPC6 expression was determined by semiquantitative analyses of immunofluorescence signals in doxorubicin (DOXO)–exposed and contralateral control (CTR) kidneys, 6, 18, and 30 weeks after treatment (B). Correlation between glomerular TRPC6 expression in the DOXO-exposed kidney and proteinuria in the unilateral DN model (C). Correlation between semiquantitative scoring for TRPC6 protein level and the FGS score in DOXO-exposed (closed dots) and contralateral control (open dots) kidneys (D). TRPC6 protein (E) and mRNA levels (F) in bilateral DN. Co-localization studies for TRPC6 with the marker for injured podocytes desmin (G), the mesangial injury marker α-SMA (H), or HS in the GBM (I). *P < 0.05.
ARBs Prevent the Increase of TRPC6 Expression During Podocyte Injury in Vivo and in Vitro
Because of the proposed role for AngII in doxorubicin-induced glomerular injury, we evaluated the effect of ARBs on TRPC6 expression in glomeruli. When rats with bilateral doxorubicin nephropathy were treated with an ARB, glomerular TRPC6 levels were reversed to levels of untreated control animals (Figure 2, A and B). Doxorubicin exposure did not alter blood pressure, whereas ARB treatment significantly reduced blood pressure (Figure 2C) and ameliorated proteinuria (245 ± 43 mg/24 h versus 697 ± 94 mg/24 h). ARBs reduced glomerular injury as indicated by the FGS score (Figure 2D), as well as glomerular desmin (Figure 2E) and α-SMA protein levels (Figure 2F). We also studied TRPC6 expression in PAN- and doxorubicin-induced podocyte injury in vitro. Enhanced TRPC6 expression has been previously demonstrated in PAN-treated cultures podocytes.2,10 Indeed, PAN treatment of immortalized mouse podocytes significantly increased TRPC6 mRNA levels, which was ameliorated by co-incubation with the ARB losartan (Figure 3A). In addition to the evaluation of glomerular TRPC6 levels in the in vivo doxorubicin nephropathy rat model, we also studied TRPC6 expression in doxorubicin-treated podocytes in vitro. Doxorubicin also increased TRPC6 mRNA levels in cultured podocytes, which was partially prevented by ARB co-incubation (Figure 3B). To ascertain whether these transcriptional changes resulted in alterations of TRPC6 protein levels, we performed immunocytochemistry (Figure 3C) and immunoblot analyses (Figure 3D). Quantitative analyses of immunoblots showed that doxorubicin increased TRPC6 protein expression in podocytes, which was inhibited by ARB co-incubation (Figure 3E). Furthermore, ARB preincubation in addition to co-incubation completely prevented the doxorubicin-induced TRPC6 up-regulation (Figure 3F).
Figure 2.
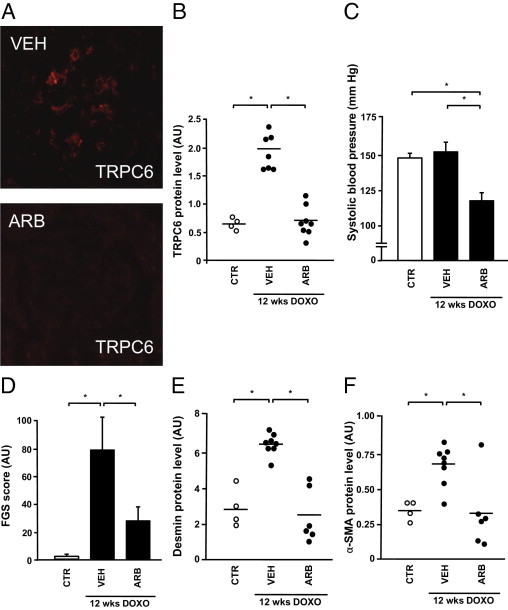
Effect of angiotensin receptor blockade on glomerular TRPC6 expression in bilateral doxorubicin nephropathy. Glomerular TRPC6 expression was determined semiquantitatively in control animals (CTR), vehicle-treated doxorubicin nephropathy rats (VEH), and L158,809-treated doxorubicin nephropathy rats (ARB) (A and B). In addition, blood pressure (C), the FGS score (D), expression of the podocyte injury marker desmin (E), and the mesangial injury marker α-SMA (F) were determined in the respective groups. DOXO, doxorubicin. *P < 0.05.
Figure 3.
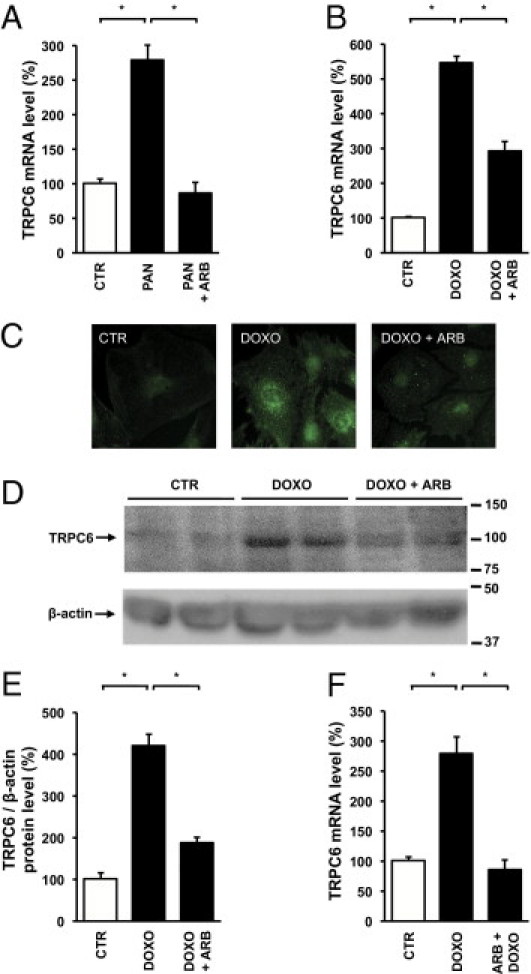
Effect of angiotensin receptor blockade on TRPC6 mRNA and protein levels in in vitro podocyte injury. TRPC6 mRNA levels were determined in untreated cultured podocytes (CTR) and podocytes incubated for 24 hours with PAN or co-incubated with losartan (PAN + ARB) (n = 5–6 separate podocyte cultures per experimental condition) (A). The effect of doxorubicin (DOXO) and co-incubation with DOXO and losartan (DOXO + ARB) on TRPC6 mRNA levels (B). TRPC6 protein expression visualized by immunocytochemistry of cultured podocytes (C). Immunoblot analysis of cell lysates derived from DOXO- and/or losartan-treated podocytes for TRPC6 and β-actin. Molecular weights indicated in kiloDaltons.(D). Intensity of TRPC6 immunoblot signals was quantified by densitometry and TRPC6 protein levels were normalized to β-actin (E). Effect of preincubation with ARB 1 hour before DOXO application (ARB + DOXO) on TRPC6 mRNA levels (F). *P < 0.05.
Elevated AngII Levels Increase TRPC6 Expression in Vitro and in Vivo
Because the data described above suggest that AngII could be an important mediator of TRPC6 expression in the podocyte, we evaluated whether in vivo and in vitro AngII application affected TRPC6 expression. When rats were infused with AngII during 21 days, semiquantitative immunohistochemistry demonstrated significantly enhanced glomerular TRPC6 expression (Figure 4A), accompanied by a mild proteinuria (110 ± 27 mg/24 h versus 15 ± 1 mg/24 h in controls). Similarly, when podocytes were cultured in the presence of AngII, TRPC6 mRNA levels were increased (Figure 4B). Subsequently, we studied glomerular TRPC6 expression in Ren2 transgenic rats, an animal model for AngII-mediated glomerular and tubulointerstitial injury.32 Glomerular TRPC6 expression was significantly increased in Ren2 transgenic rats, whereas treatment with the ARB candesartan in a nonhypotensive dose significantly reduced this effect (Figure 4C). This finding suggests that increased TRPC6 expression in Ren2 transgenic rats is AngII dependent. Alternatively, a direct effect of renin on TRPC6 expression via the (pro)renin receptor that is expressed by podocytes could cause this finding.33 However, renin application to differentiated cultured podocytes did not induce TRPC6 expression (Figure 4D). Because our results suggest that AngII regulates TRPC6 expression in podocyte injury, we evaluated the effect of lowering AngII levels in bilateral doxorubicin nephropathy. The ACEi lisinopril significantly decreased glomerular TRPC6 expression (Figure 4E). ACEi also decreased blood pressure (Figure 4F), proteinuria (289 ± 77 mg/24 h vs 641 ± 91 mg/24 h), and the FGS score (Figure 4G). Podocytes also harbor a local renin-angiotensin-aldosterone system (RAAS), whose components are synthesized on podocyte injury.34,35 However, high concentrations of the ACEi captopril and/or chymostatin, an inhibitor of non–ACE-mediated AngII production, did not affect doxorubicin-induced TRPC6 expression in cultured podocytes (Figure 4H), suggesting that injury-induced AngII production by the podocyte itself is not involved.
Figure 4.

Effects of AngII, ARBs, ACEis, and renin on TRPC6 expression in podocytes in vitro and in vivo. Glomerular TRPC6 expression was determined by semiquantitative analyses of immunofluorescence signals in vehicle-treated (VEH) and AngII-treated rats (A). TRPC6 mRNA levels in untreated differentiated cultured podocytes (CTR) and podocytes incubated with AngII (B). Glomerular TRPC6 expression was determined semiquantitatively by immunohistochemistry in Ren2 overexpressing transgenic (Ren2 Tg) rats and wild-type (WT) controls, which were treated with VEH or candesartan (ARB) (C). TRPC6 mRNA levels in CTR, podocytes incubated with doxorubicin (DOXO), and increasing concentrations of renin (REN) and losartan (ARB) (D). Glomerular TRPC6 expression in VEH and lisinopril-treated (ACEi) doxorubicin nephropathy rats (E). Systolic blood pressure (F) and the FGS score (G) in VEH and lisinopril-treated doxorubicin nephropathy rats. TRPC6 mRNA levels in CTR, podocytes incubated with DOXO, and DOXO-treated podocytes in the presence or absence of captopril (ACEi) and/or chymostatin as a blocker of non-ACE, chymase-mediated AngII production (non-ACEi) (H). n = 5 to 6 separate podocyte cultures per experimental condition. *P < 0.05.
AngII and Doxorubicin-Induced Increase of TRPC6 Expression Involves Calcineurin/NFAT Signaling
Next we evaluated whether the downstream signaling events of AngII-induced AT1R activation in podocytes involve the Ca2+-dependent calcineurin/NFAT pathway, as it does in other cell types.14,18 We demonstrated that calcineurin inhibition through application of cyclosporine prevented both doxorubicin- (Figure 5A) and AngII-induced (Figure 5B) TRPC6 expression. In other cell types it is well established that activated calcineurin dephosphorylates the transcription factor NFAT, thereby enhancing transcription of NFAT-responsive genes, including TRPC6.14,36 To analyze NFAT activity in podocytes, we used a reporter system driving luciferase expression under the control of an NFAT-responsive element. PAN administration, which increased TRPC6 expression, enhanced NFAT activation, whereas this effect was attenuated by the calcineurin inhibitors cyclosporine and tacrolimus (Figure 5C). Similarly, doxorubicin administration significantly enhanced NFAT activation, whereas cyclosporine or ARB treatment attenuated the doxorubicin-induced NFAT-controlled luciferase expression (Figure 5D). AngII treatment also enhanced NFAT-controlled luciferase expression in podocytes, and, again, both cyclosporine and ARB treatment were able to block this effect (Figure 5E). In cardiomyocytes, Ca2+ influx through TRPC6 itself induces NFAT-mediated TRPC6 transcription.14 We demonstrate that in the podocyte, enhancing TRPC6 expression through lentiviral infection with a TRPC6-FLAG construct (Figure 5F) resulted in enhanced NFAT activation (Figure 5G). In line with our hypothesis, cyclosporine administration blocked the NFAT activation secondary to exogenous TRPC6 overexpression (Figure 5G). Substantiating the in vivo significance of these in vitro findings, cyclosporine also significantly decreased doxorubicin-induced glomerular TRPC6 expression (Figure 5H) and albuminuria in rats (178 ± 58 mg/24 h versus 311 ± 46 mg/24 h).
Figure 5.
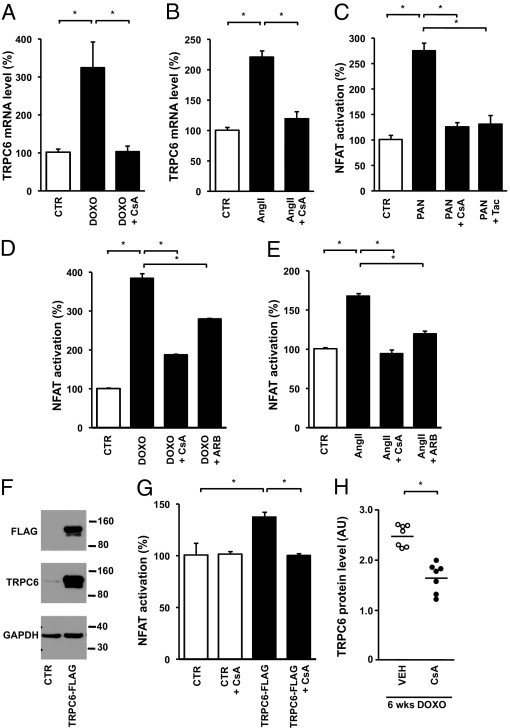
Effect of calcineurin inhibition on doxorubicin (DOXO)– and AngII-induced TRPC6 transcription and NFAT activation. TRPC6 mRNA levels in untreated cultured podocytes (CTR) and podocytes incubated with DOXO, as well as podocytes (co)-incubated with DOXO and the calcineurin inhibitor cyclosporine (CsA) (A). The effect of CsA on TRPC6 mRNA levels in AngII-exposed podocytes (B). The induction of NFAT activation in response to PAN was determined using a nuclear NFAT-responsive luciferase reporter construct; thereafter, the effects of the calcineurin inhibitors CsA and tacrolimus (Tac) were studied (C). The effect of DOXO (D) and AngII (E) treatment on NFAT-controlled luciferase expression was determined, as well as after co-treatment with CsA and losartan (ARB). Western blot analysis of total lysates of cultured podocytes infected with a TRPC6-FLAG construct or empty vector (CTR) for FLAG and TRPC6 (F). The GAPDH blot shows equal protein loading. NFAT activation was determined in podocytes infected with a TRPC6-FLAG construct or empty vector in the presence or absence of CsA (G). n = 5 to 6 separate podocyte cultures per experimental condition for all experiments. Glomerular TRPC6 expression was determined in vehicle-treated doxorubicin nephropathy rats (VEH) and CsA-treated doxorubicin nephropathy rats (H). *P < 0.05.
To study the Ca2+ influx dependency of this mechanism, we demonstrated that when doxorubicin-challenged podocytes were treated with LaCl3, which is a general blocker of Ca2+ influx across cell membranes, TRPC6 mRNA levels were significantly decreased (Figure 6A). Furthermore, the TRPC channel blocker 2-APB inhibited doxorubicin- (Figure 6B) and AngII-induced TRPC6 expression (Figure 6C). Because there are no TRPC6-specific blockers available to date, we used a TRPC6 knockdown technique using TRPC6 shRNA (Figure 6D) to evaluate the effect of reduced TRPC6 expression levels on AngII-induced NFAT activation. Indeed, TRPC6 knockdown decreased AngII-induced NFAT activation in podocytes compared with control cells (Figure 6E). This indicated that TRPC6, at least in part, mediates the effect of AngII on NFAT activation. To confirm that the TRPC channel blocker 2-APB and the TRPC6 knockdown approach indeed affected receptor-mediated Ca2+ influx, we evaluated Fura-2 ratiometry secondary to stimulation by the diacylglycerol analog OAG in podocytes. Indeed, TRPC6 knockdown in podocytes resulted in reduced OAG-stimulated Ca2+ influx compared with nontransfected podocytes or cells transfected with control shRNA (Figure 6F). Furthermore, 2-APB application significantly reduced OAG-stimulated Ca2+ influx (Figure 6F). Importantly, 2-APB did so in untransfected podocytes and cells transfected with control shRNA but to a much lesser degree in the TRPC6 knockdown situation (Figure 6G), strongly suggesting that TRPC6 is the major TRPC channel mediating the 2-APB-sensitive Ca2+ influx in cultured podocytes.
Figure 6.
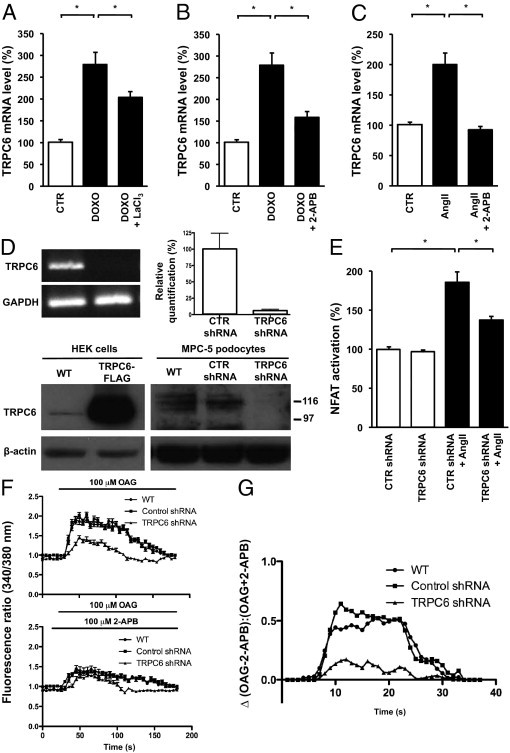
Involvement of Ca2+ influx (through TRPC6 itself) on doxorubicin (DOXO)– and AngII-induced TRPC6 transcription and NFAT activation. TRPC6 mRNA levels in untreated cultured podocytes (CTR) and podocytes incubated with DOXO and DOXO-exposed podocytes co-incubated with LaCl3 (DOXO + LaCl3), which inhibits Ca2+ influx (A). The effect of 2-APB, a TRPC channel blocker, on TRPC6 mRNA levels was evaluated in DOXO- (B) and AngII-treated (C) podocytes. Validation of a podocyte cell line stably transfected with a TRPC6 shRNA construct (D). TRPC6 knockdown was confirmed using semiquantitative RT-PCR, quantitative real-time PCR, and on the protein level Western blot analysis. We determined the effect of AngII on NFAT activation in these podocytes stably expressing a TRPC6 shRNA silencing construct or a control shRNA construct (CTR) (E). n = 3 to 6 separate podocyte cultures per experimental condition for all experiments. *P < 0.05. The OAG-induced Ca2+ response was determined by Fura-2 ratiometry in untransfected podocytes (WT), podocytes stably expressing a TRPC6 shRNA silencing construct, or a CTR shRNA construct, in either the absence or presence of 2-APB (F). The net effect of 2-APB on the OAG-induced Ca2+ response is depicted by calculating the ratio of the change in OAG-induced Ca2+ response in the absence and presence of 2-APB (G).
Podocyte-Specific Overexpression of Activated NFAT in Mice Increases TRPC6 Expression and Induces Proteinuria
Our findings suggest that in podocytes TRPC6-mediated activation of NFAT enhances TRPC6 transcription and is eventually detrimental to the glomerular filter, causing proteinuria. Because doxorubicin and AngII, two inducers of podocyte damage and proteinuria in animal models, activate this pathway and TRPC6 gain-of-function mutations and overexpression underlie human glomerular disease, it is possible that NFAT activation per se induces podocyte damage. To test this hypothesis, we induced overexpression of a constitutively active NFATc1 variant (NFATc1nuc) in podocytes of adult transgenic mice. In NFATc1nuc, the serine residues that are dephosphorylated by calcineurin are mutated to alanines, rendering this NFATc1 mutant form constitutively nuclear and insensitive to nuclear kinases.27 We crossed an inducible transgenic mouse line expressing NFATc1nuc under the control of a tetracycline-responsive element with podocin-rtTA mice, which express the rtTA, specifically in podocytes.26,28 The resulting double transgenic offspring were administered doxycycline for 4 days, and transgene expression in isolated glomeruli was monitored by RT-PCR. Only doxycycline-treated double transgenic mice expressed the NFATc1nuc transgene, whereas vehicle-treated double transgenic mice (data not shown) or doxycycline-treated single transgenic mice, both used as negative controls, did not (Figure 7A). Importantly, mice expressing the NFATc1nuc transgene developed significant proteinuria (Figure 7, B and C) and showed significantly elevated TRPC6 protein levels in isolated glomeruli when compared with control animals (Figure 7, D and E). This experiment shows that NFAT activation in podocytes per se is sufficient to induce proteinuria in mice and that NFAT regulates TRPC6 expression in podocytes in vivo.
Figure 7.
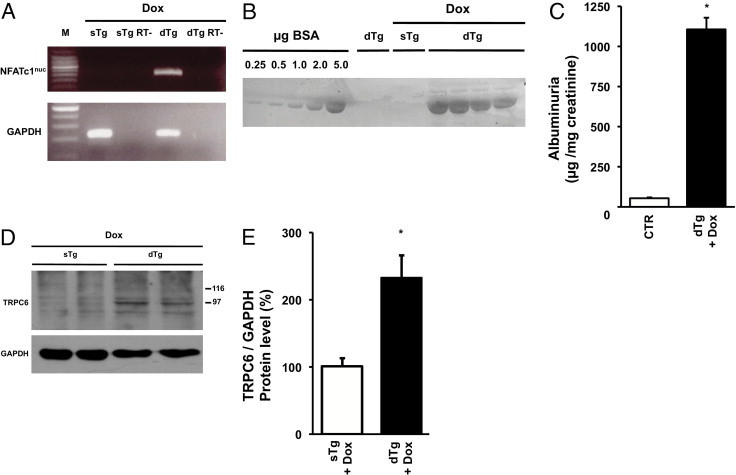
In vivo effect of NFAT activation in podocytes on TRPC6 expression and proteinuria. RT-PCR analysis of NFATc1nuc transgene expression in glomeruli of podocin-rtTA/tetO-HA-NFATc1nuc mice (A). Double transgenic mice (dTg) were treated with doxycycline (Dox) for 4 days. Single-transgenic rtTA mice (sTg) on Dox served as negative control. Per group glomeruli from three mice were pooled, and half of the RNA sample was not reverse transcribed before PCR and thereby served as no RT control (RT-). Detection of albumin in urine samples from podocin-rtTA/tetO-HA-NFATc1nuc mice by SDS-PAGE and Coomassie staining (B). As controls urine samples from two vehicle-treated dTg mice and two Dox-treated sTg rtTA mice were used. A bovine serum albumin (BSA) gradient (0.25 to 5.0 μg) was run on the same gel to quantify urine albumin signals. Dox-treated dTg mice and control (CTR) mice were analyzed, creating a pool of Dox-treated sTg rtTA mice and vehicle-treated dTg mice (n = 6) as summarized in panel C. TRPC6 and GAPDH protein expression in glomeruli as determined using Western blot analysis (D). As control, Dox-treated sTg rtTA mice were used. Intensity of immunoblot signals was quantified by densitometry and TRPC6 protein levels are depicted relative to GAPDH levels (E). *P < 0.05.
Discussion
In the present study we demonstrate that a deleterious positive feedback mechanism, in which TRPC6-mediated Ca2+ influx stimulates NFAT-dependent TRPC6 expression, is involved in AngII-associated podocyte injury. We show in different in vitro and in vivo models that AngII, a key contributor to the pathogenesis of glomerular disease, increases TRPC6 expression in podocytes. Glomerular TRPC6 expression was up-regulated by AngII infusion and in the Ren2 transgenic rat model for AngII-associated renal damage, an effect that was ameliorated by ARB treatment. TRPC6 expression was also enhanced in the doxorubicin nephropathy model for acquired progressive glomerular damage and proteinuric disease and positively correlated with markers for podocyte and glomerular damage. Blocking the effect of AngII in doxorubicin-induced in vitro podocyte injury and in vivo glomerular disease down-regulated TRPC6 expression and ameliorated glomerular damage and proteinuria. Thus, doxorubicin-induced TRPC6 expression in podocytes is mediated through AngII/AT1R signaling. Doxorubicin-induced and AngII-mediated TRPC6 transcription appears to be controlled by the Ca2+-dependent calcineurin/NFAT pathway, involving Ca2+ influx through TRPC6 itself. Indeed, we demonstrate in a novel transgenic mouse model that podocyte-specific NFAT activation induces glomerular TRPC6 expression and severe proteinuria.
Doxorubicin induces glomerular injury in rats, giving rise to progressive proteinuria and loss of renal function.37 Glomerular TRPC6 expression was enhanced in the unilateral and bilateral doxorubicin nephropathy models for acquired glomerular disease. Because doxorubicin also increased TRPC6 mRNA and protein expression in cultured podocytes, we conclude that the increased TRPC6 expression is a direct result of doxorubicin acting on the podocyte. TRPC6 was detectable in healthy control animals but maximally enhanced in segmental glomerular lesions in doxorubicin nephropathy. These glomerular segments showed evidence of injury, where TRPC6 co-localizes with desmin as a marker for podocyte injury. Accordingly, glomerular TRPC6 expression showed a significant correlation with the extent of glomerulosclerosis. Because highly sclerotic glomeruli lacked TRPC6 expression, this correlation may be lost with advanced FGS when podocyte depletion occurs. The pathological characteristics of long-term doxorubicin nephropathy, including segmental glomerulosclerosis, closely resemble those seen in human FSGS.37 Thus, increased TRPC6 expression appears crucially involved in the pathogenesis of podocyte and glomerular damage, leading to glomerulosclerosis and proteinuria in FSGS. These findings are in line with increased TRPC6 expression in human acquired FSGS and TRPC6 gain-of-function mutations leading to hereditary FSGS.2–4
We demonstrate that AngII infusion enhances glomerular TRPC6 expression in rats and that AngII application directly regulates TRPC6 expression in podocytes. Furthermore, this study is the first to demonstrate that ARBs and ACEis, counteracting the effects of AngII, decrease TRPC6 overexpression in experimental glomerular disease. Because TRPC6 expression is enhanced in acquired human glomerular disease, and in the absence of specific TRPC6 blockers for clinical use, our data suggest that treatment with ARBs and ACEis are a potential way to reduce TRPC6 expression. ARBs and ACEis are known to reduce glomerular injury, decrease proteinuria, and ameliorate renal function decline in chronic renal disease, which is partly independent of their antihypertensive action.38 We and others have previously shown that the RAAS is involved in doxorubicin nephropathy, and the key pathological involvement of AngII in glomerular disease is widely accepted.19,39,40 Accordingly, we now demonstrate that doxorubicin-induced TRPC6 expression in podocytes is AngII-mediated. In doxorubicin nephropathy, ACEis down-regulated TRPC6 expression by its effect on the systemic but not the local podocyte RAAS. Furthermore, in the Ren2 transgenic model of AngII-mediated renal injury, TRPC6 levels were significantly enhanced, which appeared to be a direct effect of AngII exemplified by the partial normalization of its expression after short-term ARB treatment. Several lines of evidence support that AngII is injurious to podocytes. For example, transgenic rats overexpressing the AT1R on their podocytes show structural podocyte damage and proteinuria, progressing to FSGS.24 AngII infusion reduces nephrin expression and provokes podocyte injury in experimental glomerulopathies, which is inhibited by RAAS blockade.30,41–45 Interestingly, adverse effects of AngII-mediated stimulation of TRPC6 have also been shown in nonrenal cells.14,18 Stimulation of TRPC6 by AngII plays an important role in vascular smooth muscle cell contraction, and TRPC6 expression in pulmonary vascular smooth muscle cell is increased in idiopathic pulmonary arterial hypertension.17,46,47 Importantly, AngII-induced Ca2+ influx through TRPC6 was shown to activate the calcineurin/NFAT signaling pathway and increase TRPC6 expression in cardiomyocytes, resulting in cardiomyocyte hypertrophy.14,18 A recent study by Eckel et al48 showed that AngII-induced albuminuria was not prevented but was significantly ameliorated in TRPC6 knockout compared with wild-type mice. Altogether, this demonstrates that AngII-induced and calcineurin/NFAT-mediated TRPC6 expression in the podocyte is an important mediator in the pathogenesis of podocyte injury and glomerular disease.
Thus, we hypothesized that the Ca2+ influx-stimulated calcineurin/NFAT pathway is involved in downstream signaling activated by AngII in the podocyte (proposed model depicted in Figure 8). The inhibitor of TRPC channel activity 2-APB reduced receptor-mediated Ca2+ influx into cultured podocytes, as well as doxorubicin-induced and AngII-mediated TRPC6 up-regulation. This demonstrates that TRPC6 expression is Ca2+-dependent and that Ca2+ influx through TRPC channels could be involved. It was previously shown that AngII induces elevation of [Ca2+]i in the podocyte by influx from the extracellular compartment, which can be blocked by ARBs.21–23 Among other downstream targets, Ca2+ influx activates the serine-threonine phosphatase calcineurin, which dephosphorylates the NFAT family of transcription factors, leading to the transcription of NFAT-responsive genes.49 We demonstrated that AngII activates NFAT in podocytes and that calcineurin inhibition and blocking of the AT1R inhibit doxorubicin-induced and AngII-mediated NFAT activation and TRPC6 expression. The TRPC6 promoter harbors two conserved NFAT-responsive sites, required for TRPC6 transcription in response to Ca2+-dependent calcineurin/NFAT signaling in cardiomyocytes.14 Schlondorff et al36 have shown that introduction of a gain-of-function TRPC6 mutant in podocytes enhances NFAT activation in vitro. Furthermore, they reported that cyclosporine, without directly affecting TRPC6 channel activity, inhibits TRPC6-mediated NFAT signaling.36 Thus, Ca2+ influx through TRPC6 itself could play a role in an AT1R-stimulated calcineurin/NFAT signaling cascade. This hypothesis is supported by our data showing that lentiviral TRPC6 overexpression in podocytes activates NFAT in an AT1R- and calcineurin-dependent manner. Importantly, we also demonstrated that TRPC6 knockdown decreases Ca2+ influx and inhibits AngII-induced NFAT activation in podocytes. In line with these data, it was recently shown that in podocytes isolated from TRPC6 knockout and wild-type mice the response to AngII significantly differs, showing a reduced Ca2+ current in the TRPC6 knockout podocytes.48 In a similar TRPC6 shRNA approach, Greka and coworkers50 recently showed that down-regulation of TRPC6 reduces AngII-evoked Ca2+ transients in podocytes, along with effects on the podocyte cytoskeleton. The residual NFAT activation in our experiments might be related to remaining TRPC6 expression but could also mean that the cascade is not solely TRPC6 dependent and other (TRPC) Ca2+ channels or influx mechanisms are involved. Expression of other TRPC channels in the podocyte has been shown, and TRP channels form heterotetramers in which different TRP subunits combined form functional channels.51–53
Figure 8.

Proposed signaling pathway that regulates the in AngII-dependent TRPC6 expression in podocytes. Results from our study together with previously published data57 suggest the existence of the following signaling pathways in podocytes mediating AngII-induced TRPC6 expression and podocyte injury. AT1R stimulation by AngII results in Ca2+ influx at least in part mediated by TRPC6. Ca2+-dependent calcineurin activation leads to activation and nuclear translocation of NFAT, which enhances transcription of NFAT-responsive genes such as TRPC6. A consequent increase in TRPC6 protein expression at the cell membrane could result in a positive feedback regulatory circuit, because Ca2+ influx through TRPC6 itself appears to be involved in the signaling pathway leading to increased TRPC6 expression. Our studies show that NFAT activation per se is sufficient to induce TRPC6 transcription and proteinuria. Hypothetically, the proposed feedback mechanism could also result in a persistent calcineurin activation, which was previously demonstrated to lead to the dephosphorylation and degradation of the actin-binding protein synaptopodin (dashed box).57 The latter was shown to result in cytoskeletal rearrangement and, eventually, proteinuria. Possibly, the latter pathway is involved in the generation of podocyte injury subsequent to AngII-induced TRPC6 expression.
Altogether, our data are the first to demonstrate that AngII induces a calcineurin/NFAT pathway in podocytes that appears to be dependent on Ca2+ influx through TRPC6 and, in addition, leads to enhanced expression of TRPC6 itself, thus forming a potentially deleterious feedback loop. Zhang et al54 previously suggested that AngII-induced TRPC6 expression might involve MAPK, ERK, JNK, and NF-κB. In cardiomyocytes, those pathways were also described, and receptor-activated TRPC6-mediated NFAT activation was shown to inhibit JNK and ERK.55 To demonstrate that NFAT activation in podocytes is independently capable of enhancing TRPC6 expression in vivo and inducing proteinuria, we generated a transgenic mouse model in which a constitutively active NFAT mutant could be induced in a podocyte-specific manner. As originally hypothesized, we demonstrated that induction of NFAT results in increased glomerular TRPC6 expression and the development of severe proteinuria. On submission of this work another article appeared56 that also describes the effect of NFAT overexpression and the subsequent development of proteinuria; however, the involvement of TRPC6 was not specifically studied in that article. Our data confirm that the activation of NFAT in podocytes is sufficient to induce proteinuria and substantiate that a positive feedback loop involving NFAT-induced TRPC6 expression contributes to the induction and/or maintenance of proteinuric disease.
The current findings add an additional downstream pathway secondary to calcineurin signaling in podocytes, in addition to the dephosphorylation of the actin-binding protein synaptopodin by calcineurin as we previously reported.57 Synaptopodin is vitally important in maintaining the podocyte actin cytoskeleton, and its dephosphorylation results in subsequent cathepsin l–mediated degradation, cytoskeletal disorganization, and proteinuria.57,58 Thus, our data contribute to a hypothetical mechanism by which AngII-induced and NFAT-mediated TRPC6 overexpression, as addressed in our experiments, fuel persistent calcineurin activation, leading to synaptopodin degradation as described previously, eventually perpetuating podocyte injury (Figure 8). For this hypothesis to be proven, it remains to be established whether Ca2+ influx through TRPC6 directly induces calcineurin-mediated dephosphorylation of synaptopodin.
In conclusion, we have demonstrated an AngII-induced, TRPC6-dependent, NFAT-mediated feedback mechanism driving TRPC6 transcription in podocytes. This finding underlines the crucial role of TRPC6 in the pathogenesis of podocyte injury and proteinuria.
Footnotes
Supported by a Kolff Career Stimulation Grant from the Dutch Kidney Foundation (KJPB 07.0001), a grant from the Genzyme Renal Innovations Program and a Ruby Diabetes Research Grant (2009.80.118) (T.N.), a EURYI award (J.H.), a grant from the Swiss National Science Foundation (PBZHP3-128278) and an Amgen-FROMO fellowship (A.D.K.), grants from the US National Institutes of Health (grants DK073495 and DK089394) (J.R.), and a Young Investigator Career Development Grant from the NephCure Foundation and a National Scientist Development Grant from the American Heart Association (C.F.).
T.N., A.J.S., J.G.J.H., and J.F. contributed equally to this work.
Contributor Information
Christian Faul, Email: cfaul@med.miami.edu.
Johan van der Vlag, Email: J.vanderVlag@nier.umcn.nl.
References
- 1.Faul C., Asanuma K., Yanagida-Asanuma E., Kim K., Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Reiser J., Polu K.R., Moller C.C., Kenlan P., Altintas M.M., Wei C., Faul C., Herbert S., Villegas I., Avila-Casado C., McGee M., Sugimoto H., Brown D., Kalluri R., Mundel P., Smith P.L., Clapham D.E., Pollak M.R. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winn M.P., Conlon P.J., Lynn K.L., Farrington M.K., Creazzo T., Hawkins A.F., Daskalakis N., Kwan S.Y., Ebersviller S., Burchette J.L., Pericak-Vance M.A., Howell D.N., Vance J.M., Rosenberg P.B. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 4.Moller C.C., Wei C., Altintas M.M., Li J., Greka A., Ohse T., Pippin J.W., Rastaldi M.P., Wawersik S., Schiavi S., Henger A., Kretzler M., Shankland S.J., Reiser J. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 5.Krall P., Canales C.P., Kairath P., Carmona-Mora P., Molina J., Carpio J.D., Ruiz P., Mezzano S.A., Li J., Wei C., Reiser J., Young J.I., Walz K. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS One. 2010;5:e12859. doi: 10.1371/journal.pone.0012859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clapham D.E. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 7.Hsu Y.J., Hoenderop J.G., Bindels R.J. TRP channels in kidney disease. Biochim Biophys Acta. 2007;1772:928–936. doi: 10.1016/j.bbadis.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Nijenhuis T., Vallon V., van der Kemp A.W., Loffing J., Hoenderop J.G., Bindels R.J. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115:1651–1658. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woudenberg-Vrenken T.E., Bindels R.J., Hoenderop J.G. The role of transient receptor potential channels in kidney disease. Nat Rev Nephrol. 2009;5:441–449. doi: 10.1038/nrneph.2009.100. [DOI] [PubMed] [Google Scholar]
- 10.Moller C.C., Flesche J., Reiser J. Sensitizing the slit diaphragm with TRPC6 ion channels. J Am Soc Nephrol. 2009;20:950–953. doi: 10.1681/ASN.2008030329. [DOI] [PubMed] [Google Scholar]
- 11.Zhu B., Chen N., Wang Z.H., Pan X.X., Ren H., Zhang W., Wang W.M. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat Res. 2009;664:84–90. doi: 10.1016/j.mrfmmm.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 12.Santin S., Ars E., Rossetti S., Salido E., Silva I., Garcia-Maset R., Gimenez I., Ruiz P., Mendizabal S., Nieto J.L., Pena A., Camacho J.A., Fraga G., Cobo M.A., Bernis C., Ortiz A., de Pablos A.L., Sanchez-Moreno A., Pintos G., Mirapeix E., Fernandez-Llama P., Ballarin J., Torra R. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009;24:3089–3096. doi: 10.1093/ndt/gfp229. [DOI] [PubMed] [Google Scholar]
- 13.Hofmann T., Obukhov A.G., Schaefer M., Harteneck C., Gudermann T., Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 14.Kuwahara K., Wang Y., McAnally J., Richardson J.A., Bassel-Duby R., Hill J.A., Olson E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruggenenti P., Perna A., Gherardi G., Garini G., Zoccali C., Salvadori M., Scolari F., Schena F.P., Remuzzi G. Renoprotective properties of ACE-inhibition in non-diabetic nephropathies with non-nephrotic proteinuria. Lancet. 1999;354:359–364. doi: 10.1016/S0140-6736(98)10363-X. [DOI] [PubMed] [Google Scholar]
- 16.Ruggenenti P., Perna A., Loriga G., Ganeva M., Ene-Iordache B., Turturro M., Lesti M., Perticucci E., Chakarski I.N., Leonardis D., Garini G., Sessa A., Basile C., Alpa M., Scanziani R., Sorba G., Zoccali C., Remuzzi G. Blood-pressure control for renoprotection in patients with non-diabetic chronic renal disease (REIN-2): multicentre, randomised controlled trial. Lancet. 2005;365:939–946. doi: 10.1016/S0140-6736(05)71082-5. [DOI] [PubMed] [Google Scholar]
- 17.Saleh S.N., Albert A.P., Peppiatt C.M., Large W.A. Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J Physiol. 2006;577:479–495. doi: 10.1113/jphysiol.2006.119305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Onohara N., Nishida M., Inoue R., Kobayashi H., Sumimoto H., Sato Y., Mori Y., Nagao T., Kurose H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006;25:5305–5316. doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van den Hoven M.J., Waanders F., Rops A.L., Kramer A.B., van Goor H., Berden J.H., Navis G., van der Vlag J. Regulation of glomerular heparanase expression by aldosterone, angiotensin II and reactive oxygen species. Nephrol Dial Transplant. 2009;24:2637–2645. doi: 10.1093/ndt/gfp182. [DOI] [PubMed] [Google Scholar]
- 20.Harrison-Bernard L.M., Navar L.G., Ho M.M., Vinson G.P., el-Dahr S.S. Immunohistochemical localization of ANG II AT1 receptor in adult rat kidney using a monoclonal antibody. Am J Physiol. 1997;273:F170–F177. doi: 10.1152/ajprenal.1997.273.1.F170. [DOI] [PubMed] [Google Scholar]
- 21.Henger A., Huber T., Fischer K.G., Nitschke R., Mundel P., Schollmeyer P., Greger R., Pavenstadt H. Angiotensin II increases the cytosolic calcium activity in rat podocytes in culture. Kidney Int. 1997;52:687–693. doi: 10.1038/ki.1997.383. [DOI] [PubMed] [Google Scholar]
- 22.Nitschke R., Henger A., Ricken S., Gloy J., Muller V., Greger R., Pavenstadt H. Angiotensin II increases the intracellular calcium activity in podocytes of the intact glomerulus. Kidney Int. 2000;57:41–49. doi: 10.1046/j.1523-1755.2000.00810.x. [DOI] [PubMed] [Google Scholar]
- 23.Gloy J., Henger A., Fischer K.G., Nitschke R., Mundel P., Bleich M., Schollmeyer P., Greger R., Pavenstadt H. Angiotensin II depolarizes podocytes in the intact glomerulus of the rat. J Clin Invest. 1997;99:2772–2781. doi: 10.1172/JCI119467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann S., Podlich D., Hahnel B., Kriz W., Gretz N. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol. 2004;15:1475–1487. doi: 10.1097/01.asn.0000127988.42710.a7. [DOI] [PubMed] [Google Scholar]
- 25.Huby A.C., Rastaldi M.P., Caron K., Smithies O., Dussaule J.C., Chatziantoniou C. Restoration of podocyte structure and improvement of chronic renal disease in transgenic mice overexpressing renin. PLoS One. 2009;4:e6721. doi: 10.1371/journal.pone.0006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winslow M.M., Pan M., Starbuck M., Gallo E.M., Deng L., Karsenty G., Crabtree G.R. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell. 2006;10:771–782. doi: 10.1016/j.devcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 27.Beals C.R., Clipstone N.A., Ho S.N., Crabtree G.R. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 1997;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- 28.Shigehara T., Zaragoza C., Kitiyakara C., Takahashi H., Lu H., Moeller M., Holzman L.B., Kopp J.B. Inducible podocyte-specific gene expression in transgenic mice. J Am Soc Nephrol. 2003;14:1998–2003. doi: 10.1681/ASN.V1481998. [DOI] [PubMed] [Google Scholar]
- 29.van den Born J., van den Heuvel L.P., Bakker M.A., Veerkamp J.H., Assmann K.J., Berden J.H. A monoclonal antibody against GBM HS induces an acute selective proteinuria in rats. Kidney Int. 1992;41:115–123. doi: 10.1038/ki.1992.15. [DOI] [PubMed] [Google Scholar]
- 30.Raats C.J., Bakker M.A., Hoch W., Tamboer W.P., Groffen A.J., van den Heuvel L.P., Berden J.H., van den Born J. Differential expression of agrin in renal basement membranes as revealed by domain-specific antibodies. J Biol Chem. 1998;273:17832–17838. doi: 10.1074/jbc.273.28.17832. [DOI] [PubMed] [Google Scholar]
- 31.Shankland S.J., Pippin J.W., Reiser J., Mundel P. Podocytes in culture: past, present, and future. Kidney Int. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 32.Lee M.A., Bohm M., Paul M., Bader M., Ganten U., Ganten D. Physiological characterization of the hypertensive transgenic rat TGR(mREN2)27. Am J Physiol. 1996;270:E919–E929. doi: 10.1152/ajpendo.1996.270.6.E919. [DOI] [PubMed] [Google Scholar]
- 33.Ichihara A., Kaneshiro Y., Takemitsu T., Sakoda M., Itoh H. The (pro)renin receptor and the kidney. Semin Nephrol. 2007;27:524–528. doi: 10.1016/j.semnephrol.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Durvasula R.V., Petermann A.T., Hiromura K., Blonski M., Pippin J., Mundel P., Pichler R., Griffin S., Couser W.G., Shankland S.J. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 35.Durvasula R.V., Shankland S.J. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol. 2008;294:F830–F839. doi: 10.1152/ajprenal.00266.2007. [DOI] [PubMed] [Google Scholar]
- 36.Schlondorff J., Del Camino D., Carrasquillo R., Lacey V., Pollak M.R. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am J Physiol Cell Physiol. 2009;296:C558–C569. doi: 10.1152/ajpcell.00077.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pippin J.W., Brinkkoetter P.T., Cormack-Aboud F.C., Durvasula R.V., Hauser P.V., Kowalewska J., Krofft R.D., Logar C.M., Marshall C.B., Ohse T., Shankland S.J. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009;296:F213–F229. doi: 10.1152/ajprenal.90421.2008. [DOI] [PubMed] [Google Scholar]
- 38.Reiser J., Mundel P. Dual effects of RAS blockade on blood pressure and podocyte function. Curr Hypertens Rep. 2007;9:403–408. doi: 10.1007/s11906-007-0074-7. [DOI] [PubMed] [Google Scholar]
- 39.Kramer A., van den Hoven M., Rops A., Wijnhoven T., van den Heuvel L., Lensen J., van Kuppevelt T., van Goor H., van der Vlag J., Navis G., Berden J.H. Induction of glomerular heparanase expression in rats with adriamycin nephropathy is regulated by reactive oxygen species and the renin-angiotensin system. J Am Soc Nephrol. 2006;17:2513–2520. doi: 10.1681/ASN.2006020184. [DOI] [PubMed] [Google Scholar]
- 40.Kramer A.B., van der Meulen E.F., Hamming I., van Goor H., Navis G. Effect of combining ACE inhibition with aldosterone blockade on proteinuria and renal damage in experimental nephrosis. Kidney Int. 2007;71:417–424. doi: 10.1038/sj.ki.5002075. [DOI] [PubMed] [Google Scholar]
- 41.Hiramatsu N., Hiromura K., Shigehara T., Kuroiwa T., Ideura H., Sakurai N., Takeuchi S., Tomioka M., Ikeuchi H., Kaneko Y., Ueki K., Kopp J.B., Nojima Y. Angiotensin II type 1 receptor blockade inhibits the development and progression of HIV-associated nephropathy in a mouse model. J Am Soc Nephrol. 2007;18:515–527. doi: 10.1681/ASN.2006030217. [DOI] [PubMed] [Google Scholar]
- 42.Ideura H., Hiromura K., Hiramatsu N., Shigehara T., Takeuchi S., Tomioka M., Sakairi T., Yamashita S., Maeshima A., Kaneko Y., Kuroiwa T., Kopp J.B., Nojima Y. Angiotensin II provokes podocyte injury in murine model of HIV-associated nephropathy. Am J Physiol Renal Physiol. 2007;293:F1214–F1221. doi: 10.1152/ajprenal.00162.2007. [DOI] [PubMed] [Google Scholar]
- 43.Benigni A., Tomasoni S., Gagliardini E., Zoja C., Grunkemeyer J.A., Kalluri R., Remuzzi G. Blocking angiotensin II synthesis/activity preserves glomerular nephrin in rats with severe nephrosis. J Am Soc Nephrol. 2001;12:941–948. doi: 10.1681/ASN.V125941. [DOI] [PubMed] [Google Scholar]
- 44.Remuzzi A., Gagliardini E., Sangalli F., Bonomelli M., Piccinelli M., Benigni A., Remuzzi G. ACE inhibition reduces glomerulosclerosis and regenerates glomerular tissue in a model of progressive renal disease. Kidney Int. 2006;69:1124–1130. doi: 10.1038/sj.ki.5000060. [DOI] [PubMed] [Google Scholar]
- 45.Kawachi H., Koike H., Shimizu F. Molecular structure and function of the slit diaphragm: expression of nephrin in proteinuric states and in developing glomeruli. Nephrol Dial Transplant. 2002;17(Suppl 9):20–22. doi: 10.1093/ndt/17.suppl_9.20. [DOI] [PubMed] [Google Scholar]
- 46.Weissmann N., Dietrich A., Fuchs B., Kalwa H., Ay M., Dumitrascu R., Olschewski A., Storch U., Mederos y Schnitzler M., Ghofrani H.A., Schermuly R.T., Pinkenburg O., Seeger W., Grimminger F., Gudermann T. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A. 2006;103:19093–19098. doi: 10.1073/pnas.0606728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu Y., Fantozzi I., Remillard C.V., Landsberg J.W., Kunichika N., Platoshyn O., Tigno D.D., Thistlethwaite P.A., Rubin L.J., Yuan J.X. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2004;101:13861–13866. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eckel J., Lavin P.J., Finch E.A., Mukerji N., Burch J., Gbadegesin R., Wu G., Bowling B., Byrd A., Hall G., Sparks M., Zhang Z.S., Homstad A., Barisoni L., Birbaumer L., Rosenberg P., Winn M.P. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol. 2011;22:526–535. doi: 10.1681/ASN.2010050522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gwack Y., Feske S., Srikanth S., Hogan P.G., Rao A. Signalling to transcription: store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium. 2007;42:145–156. doi: 10.1016/j.ceca.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 50.Tian D., Jacobo S.M., Billing D., Rozkalne A., Gage S.D., Anagnostou T., Pavenstadt H., Hsu H.H., Schlondorff J., Ramos A., Greka A. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal. 2010;3:ra77. doi: 10.1126/scisignal.2001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goel M., Sinkins W.G., Zuo C.D., Estacion M., Schilling W.P. Identification and localization of TRPC channels in the rat kidney. Am J Physiol Renal Physiol. 2006;290:F1241–F1252. doi: 10.1152/ajprenal.00376.2005. [DOI] [PubMed] [Google Scholar]
- 52.Hoenderop J.G., Voets T., Hoefs S., Weidema F., Prenen J., Nilius B., Bindels R.J. Homo- and heterotetrameric architecture of the epithelial Ca2+ channels TRPV5 and TRPV6. EMBO J. 2003;22:776–785. doi: 10.1093/emboj/cdg080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hofmann T., Schaefer M., Schultz G., Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H., Ding J., Fan Q., Liu S. TRPC6 up-regulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-{kappa}B translocation. Exp Biol Med (Maywood) 2009;234:1029–1036. doi: 10.3181/0901-RM-11. [DOI] [PubMed] [Google Scholar]
- 55.Nishida M., Onohara N., Sato Y., Suda R., Ogushi M., Tanabe S., Inoue R., Mori Y., Kurose H. Galpha12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J Biol Chem. 2007;282:23117–23128. doi: 10.1074/jbc.M611780200. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y., Jarad G., Tripathi P., Pan M., Cunningham J., Martin D.R., Liapis H., Miner J.H., Chen F. Activation of NFAT signaling in podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2010;21:1657–1666. doi: 10.1681/ASN.2009121253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faul C., Donnelly M., Merscher-Gomez S., Chang Y.H., Franz S., Delfgaauw J., Chang J.M., Choi H.Y., Campbell K.N., Kim K., Reiser J., Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asanuma K., Kim K., Oh J., Giardino L., Chabanis S., Faul C., Reiser J., Mundel P. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest. 2005;115:1188–1198. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]