

Abstract
Oculopharyngeal muscular dystrophy (OPMD) is an autosomal dominant disease caused by an alanine tract expansion mutation in poly(A) binding protein nuclear 1 (expPABPN1). To model OPMD in a myogenic and physiological context, we generated mouse myoblast cell clones stably expressing either human wild type (WT) or expPABPN1 at low levels. Transgene expression is induced on myotube differentiation and results in formation of insoluble nuclear PABPN1 aggregates that are similar to those observed in patients with OPMD. Quantitative analysis of PABPN1 in myotube cultures revealed that expPABPN1 accumulation and aggregation is greater than that of the WT protein. We found that aggregation of expPABPN1 is more affected than WT PABPN1 by inhibition of proteasome activity. Consistent with this, in myotube cultures expressing expPABPN1, deregulation of the proteasome was identified as the most significantly perturbed pathway. Differences in the accumulation of soluble WT and expPABPN1 were consistent with differences in ubiquitination and rate of protein turnover. This study demonstrates, for the first time to our knowledge, that, in myotubes, the ratio of soluble/insoluble expPABPN1 is significantly lower compared with that of the WT protein. We suggest that this difference can contribute to muscle weakness in OPMD.
Single amino acid (or trinucleotide) repeat expansions are recognized to be a common cause of human disease. Although most of these expansion mutations generate increasing lengths of polyglutamine tracts that cause progressive neurodegenerative disorders, such as Huntington's disease,1 polyalanine tract expansions have also been identified as underlying several monogenic disorders.2,3 The expanded mutant proteins often accumulate in salt-resistant insoluble bodies and, thus, can also cause diseases classified as protein aggregation disorders, which include progressive neurodegenerative diseases, such as Alzheimer's and Parkinson's diseases.4 Among these mutant proteins, a polyalanine tract expansion in the gene encoding for poly(A) binding protein nuclear 1 (PABPN1) causes oculopharyngeal muscular dystrophy (OPMD).5 PABPN1 is a nuclear protein that is normally localized to nuclear interchromatin granules (speckles). Together with poly(A) polymerase, it regulates the elongation of mRNA poly(A) tails.6,7 Despite the ubiquitous expression of PABPN1, patients with OPMD show late-onset symptoms in restricted subsets of muscles.8–10 A key pathological hallmark of OPMD is the presence of aggregated mutant PABPN1 (expPABPN1) in insoluble intranuclear inclusions (INIs) in the nuclei of skeletal muscle fibers.11,12 Naturally occurring wild-type (WT) PABPN1 inclusions with a fibrillar-type structure have also been reported.13,14 In contrast to INI in OPMD myofibers, inclusions of the WT protein are not associated with disease.15–17 The mechanisms by which expPABPN1 causes muscle weakness are not well understood.
ExpPABPN1 overexpression precedes muscle weakness and INI formation in animal models.17–20 In these animal model systems, anti-aggregation treatments reduced muscle symptoms and INI formation19,21,22; however, the molecular mechanisms by which expPABPN1 is toxic to cells have not been fully elucidated. Increased frequency of cell death is found in animal and cellular models with expPABPN1 overexpression,9 but cell death and overexpression of expPABPN1 have not been reported in heterozygous patients with OPMD. In addition, muscle atrophy was found in an OPMD mouse model with high overexpression of expAPBPN1.23 In humans, muscle atrophy was reported in rare homozygous patients,24 whereas in heterozygous patients, muscle atrophy is not a common pathological characteristic of the disease in its early stages.18 In a mouse model with low and constitutive expPABPN1 expression, only minor muscle defects were reported without obvious muscle atrophy.25 Because PABPN1 plays an essential role in diverse cellular functions, such as proliferation and differentiation, manipulating PABPN1 expression beyond physiological concentrations can cause myogenic defects,26–28 which may not be reflective of OPMD pathophysiological features.
To study the specific effect of expPABPN1 in muscle cells, we generated mouse myotube cultures that stably express either a 10-residue alanine tract (Ala10), which represents the WT allele, or a 17-residue alanine tract (Ala17), which represents expPABPN1. Both transgenes were expressed at low physiological levels that are similar to endogenous murine Pabpn1. We used the human desmin (DES) locus control region (LCR) and promoter to ensure myotube-specific expression of the human PABPN1 transgene. Comparative transcriptome analysis indicated molecular pathways that are specifically deregulated because of expPABPN1 expression. Among these pathways, proteasome deregulation was also found in patients with OPMD, which also affects PABPN1 protein accumulation. In this myotube cell culture model, quantitative differences between aggregation of WT and expPABPN1 were identified. These differences are associated with accumulation of the soluble protein, which is regulated by the proteasome. Based on our observations, we suggest that expression of expPABPN1 protein in physiologically relevant amounts is not toxic per se, but the reduced amount of the soluble form is likely to be causally involved in muscle weakness.
Materials and Methods
Plasmid Vector Constructions
The human β-globin (HBB) gene micro–LCR-based expression vector, pEV,30 was used as a starting point for the construction of a muscle-specific expression vector based on the human DES gene LCR.31 The HBB-LCR/promoter fragment was removed by digestion with AatII/NotI and replaced by a synthetic polylinker with the following restriction enzyme sites: 5′-AatII-PmeI-XhoI-SnaBI-NheI-ClaI-PmlI-EcoRV-AgeI-NotI-3′. The DES-LCR/promoter transcriptional control region consists of a 7.7-kb fragment from the -16.3-kb XbaI to the -8.6-kb BglII sites, spanning DES-LCR elements HS1-4 linked to the DES promoter region extending from the -1.7-kb XhoI site to 30 bp from the DES transcriptional start site.31 The DES-LCR/promoter combination was isolated as a 9.4-kb fragment (from plasmid construction MA590) using XbaI/XhoI and blunt end ligated into the SnaBI site of the polylinker, to generate the muscle-specific expression vector, DesLCR-EV. Human cDNAs encoding either WT or expPABPN1 were a gift from Prof. Maria Carmo-Fonseca (Institute of Molecular Medicine, Lisbon, Portugal), cloned into the pTRE2Hyg vector (Clontech, Mountain View, CA). These cDNAs were tagged at their C-terminus with the FLAG epitope (DYKDDDDK)32 by PCR using the primers FLAG (5′-TTACTTGTCATCGTCGTCCTTGTAGTCGTAAGGGGAGTGCCATGATGTCG-3′) and PABPN1 (5′-CACGCTGTTTTGACCTCCATAGAAGAC-3′) and cloned into the pCRII TOPO vector (Invitrogen, Carlsbad, CA). These tagged cDNAs were then subcloned into pBluescript (Stratagene, La Jolla, CA) between the Acc65I (KpnI) and SpeI sites. FLAG-tagged cDNAs were finally cloned into DesLCR-EV as Acc65I (blunted)/NotI fragments between the PmlI and NotI sites within the polylinker to generate the PABPN1 expression constructs pDWT (WT FLAG-tagged PABPN1) and pD7 (FLAG-tagged expPABPN1) (Figure 1A). The expPABPN1–green fluorescent protein fusion construct was a gift from Dr. Theo Verrips (Utrecht University, The Netherlands).
Figure 1.
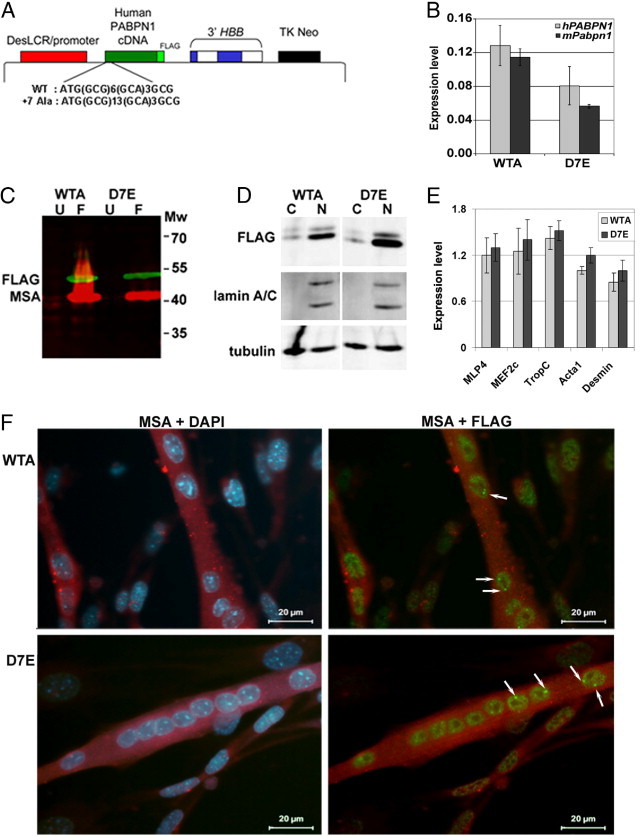
Stably transfected myoblast cell lines expressing either WT or mutant 7Ala-expanded PABPN1. A: Illustration of the muscle-specific DesLCR-EV expression vector containing cDNAs (green box) coding for either human WT (10 Ala; WTA) or 7Ala-expanded (17Ala; D7E) PABPN1. The PABPN1 in both cases is tagged at its C-terminus with the FLAG epitope (DYKDDDDK). The 5′ sequence of the PABPN1 cDNA showing the WT and the mutated poly-Ala track (GCG, GCA) is indicated. The DesLCR/promoter, a muscle-specific element, drives transgene expression in myotubes. The 3′ half of HBB at the 3′ of the expression cassette confers efficient processing, transport, and stability of the mRNA. A neomycin-resistance gene under the control of an HSV-TK promoter allows selection of stably transfected clones. B: A histogram showing expression levels of murine mPabpn1 and human hPABPN1 mRNA expression in WTA and D7E that were induced for myotube fusion for 4 days. The expression level is calculated from ΔΔCT values after normalization to DES and to IM2 parental cells. Error bars are calculated from three replicates. C: PABPN1-FLAG expression in myotubes. Western blot analysis of total protein extracts of unfused (U) and 4 days after myoblast fusion (F) of WTA and D7E cell lines. Molecular weights (Mw) are indicated in kDa. D: Western blot analysis of cytoplasmic (C) and nuclear (N) fractions isolated from 4-day fused WTA and D7E cell lines. Anti-lamin A marks the nuclear fraction, and tubulin was used as a loading control for both fractions. The blot is a representative of three independent experiments. E: RT-qPCR analysis of muscle differentiation markers in WTA and D7E cell lines. Expression levels were calculated from ΔΔCT values after normalization to murine Hrpt and to IM2 parental cells. Error bars are calculated from three replicates. F: IHC of PABPN1-FLAG in WTA and D7E cell lines 4 days after fusion. PABPN1-FLAG is detected with an anti-FLAG antibody, and differentiated myotubes are detected with an MSA antibody. Nuclei are counterstained with DAPI. Arrows show INIs.
Cell Culture and Generation of Stably Transfected IM2 Cell Lines
Primary mouse myoblasts (clone IM2) conditionally immortalized with a temperature-sensitive SV40 large T-antigen (tsA58) transgene and derived from the ImmortoMouse33 were maintained in Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 20% fetal bovine serum (Invitrogen), 0.5% chicken embryo extract (PAA Laboratories, Somerset, UK), 100 U/mL penicillin-streptomycin, 2 mmol/L l-glutamine, and 20 U/mL interferon-γ (HyCult Biotech, Plymouth Meeting, PA) at 33°C in a humidified 10% CO2 air atmosphere. Terminal differentiation of myoblast cultures to myotubes was achieved by plating cells at a high density, allowing growth to confluency, changing to nonmitotic fusion media (Dulbecco's modified Eagle's medium containing 5% horse serum without interferon-γ), transferring the flasks or plates to the nonpermissive temperature of 37°C in a humidified 5% CO2 air atmosphere, and culturing for up to 5 days. Cell treatment with 5 μmol/L MG132 (Sigma-Aldrich, St. Louis, MO) was performed with 4-day myotube cultures. MG132 was added to the fusion medium 8 hours before harvesting. Dimethylsulfoxide treatment was used as a control.
To establish the IM2-derived stable cell lines containing either the pWT or pD7 constructs, 107 cells were transfected with 25 μg of PvuI-linearized plasmid by electroporation using a BioRad Gene Pulser (BioRad, Hercules, CA), set to deliver a single pulse at 960 mF at 250 V. The transfected cells were left for 24 hours to recover, after which antibiotic (Geneticin sulfate, G418; Invitrogen) was added to a concentration of 500 μg/mL. Clonal cell lines were then isolated by serial dilution of G418-resistant pools of cells obtained 10 days after application of selection by replating in 24-well tissue culture plates at a low density to avoid cell cross contamination. G418-resistant clones were subcultured and maintained in 500 μg/mL G418. Individual clones were analyzed for copy number and transgene integrity by Southern blot analysis of BamHI-digested genomic DNA. DNA from untransfected IM2 cells was used as a negative control. Blots were hybridized with a 1-kb probe extending from the -16.3-kb XbaI to the -15.3-kb XmaI site, spanning HS4 of the DES LCR region to determine copy number and integrity. Clones with single copy number were assayed for transgene expression level using quantitative PCR (qPCR). Clones with an expression level of human PABPN1 that was similar to that of the endogenous mouse PABPN1 were selected for further studies.
RT-qPCR
Total RNA was extracted from myotube cultures after 4-day differentiation using TRIzol reagent (Invitrogen), according to the manufacturer's instructions. First-strand cDNA was synthesized with random hexamer oligonucleotides and Moloney murine leukemia virus reverse transcriptase (First Strand kit; Fermentas, Burlington, Ontario, Canada), according to the manufacturer's instructions. A 3.6-ng aliquot of cDNA was used for qPCR analysis using SYBR Green mix buffer (BioRad), in a 15-μL reaction volume. The PCR was performed as follows: 4 minutes at 95°C, followed by 40 cycles of 10 seconds at 95°C and 60 seconds at 60°C. The program was ended with 1 minute at 60°C. Primer sets used in this study are indicated in Table 1. The specificity of RT-PCR products was determined by melting curve analysis for each of the primer sets. Expression levels were calculated according to the ΔΔCT method, normalized to murine Hrpt and IM2 parental cells. Statistical significance was determined with the Student's t-test.
Table 1.
Primers Used for RT-qPCR Analysis
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Mef2C | 5′-TTGAAAACTGTGAAGTAACCTC-3′' | 5′-TTCCCAAATCCCTGCATTCG-3′ |
| Mlp4 | 5′-CAAGGACTCTGCCTTTGACC-3′ | 5′-ATCTCTCCCGTTGGAGTCC-3′ |
| TropC | 5′-GTTCGGTGCATGAAGGACG-3′ | 5′-CTCATCTAAGTCAATGTAGCC-3′ |
| Acta1 | 5′-CGAGGTATCCTGACCCTGAA-3′ | 5′-AGGTGTGGTGCCAGATCTTC-3′ |
| Desmin | 5′-TCGCGGCTAAGAACATCTCT-3′ | 5′-GCATCAATCTCGCAGGTGTA-3′ |
| mHprt | 5′-CGTCGTGATTAGCGATGATG-3′ | 5′-TTTTCCAAATCCTCGGCATA-3′ |
| PABPN1-Flag | 5′-CGTTGGCAATGTGGACTATG-3′ | 5′-ACACGGTTGACTGAACCACA-3′ |
| Mouse Pabpn1 | 5′-CGCTCTCGATTCTACAGTGGTTT-3′ | 5′-AGGAGAGAGAGGAGGATTATGTGTGAA-3′ |
| Psme2 | 5′-CCTGGAGAGTGAAAGCGAAA-3′ | 5′-GTCATCAGCCTCCTGGAAAA-3′ |
| Psme3 | 5′-GCGAAGGTCAAACCCATAGA-3′ | 5′-GAAAGTGATGCATCCCAGGT-3′ |
| PsmB5 | 5′-CCTCCAAACTGCTCGCTAAC-3′ | 5′-CTGTTCCCCTCGCTGTCTAC-3′ |
| PsmB7 | 5′-CCTCCAAACTGCTCGCTAAC-3′ | 5′-CTGTTCCCCTCGCTGTCTAC-3′ |
| PsmB10 | 5′-ATTTGCTCCTGGAACCACC-3′ | 5′-CCACTTCATTCCACCTCCAT-3′ |
| PsmD14 | 5′-CACCTGAACAGCTGGCAATA-3′ | 5′-GAGCATTGGGAACGAAGAAG-3′ |
Microarray and Transcriptome Analysis
RNA from 4-day fused WTA and D7E myotube cultures (in triplicate) with RIN >9 were used for labeling with the Illumina TotalPrep RNA Amplification kit (Ambion, Carlsbad, CA) and subsequently hybridized to Illumina (San Diego, CA) mouse v2 Bead arrays. Microarray analysis and normalization were performed as previously described.34 The Microarray data sets are available [Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo, last accessed September 1, 2011; accession no. GSE29909]. A list of D7E-deregulated genes was obtained with a P = 0.05 cutoff. The global test35 was used to identify significant Gene Ontology categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in D7E cells. Clusters with the multiple testing adjusted (Holm's method, P < 0.05) were selected as significant.
Protein Analyses
For Western blot analysis of total protein, cell pellets were dissolved in one times SDS loading buffer [50 mmol/L Tris (pH 6.8), 10 mmol/L dithiothreitol (freshly added), 2% SDS, 0.1% bromophenol blue, and 20% glycerol], followed by heat denaturation for 5 minutes before resolution by SDS-PAGE. The soluble fraction of proteins was obtained with an extraction buffer composed of 20 mmol/L Tris (pH 7.5), 10% glycerol, 150 mmol/L NaCl, and 5 mmol/L EDTA, complemented with protease inhibitor (SigmaFAST protease inhibitor; Sigma-Aldrich). Fractionation of cytoplasmic and nuclear fractions was performed with the NuCLEAR extraction kit (Sigma-Aldrich), according to the manufacturer's protocol. For immunoprecipitation and pull-down experiments, 0.1% NP40 (Sigma-Aldrich, St. Louis, MO) was added to the extraction buffer, and lysates were sonicated for 5 seconds. For de-ubiquitinating enzyme (DUB) inhibition, 10 mmol/L iodoacetamide (Sigma-Aldrich) or 5 nmol/L PR619 (TEBU-Bio, Le Perray, France) was added to the extraction buffer. Immunoprecipitation and pull down were conducted with protein aliquots of 300 to 400 and 700 μg, respectively. Rabbit anti-FLAG antibody (Sigma-Aldrich) was used for immunoprecipitation of either WT or expPABPN1-FLAG, with the immune complex isolated with Protein A Sepharose for Fast Flow (GE Healthcare, Waukesha, WI). Pull down of poly-ubiquitinated proteins with tandem ubiquitin-binding entities (TUBEs; TEBU-Bio) was performed overnight according to the manufacturer's protocol. After extensive washing of the beads, proteins were heat denaturated and resolved by SDS-PAGE on 10% polyacrylamide gels, followed by Western blot analysis using the following primary antibodies: mouse monoclonal anti-FLAG M2 (Sigma-Aldrich), mouse monoclonal anti-muscle actin (MSA; Novocastra, Newcastle on Tyne, UK), mouse monoclonal anti-tubulin or mouse anti-actin (Sigma-Aldrich), anti-ubiquitin (FD41; Cell Signaling Technology, Danvers, MA), and mouse monoclonal anti-lamin A (Santa Cruz), used at a dilution of 1:1000 to 1:3000. Detection of the first antibodies was conducted with the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE) and suitable secondary antibodies. Western blot quantification was performed with the ImageJ image processing program (http://rsbweb.nih.gov/ij, last accessed April 8, 2011). Statistical analysis was performed with Microsoft Excel software (Redmond, WA).
Immunofluorescence and Fluorescence in Situ Hybridization
Cells were cultured on glass microscope slide coverslips, and immunofluorescence analysis was performed as previously described.36 Immunolabeled cells were mounted in Vectashield (Vector, Peterborough, UK) or Vectashield containing DAPI (Sigma-Aldrich) at a concentration of 3 ng/L. The primary antibodies used were as follows: mouse monoclonal anti-FLAG M2 and rabbit anti-FLAG (Sigma-Aldrich); human anti-SC35 serum (a gift from Prof. Maria Carmo-Fonseca, Institute of Molecular Medicine, Lisbon, Portugal); rabbit anti-20S proteasome (donated by Dr. John C. Morris, Washington University School of Medicine, St. Louis, MO); sheep anti-human ubiquitin (Abcam, Cambridge, MA); rabbit polyclonal anti-poly(A) polymerase (AutogenBioclear, Wiltshire, UK); Myc-tagged 3F5 llama single-chain antibody,37 which is recognized by mouse anti-Myc clone SE50 (Sigma-Aldrich); mouse anti-MSA (Novocastra); and rabbit cleaved caspase 3–specific antibody (R&D Systems, Minneapolis, MN). Secondary antibodies, conjugated to Alexa 488, Alexa 430, or Alexa 594, were obtained from Molecular Probes, Invitrogen, Carlsbad, CA. Anti-rabbit horseradish peroxidase conjugate was visualized after incubation with 3,3 diaminobenzidine substrate.
Fluorescence in situ hybridization was performed using biotinylated 2′-O-alkyl polyuridine, as previously described.10 Images were recorded with a Zeiss Axiovert, Oberkochen, Germany (model 135TV) fluorescence microscope equipped with a 100-W mercury arc lamp and a ×40/1.3 NA plan Apo objective. Single confocal sections were obtained with a Leica TCS/SP2 confocal microscope system equipped with a ×100/1.4 NA plan Apo objective.
Results
Expression of WT or expPABPN1 in Mouse Myotube Cultures at Low Expression Levels Leads to INI Formation
Previous models of OPMD have demonstrated that high overexpression of expPABPN1 leads to INI formation.10,27,28,38–42 However, so far, it has not been investigated as to how expPABPN1 leads to muscle weakness at a low expression level. This question is relevant, because high overexpression is not observed to occur in heterozygous patients with OPMD. We generated a muscle-cell model for OPMD, in which transgene expPABPN1 is expressed at a similar level to endogenous PABPN1 mRNA, and, therefore, can be considered as being more representative of the true physiological condition in OPMD. To study the specific effect of mutant expPABPN1, a clone expressing the WT (Ala10) PABPN1 at a low expression level was also generated. The human PABPN1 transgene was expressed under the control of the human Des-LCR and promoter31 (Figure 1A), and mRNA was stabilized with the use of the 3′ half of the HBB gene.43–46 Stable clones of ImmortoMouse myoblasts (clone IM2)33 were generated, and the transgene mRNA expression level was determined using RT-qPCR. The D7E and WTA clones, expressing the Ala17 or Ala10 alleles, respectively (Figure 1, A and B), were selected for further study. Within these clones, the transgene mRNA expression levels were similar to that of endogenous Pabpn1 (Figure 1B); therefore, we consider the transgene expression levels as physiologically relevant.
The PABPN1 transgene was tagged at the 3′ end with sequences that express the FLAG epitope, allowing specific detection of the transgene mRNA and protein products. The expression of PABPN1-FLAG in myotubes was verified by using Western blot analysis (Figure 1C). Its expression was only residual in unfused myoblast cells. The muscle-actin (MSA) protein was used as a marker for fused myoblasts (Figure 1C). In these clones, the human PABPN1 protein product was mostly localized to the nucleus, and only a small fraction of the protein was found in the cytosol (Figure 1D). More important, no differences in subcellular accumulation were found between PABPN1 in the WTA and D7E myotubes (Figure 1D). Although we cannot exclude an effect of FLAG on PABPN1 protein distribution, the predominant nuclear localization of PABPN1-FLAG indicates a similar localization to that of endogenous PABPN1. In addition, in a recent study,29 we demonstrated that green fluorescent protein tagging affected neither PABPN1 nuclear localization nor its mobility. Thus, green fluorescent protein or FLAG epitope tagging of PABPN1 seems to have little effect on its localization and mobility. Because the subcellular localization of WT and expPABPN1 were similar in this cell model, we conclude that if FLAG tagging has an effect on PABPN1 protein, this would be similar on both WT and expPABPN1.
To determine whether, at low expression levels, WT or expPABPN1 leads to myogenic defects, the expression of Mef2C, Mlp4, TropC, Acta1, and Des was compared using RT-qPCR. The expression of these genes significantly changed on differentiation of myoblasts (see Supplemental Figure S1 at http://ajp.amjpathol.org); thus, the expression profile of these genes can mark skeletal myoblast muscle differentiation. The level of expression of these genes did not significantly change between WTA and D7E myotube cultures (Figure 1E). In addition, immunohistochemical (IHC) analysis of myotubes did not reveal obvious morphological differences between WTA and D7E (Figure 1F). These results indicate that if myogenic differences occur between the WTA and D7E clones, they are minor.
Nuclear expPABPN1 localizes to nuclear speckles, which are marked with the splicing factor SC35.16 Also, in our cell model, PABPN1-FLAG colocalized with SC35 in unfused cells that were incubated in fusion conditions for only 1 day (Figure 2A). This further indicates that FLAG tagging did not disrupt the nuclear localization of the PABPN1 transgene products. After 4 days of myoblast fusion, intense fluorescent foci were detected (INIs, Figure 1F and Figure 2A) and were visualized by either anti-FLAG or the single-domain antibody fragment to PABPN1, VHH-3F5. In contrast to myotubes from the WTA or D7E clones, in the IM2 clone, fluorescent foci of PABPN1 were not detected (Figure 2A). This indicates that PABPN1 foci are formed by the expression of the transgene protein product. PABPN1-FLAG foci were resistant to high salt extraction with 1 mol/L KCl (Figure 2B), indicating that these PABPN1 foci contained aggregated PABPN1. In addition, PABPN1 foci colocalized with ubiquitin (Figure 2, A and B), suggesting that the PABPN1 foci are related to INIs observed in vivo. Furthermore, these PABPN1 foci in our cell model system also colocalized with poly(A)-RNA and poly(A) polymerase but not with SC35 (Figure 2C). Taken together, these observations suggest that the expression of PABPN1 in myotube cultures of the WTA or D7E clones led to protein accumulation in insoluble nuclear structures that resembled the INIs that are formed in affected patient muscles.10
Figure 2.
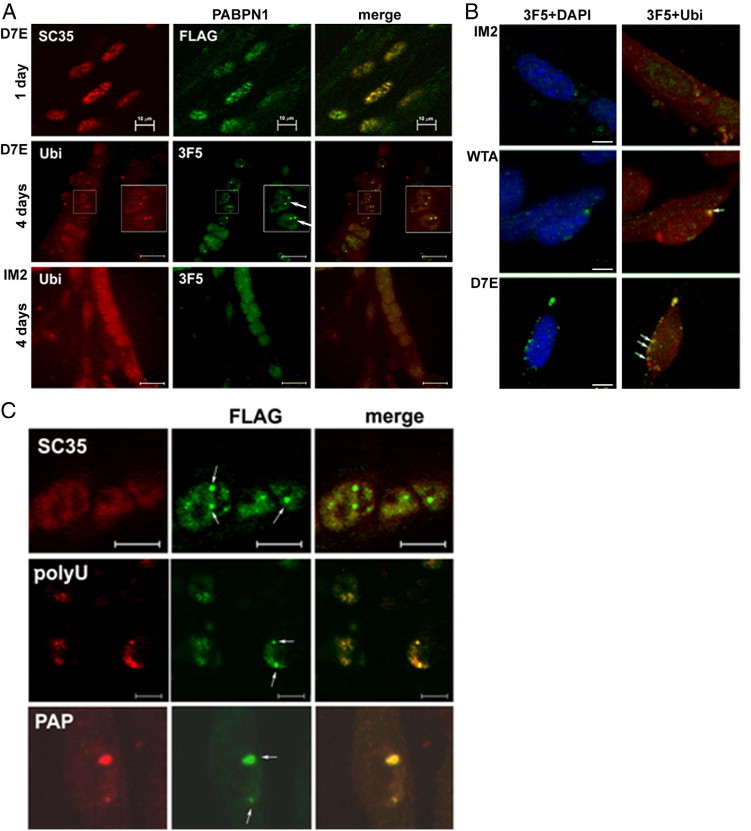
INIs are formed in WTA and D7E cells lines. A: INIs are formed in D7E myotubes but not in myoblasts or in IM2 myotubes. In unfused cells (day 1), PABPN1-FLAG colocalizes with SC35, indicating its localization in speckles. Scale bar = 10 μm. In 4-day myotube cultures of D7E, but not of IM2 PABPN1, fluorescent foci, labeled with VHH-3F5, colocalize with ubiquitin (Ubi). Scale bar = 20 μm. Original magnification, ×1.5 (of nuclei with INIs, highlighted in boxes). B: INIs in D7E and WTA are KCl resistant. At 4 days after fusion, cultures were treated with 1 mol/L KCl before IHC with VHH-3F5 (green) and Ubi (red). Nuclei were counterstained with DAPI. Arrows, INI. Scale bar = 5 μm. C: INIs colocalize with polyU and poly(A) polymerase (PAP) but not with SC35. Myotubes of D7E were coimmunolabeled with SC35, polyU, or PAP. The PABPN1-FLAG protein is labeled with an anti-FLAG antibody. Scale bar = 5 μm. Arrows, INI.
Comparative Transcriptome Analysis of D7E and WTA Myotube Cultures Reveals Similar Deregulated Pathways as in the OPMD Mouse Model and in the Muscles of Patients with OPMD
Next, we investigated the physiological relevance of our muscle-cell model to OPMD by conducting a comparative transcriptome study. This was performed on microarrays that were hybridized with RNA from myotube cultures of the WTA or D7E clones. Deregulated KEGG pathways were identified by global test analysis, with a significance level of P < 0.05. In addition, the D7E-deregulated KEGG pathways were considered as significant when more than five genes are deregulated and represented >10% of the genes in a given pathway (see Supplemental Table S1 at http://ajp.amjpathol.org). In a study comparing D7E with WTA, a total of 1043 genes were deregulated. These were mapped to only 37 KEGG pathways (Figure 3). The relevance of the D7E transcriptome to OPMD was further addressed by comparing the D7E transcriptome with that obtained from skeletal muscles of the mouse model for OPMD (A17.1)23 and patients with OPMD.29 High overlap was found between the D7E-deregulated pathways and the A17.1- and human OPMD-deregulated pathways (see Supplemental Table S1 at http://ajp.amjpathol.org). Most differences were found in the metabolic cluster, likely reflecting differences in growth and environment between the in vivo and in vitro conditions. In contrast to the moderate gene deregulation in the D7E myotube culture, the mouse model exhibited massive gene deregulation, with 6342 genes being differentially expressed between OPMD (A17.1) and WT mice. This amount of differential gene expression is more than sixfold greater than in our murine cell culture model. Because the mouse model was generated with a high overexpression of PABPN1, it is possible that the difference in the amount of deregulated genes between the two model systems is, at least in part, because of differences in expPABPN1 expression levels. To explore the possibility that overexpression of PABPN1 affects gene expression, two clones that express WT-PABPN1 at different levels (Named as WTA and WTD. In the WTD clone PABPN1-FLAG fold change is 6-fold lower compared with the expression in the WTA clone; see Supplemental Figure S2 at http://ajp.amjpathol.org) were also subjected to transcriptome analysis. A total of 1047 genes that were mapped to 41 KEGG pathways were deregulated because of differences in PABPN1 transgene expression levels (see Supplemental Table S2 at http://ajp.amjpathol.org). These WTA-deregulated genes were mapped to 41 KEGG pathways, and 40 were also deregulated in the A17.1 mouse model of OPMD (see Supplemental Table S2 at http://ajp.amjpathol.org). Therefore, these KEGG pathways could reflect transcriptional changes that are solely attributable to PABPN1 expression levels.
Figure 3.
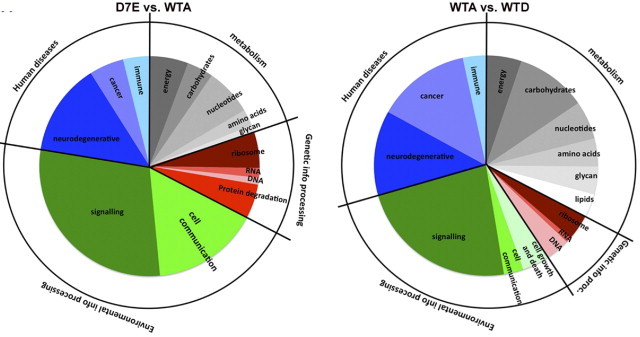
Transcriptome study in D7E and WTA myotubes. Pie chart analysis of KEGG pathways that are deregulated in D7E (left) or WTA (right) myotubes. The inner pie shows the subcategories, and the outer ring shows the major categories.
To identify transcriptional changes that are specifically caused by expPABPN1, we compared these two transcriptome studies in myotube cultures. The cell cycle category and Wnt signaling pathway were solely deregulated because of PABPN1 expression levels (Table 2). In addition, the proportion of deregulated genes in metabolic pathways is significantly larger in the study comparing the effect of expression level (Figure 3). In the A17.1 mouse, apoptosis was listed among the top 20 deregulated pathways.23 The activation of apoptosis in A17.1 and in cellular models with high overexpression of OPMD has been subjected to extensive studies.22,47,48 In our D7E clone transcriptome analysis, the apoptosis pathway was not identified as being significantly deregulated (see Supplemental Table S1 at http://ajp.amjpathol.org). The activation of apoptosis was not detected in D7E myotube cultures by TUNEL assay or with antibody staining against cleaved caspase-3 (see Supplemental Figure S3 at http://ajp.amjpathol.org). Therefore, we conclude that apoptosis is activated by high overexpression of expPABPN1 but not at low physiologically relevant levels. This conclusion is consistent with our recent studies29 in U2OS cells, in which we demonstrated that only at high, but not at low, expression levels, expPABPN1 leads to caspase-3 activation. Because apoptosis has not been reported in affected muscles of patients with OPMD, we suggest that, at low expression levels, cell toxicity is most likely caused by other mechanisms.
Table 2.
KEGG Pathways That Are Specific for the WTA-WTD or D7E-WTA Studies
| KEGG ID | KEGG pathway | P value | No. of genes/pathway | Deregulated genes |
|
|---|---|---|---|---|---|
| No. | % | ||||
| D7E deregulated | |||||
| 3050 | Proteasome | 8.54E-05 | 45 | 10 | 22.2 |
| 4120 | Ubiquitin-mediated proteolysis | 2.45E-02 | 131 | 15 | 11.5 |
| 4530 | Tight junction | 8.17E-03 | 121 | 17 | 14.0 |
| 4520 | Adherens junction | 4.19E-02 | 73 | 10 | 13.7 |
| 4510 | Focal adhesion | 4.21E-02 | 186 | 21 | 11.3 |
| 4810 | Regulation of actin cytoskeleton | 2.94E-02 | 199 | 24 | 12.1 |
| 4910 | Insulin signaling pathway | 1.04E-02 | 132 | 16 | 12.1 |
| 4912 | GnRH signaling pathway | 1.23E-02 | 96 | 12 | 12.5 |
| WTA deregulated | |||||
| 4110 | Cell cycle | 9.47E-03 | 111 | 15 | 13.5 |
| 4310 | Wnt signaling pathway | 1.11E-02 | 143 | 17 | 11.9 |
For each KEGG pathway P value, the number of deregulated genes and the corresponding percentage from the total annotated genes are indicated.
In this comparative study, few pathways were identified as being specific to the expPABPN1-expressing D7E cell model (Table 2). The D7E-specific deregulated pathways can be categorized into three classes: pathways that are associated with the muscle-cell structure (pathways of tight and adherens junctions, focal adhesion, and the cytoskeleton), the insulin-signaling pathway, and the ubiquitin proteasome system (Table 2). More important, these pathways are commonly deregulated in patients with D7E and OPMD (see Supplemental Table S1 at http://ajp.amjpathol.org). This strongly suggests that they are involved in OPMD disease etiology.
expPABPN1 Aggregation Is Regulated by the Proteasome
The ubiquitin proteasome system was recently identified as the most consistently and significantly deregulated pathway in a cross-species study29 for OPMD. In the D7E transcriptome, the proteasome pathway had the lowest P value, suggesting the strongest association with expPABPN1 expression (see Supplemental Table S1 at http://ajp.amjpathol.org). Approximately 20% of the proteasome-encoding genes that are annotated to KEGG or Gene Ontology were deregulated in D7E myotubes, among which 80% were down-regulated. Validation of the microarray was performed by RT-qPCR (Figure 4A).
Figure 4.
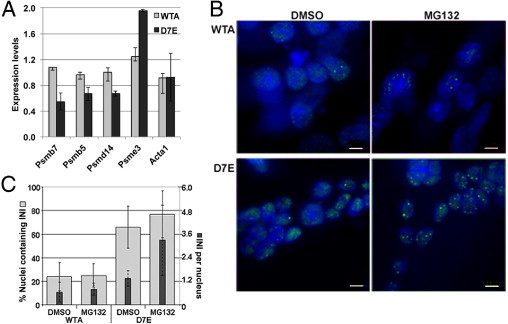
Proteasome down-regulation is associated with protein aggregation in D7E. A: RT-qPCR analysis of proteasome-encoding genes in 4-day myotube cultures of WTA and D7E. Expression levels were calculated from ΔΔCT values after normalization to murine Hrpt and to IM2 parental cells. Error bars are calculated from two biological and six technical replicates. B: The effect of 5 μmol/L MG132 treatment on PABPN1 aggregation. Myotube cultures of WTA and D7E at 4 days after fusion were treated with 5 μmol/L MG132 for 8 hours. Dimethylsulfoxide (DMSO) treatment was used as a control. Protein aggregation and INI formation are detected by immunofluorescence using an anti-FLAG antibody, and nuclei are counterstained with DAPI. Scale bar = 5 μm. C: Histograms show the percentage of nuclei containing PABPN1 INI (gray bars) and the average number of INIs per nucleus (black bars). SDs represent 10 myotubes containing 90 nuclei in total.
We further assessed whether aggregation of PABPN1 is regulated by the proteasome. In this experiment, PABPN1 aggregation was evaluated by INI formation in myonuclei (Figure 4B). More myonuclei containing INIs were found in D7E compared with WTA (Figure 4C). This observation confirms our previous study29 and indicates that, when PABPN1 is expressed at low levels, the aggregation potential of the Ala-expanded PABPN1 is significantly higher than of WT PABPN1 (Figure 4C). The effect of the proteasome on INI formation was analyzed by treatment of myotube cultures with the proteasome inhibitor, MG132. We observed an approximate 20% increase in nuclei containing INIs in D7E myotubes, whereas the effect in WTA myotubes was less pronounced (Figure 4B). Of greater significance, the number of INIs per nucleus was higher after treatment with MG132 in D7E, but not in WTA, myotubes (Figure 4C). This suggests that aggregation of WT and expPABN1 is differentially regulated by the proteasome. Because proteasome inhibition affects expPABPN1 aggregation in non-muscle cells,38 proteasome regulation of expPABPN1 accumulation may not be muscle specific.
Differences between expPABPN1 and WT PABPN1 Protein Accumulation
Protein aggregation can result from protein accumulation. The comparable expression levels of expPABPN1 and WT PABPN1 mRNA in our cell models facilitate a system for accurate quantification of protein accumulation. Aggregated PABPN1 proteins are resistant to Triton X-100 treatment,29 and when extracted with SDS-PAGE loading buffer, the aggregated PABPN1 accumulates in the stacking gel (Figure 5A). A clear signal in the stacking gel was found in protein extracts from D7E myotubes, whereas that from WTA was minor. Quantitative analysis demonstrated a consistent and significant accumulation of expPABPN1 compared with the WT protein (Figure 5B). Surprisingly, the accumulation of the soluble 53-kDa PABPN1 was also higher in D7E compared with the WT protein in WTA myotube cultures (Figure 5A). After normalization to MSA expression, which controls for fusion variation between experiments, revealed that differences between WT and expPABPN1 protein accumulation are significant (Figure 5B).
Figure 5.
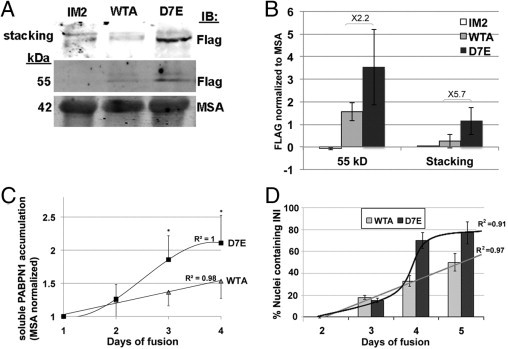
expPABPN1 aggregation potency is higher than WT-PABPN1 in myotubes. A: Western blot analysis of total protein extracts isolated from IM2, WTA, and D7E myotubes. PABPN1-FLAG is detected with an anti-FLAG antibody. Aggregated PABPN1-FLAG is retained in the stacking gel, and the soluble protein migrates at 53 kDa. Muscle skeletal actin (MSA) is used as a loading control. B: Quantification of PABPN1-FLAG accumulation in IM2, WTA, and D7E myotubes at 4 days after fusion. The histogram shows the FLAG signal after normalizing to actin. Averages are calculated from six independent experiments. C: Quantification of soluble PABPN1-FLAG protein accumulation in WTA and D7E during cell fusion. Values show the FLAG signal normalized to MSA. Averages are calculated from six replicates. *P < 0.05. Linear or sigmoid curve fits of PABPN1-FLAG accumulation in WTA or D7E, respectively (R2 values are indicated). D: INI formation in WTA and D7E cell lines during myoblast cell fusion. INIs were visualized with an anti-FLAG antibody. Histograms show the percentage of nuclei containing PABPN1 INI from 80 to 100, counted in 20 myotubes at 2, 3, 4, and 5 days after fusion. A linear or sigmoid curve fits INI formation in WTA or D7E, respectively (R2 values are indicated).
Next, we investigated whether the difference in accumulation of soluble WT and expPABPN1 is time-dependent. Similar levels of protein accumulation were found in cultures at 2 days after fusion, but a significant increase in accumulation of expPABPN1 was observed at days 3 and 4 (Figure 5C). Curve fitting indicated that WT protein accumulation is linear over time, whereas that of expPABPN1 fits a sigmoid curve. This temporal difference suggests differential regulation of soluble WT and expPABPN1 protein accumulation.
To further investigate the temporal differences between WTA and D7E, we compared INI formation during myotube formation. Nuclear foci of PABPN1-FLAG were quantified in myotubes between 2 and 4 days after fusion conditions by immunofluorescence using anti-FLAG antibodies. INIs in D7E myotubes were more abundant than in the WTA equivalent (Figure 5D). INI formation in WTA during myotube formation fits a linear curve, whereas that of D7E fits a sigmoid curve. This result suggests that the rate of INI formation reflects the accumulation pattern of soluble PABPN1 protein.
Soluble expPABPN1 Protein Turnover Is Regulated by the Proteasome
Because differences between WT and expPABPN1 INI formation seem to be the result of differential accumulation of the soluble protein, we next investigated whether differences in protein turnover are present. Myotube cultures at 4 days after fusion were treated with cyclohexamide for 6 and 19 hours. During this incubation period, the myotubes remained intact, whereas a longer incubation period (24 hours) was toxic. Although the accumulation of soluble WT PABPN1 was dramatically reduced by 6 hours of cyclohexamide treatment (Figure 6A), the accumulation of expPABPN1 was essentially unaffected, even after 19 hours of treatment (Figure 6B). Based on this analysis, the protein turnover for WT PABPN1 is significantly shorter than that of expPABPN1. This difference in protein turnover can explain the differences in accumulation and aggregation of WT and expPABPN1 proteins.
Figure 6.
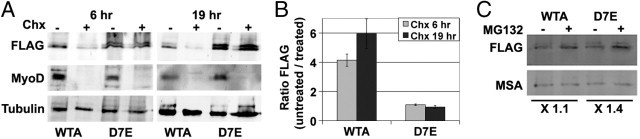
Soluble expPABPN1 protein has slow turnover, and its accumulation is affected by proteasome inhibition. A: Cyclohexamide treatment (Chx; 10 μmol/L) of 3-day myotube cultures was conducted for 6 or 19 hours. Extracts of soluble protein were resolved by SDS-PAGE. The blot image shows the 53-kDa PABPN1-FLAG. MyoD is a control for cyclohexamide treatment, and tubulin is a loading control. B: Histograms show the ratio of FLAG signal between cyclohexamide untreated and treated WTA and D7E myotube cultures. SDs represent two experiments. C: Myotube cultures of WTA and D7E at 4 days after fusion were treated with 5 μmol/L MG132 for 8 hours. Dimethylsulfoxide treatment was used as a control. The soluble PABPN1 protein was detected by using Western blot analysis. PABPN1-FLAG was detected with anti-FLAG antibody; MSA is used as a fusion marker and as a loading control. The fold change of the FLAG signal between treated and untreated WTA and D7E myotubes is indicated.
Because aggregation of PABPN1 is regulated by the proteasome, we next studied whether the proteasome also affects accumulation of the soluble protein. Treatment with MG132 causes an increase in accumulation of soluble PABPN1 (Figure 6C). However, the effect of MG132 treatment on expPABPN1 accumulation was more pronounced compared with that of the WT protein. This suggests that turnover of soluble PABPN1 protein is regulated by the ubiquitin proteasome system.
Soluble WT-PABPN1 Is Poly-Ubiquitinated to a Higher Degree Compared with expPABPN1
If the accumulation of soluble PABPN1 is regulated by the proteasome, variations in PABPN1 ubiquitination could possibly explain the differences in accumulation of soluble WT and expPABPN1. Consistent with the higher accumulation of soluble expPABPN1 in myotube cultures, more immunoprecipitated PABPN1 was obtained from D7E compared with WTA cells (Figure 7A). Immunoprecipitation with the FLAG antibody was specific to the FLAG-tagged protein because no PABPN1 signal was detected in control parental IM2 myotubes (Figure 7A). The ubiquitinated PABPN1 protein was detected in cell extracts prepared in the presence of iodoacetamide, an inhibitor of protein DUB. In contrast to the protein extraction conditions without DUB inhibition, in the presence of iodoacetamide, more PABPN1 protein was immunoprecipitated from WTA than from D7E extracts (with or without iodoacetamide, respectively; IB: FLAG; Figure 7B). This suggests that accumulation of soluble WT PABPN1 is regulated by DUB. Western blot analysis of the immunoprecipitated proteins with an anti-ubiquitin antibody detects PABPN1 at a molecular weight close to 58 kDa and the poly-ubiquitinated species (Figure 7B). Because the molecular weight of PABPN1-FLAG is 53 kDa, it is possible that the 58-kDa protein represents a mono-ubiquitinated form of PABPN1. More important, in WTA, increased levels of mono- and poly-ubiquitinated WT PABPN1 species were detected with the anti-ubiquitin antibody compared with D7E (Figure 7B). This implies that the higher rate of turnover of soluble WT PABPN1 results from its higher degree of ubiquitination compared with soluble expPABPN1 protein.
Figure 7.
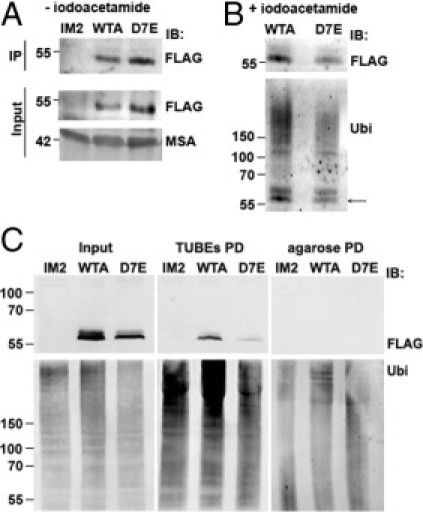
Ubiquitination of soluble PABPN1. Ubiquitination of soluble PABPN1 protein was compared between extracts of soluble proteins in the absence (A) or presence (B) of iodoacetamide. Protein extracts were prepared from myotube cultures of WTA, D7E, or IM2 at 4 days after fusion. Aliquots were used for immunoprecipitation (IP) with rabbit anti-FLAG antibody. Western blot analysis was conducted with a mouse anti-FLAG antibody. Protein input is indicated in the input fraction, and MSA is used as a loading control. FLAG-immunoprecipitated ubiquitinated proteins were detected with anti-ubiquitin antibody (Ubi) (B). Arrow, PABPN1. C: Detection of ubiquitinated PABPN1 in D7E and WTA myotubes was performed by TUBE pull down (PD). Myotubes were mock treated (-MG132) or treated with 5 μmol/L MG132 for 8 hours (+MG132). Protein aliquots are shown in the input fraction. Pull down of naked agarose beads and extracts from myotubes of IM2 parental cells were used as negative controls. Immunoblot (IB) of PABPN1 was performed with mouse anti-FLAG antibody, and ubiquitinated proteins were detected with an anti-uniquitin (UBi) antibody.
To validate this observation, the TUBE-agarose beads49 were used to pull down ubiquitinated proteins in protein extracts derived from WTA or D7E myotube cultures. Protein extracts were prepared in the presence of PR619, a specific DUB inhibitor. Protein extracts from IM2 myotubes were used as a negative control. The 58-kDa PABPN1-FLAG protein was pulled down with TUBEs from WTA and D7E extracts (Figure 7C) and was absent from pull-down experiments with the IM2 control protein extract and from pull down with naked agarose beads (Figure 7C). Enrichment in binding of ubiquitinated proteins to TUBEs was demonstrated with anti-ubiquitin antibody. These data show that the binding of ubiquitinated PABPN1 to TUBEs is specific and confirms that, in WTA culture, PABPN1 is more ubiquitinated compared with D7E culture (Figure 7C). The TUBEs pulled down did not reveal the ladder of ubiquitinated PABPN1 species, as observed with immunoprecipitation (Figure 7B). In all likelihood, this is because protein extractions in each of these procedures were conducted with different DUB inhibitors with different specificities, resulting in variance in the detection of PABPN1 ubiquitination species. Nevertheless, these data sets consistently demonstrate that WT PABPN1 is ubiquitinated to a higher degree than the mutant protein. More important, the greater poly-ubiquitinated WT PABPN1 is consistent with the observed higher rate of turnover of the soluble form of this protein.
Discussion
PABPN1 is ubiquitously expressed in all cell types and plays an indispensable role in different cellular processes. PABPN1 regulates poly(A) tail length and is involved in nuclear-cytoplasmic RNA transport and post-transcriptional gene regulation.50 Therefore, manipulation of PABPN1 expression levels would have a broad effect on diverse cellular functions. To study the effect of expPABPN1 at low and physiologically relevant expression levels, we generated a murine muscle myoblast/myotube cell model in which mRNA levels of expPABPN1 are comparable to endogenous Pabpn1. We investigated the specific effect of expPABPN1 in myotube cultures compared with that of WT PABPN1, which was expressed at a similar level. In this cell culture model, PABPN1 aggregates to form INIs in expPABPN1-expressing D7E myotubes that are similar to those reported in patients with OPMD. Myotubes of WTA or D7E clones did not show major myogenic differences. Because high overexpression and down-regulation of PABPN1 lead to myogenic defects,26–28 PABPN1's effect on myogenesis may be concentration dependent. The relevance of D7E as a cell model for OPMD was further evaluated by a transcriptome study. We show that most D7E deregulated pathways were also identified as significantly deregulated in skeletal muscles of patients with OPMD. These data suggest that the involvement of these pathways in OPMD pathological features, which are commonly deregulated in D7E and OPMD, can be studied in our cell model using molecular and biochemical tools.
Only a few KEGG pathways were identified as D7E specific because they were not found in the transcriptome study that addressed PABPN1 overexpression. Of relevance to OPMD, these D7E-specific KEGG pathways are also deregulated in patients with OPMD. Among the pathways, the insulin signaling pathway and the proteasome have been associated with muscle aging.34,51 Because OPMD is a late-onset disorder, the deregulated genes in these pathways could be associated with disease onset. In a recent study,29 we identified the OPMD-deregulated proteasome genes as candidates for association with disease onset. The involvement of these pathways in OPMD onset could be a subject for further investigations.
The aggregation potency of expPABPN1 protein has been addressed in many studies, typically in the context of high overexpression.18,52 We found greater accumulation of the soluble mutant compared with that of the WT protein. The greater accumulation of soluble expPABPN1 results from a slower protein turnover and reduced poly-ubiquitination, suggesting that they are differentially ubiquitinated or that the ubiquitination/de-ubiquitination ratio differs. Because expression of several E3 ligases and DUBs is specifically deregulated in OPMD,29 we expect to identify the enzymes that regulate the differential poly-ubiquitination in WTA and D7E myotubes.
The reason for muscle-cell dysfunction in patients with OPMD is unclear. Our study shows that a significant difference in turnover of the soluble form is found between WT and expPABPN1. In a recent in vitro study,53 the soluble PABPN1 was found to elongate mRNA poly(A) tails. To date, no differences between WT and expPABPN1 in efficiency of poly(A) elongation have been found.52 However, because the in vitro activity of PABPN1 is concentration-dependent53 and the aggregation potency of expPABPN1 is higher than that of the WT protein, our data suggest that, in the heterozygous situation, the active form of PABPN1 is reduced. In a recent study, 29 we found that a pre-aggregated form of expPABPN1, but not of WT PABPN1, can be found in the nucleus. Pre-aggregated expPABPN1 could be reversed to a soluble mobile protein form, whereas the aggregated PABPN1 in INIs is not reversible.29 We propose that expPABPN1 is not toxic per se but that the reduced availability of the soluble protein is likely to be central to this muscular dystrophy. This concept of a misbalance between soluble and insoluble protein species could also apply to other aggregation disorders, such as dystrophia myotonia type 154 and Huntington's disease.55 Thus, increased accumulation of the mutant protein, which is aggregation-prone, would lead to reduction in the functional form.
Footnotes
Supported by the European Commission (TRI-EX QLG2-CT-2001-01673 and POLYALA LSHM-CT-2005-018675) and the Muscular Dystrophy Association (68015).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.06.044.
Supplementary data
Fold-change of fusion markers between IM2 myoblasts and myotubes. Histograms show the fold change in expression of MLP4, MEF2C, TropC, MyoD, and MyoG between unfused (myoblasts) and fused myotubes of IM2 cells. ΔCT was normalized to cyclophilin B housekeeping gene, and ΔΔCT was normalized to unfused cells. SDs represent two biological experiments. The fold change of the well-known MyoD and MyoG differentiation markers was approximately 10- to 100-fold lower than that obtained for MLP4, MEF2C, and TropC.
WT-PABPN1-FLAG expression levels in WTA and WTD clones. qPCR of WT hPABPN1 in stable IM2 clones, WTA, and WTD. Expression levels were normalized to mDES.
Detection of apoptosis in D7E myotubes. Myotubes of WTA, D7E, or IM2 parental were subjected to TUNEL assay (Roche) (A) or immunofluorescence with anti-cleaved caspase 3 (B). A: The nuclear localization of fluorerescein-dUTP, which marks apoptotic cells, was studied in the merge image with bright field. Nuclear TUNEL staining was detected only after DNaseI treatment was served as a positive control. B: The anti-cleaved caspase-3 antibody (R&D Systems, Minneapolis, MN) was visualized with Alexa 594, and PABPN1-FLAG was visualized with Alexa 488. Nuclei were counterstained with DAPI. Together, these experiments reveal activation of apoptosis in myotubes expressing WT or expPABPN1. For the TUNEL staining, 4-day fused myotubes were labeled with UTP-fluorescein (Cell Death Detection Kit; Roche, Indianapolis, IN) for 15 minutes. Cells were washed with PBS, fixed with 2% formaldehyde, and imaged with a 485- to 535-nm filter. DNaseI-treated cells (according to the manufacturer's protocol) were used as a positive control.
References
- 1.Cummings C.J., Zoghbi H.Y. Fourteen and counting: unraveling trinucleotide repeat diseases. Hum Mol Genet. 2000;9:909–916. doi: 10.1093/hmg/9.6.909. [DOI] [PubMed] [Google Scholar]
- 2.Albrecht A., Mundlos S. The other trinucleotide repeat: polyalanine expansion disorders. Curr Opin Genet Dev. 2005;15:285–293. doi: 10.1016/j.gde.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Brown L.Y., Brown S.A. Alanine tracts: the expanding story of human illness and trinucleotide repeats. Trends Genet. 2004;20:51–58. doi: 10.1016/j.tig.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Ross C.A., Poirier M.A. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 5.Brais B., Bouchard J.P., Xie Y.G., Rochefort D.L., Chretien N., Tome F.M., Lafreniere R.G., Rommens J.M., Uyama E., Nohira O., Blumen S., Korczyn A.D., Heutink P., Mathieu J., Duranceau A., Codere F., Fardeau M., Rouleau G.A. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 6.Wahle E. A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell. 1991;66:759–768. doi: 10.1016/0092-8674(91)90119-j. [DOI] [PubMed] [Google Scholar]
- 7.Wahle E. 3′-end cleavage and polyadenylation of mRNA precursors. Biochim Biophys Acta. 1995;1261:183–194. doi: 10.1016/0167-4781(94)00248-2. [DOI] [PubMed] [Google Scholar]
- 8.Bouchard J.P., Brais B., Brunet D., Gould P.V., Rouleau G.A. Recent studies on oculopharyngeal muscular dystrophy in Quebec. Neuromuscul Disord. 1997;7(Suppl 1):S22–S29. doi: 10.1016/s0960-8966(97)00077-1. [DOI] [PubMed] [Google Scholar]
- 9.Taylor E.W. Progressive vagus-glossopharyngeal paralysis with ptosis: contribution the group of family diseases. J Nerv Ment Dis. 1915;42:129–139. [Google Scholar]
- 10.Calado A., Tome F.M., Brais B., Rouleau G.A., Kuhn U., Wahle E., Carmo-Fonseca M. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum Mol Genet. 2000;9:2321–2328. doi: 10.1093/oxfordjournals.hmg.a018924. [DOI] [PubMed] [Google Scholar]
- 11.Tome F.M., Fardeau M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol. 1980;49:85–87. doi: 10.1007/BF00692226. [DOI] [PubMed] [Google Scholar]
- 12.Tome F.M., Chateau D., Helbling-Leclerc A., Fardeau M. Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul Disord. 1997;7(Suppl 1):S63–S69. doi: 10.1016/s0960-8966(97)00085-0. [DOI] [PubMed] [Google Scholar]
- 13.Berciano M.T., Villagra N.T., Ojeda J.L., Navascues J., Gomes A., Lafarga M., Carmo-Fonseca M. Oculopharyngeal muscular dystrophy-like nuclear inclusions are present in normal magnocellular neurosecretory neurons of the hypothalamus. Hum Mol Genet. 2004;13:829–838. doi: 10.1093/hmg/ddh101. [DOI] [PubMed] [Google Scholar]
- 14.Villagra N.T., Bengoechea R., Vaque J.P., Llorca J., Berciano M.T., Lafarga M. Nuclear compartmentalization and dynamics of the poly(A)-binding protein nuclear 1 (PABPN1) inclusions in supraoptic neurons under physiological and osmotic stress conditions. Mol Cell Neurosci. 2008;37:622–633. doi: 10.1016/j.mcn.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 15.Klein A.F., Ebihara M., Alexander C., Dicaire M.J., Sasseville A.M.J., Langelier Y., Rouleau G.A., Brais B. PABPN1 polyalanine tract deletion and long expansions modify its aggregation pattern and expression. Exp Cell Res. 2008;314:1652–1666. doi: 10.1016/j.yexcr.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Tavanez J.P., Calado P., Braga J., Lafarga M., Carmo-Fonseca M. In vivo aggregation properties of the nuclear poly(A)-binding protein PABPN1. RNA. 2005;11:752–762. doi: 10.1261/rna.7217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chartier A., Benoit B., Simonelig M. A Drosophila model of oculopharyngeal muscular dystrophy reveals intrinsic toxicity of PABPN1. EMBO J. 2006;25:2253–2262. doi: 10.1038/sj.emboj.7601117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brais B. Oculopharyngeal muscular dystrophy: a polyalanine myopathy. Curr Neurol Neurosci Rep. 2009;9:76–82. doi: 10.1007/s11910-009-0012-y. [DOI] [PubMed] [Google Scholar]
- 19.Catoire H., Pasco M.Y., Abu-Baker A., Holbert S., Tourette C., Brais B., Rouleau G.A., Parker J.A., Neri C. Sirtuin inhibition protects from the polyalanine muscular dystrophy protein PABPN1. Hum Mol Genet. 2008;17:2108–2117. doi: 10.1093/hmg/ddn109. [DOI] [PubMed] [Google Scholar]
- 20.Davies J.E., Wang L., Garcia-Oroz L., Cook L.J., Vacher C., O'Donovan D.G., Rubinsztein D.C. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat Med. 2005;11:672–677. doi: 10.1038/nm1242. [DOI] [PubMed] [Google Scholar]
- 21.Chartier A., Raz V., Sterrenburg E., Verrips C.T., van der Maarel S.M., Simonelig M. Prevention of oculopharyngeal muscular dystrophy by muscular expression of Llama single-chain intrabodies in vivo. Hum Mol Genet. 2009;18:1849–1859. doi: 10.1093/hmg/ddp101. [DOI] [PubMed] [Google Scholar]
- 22.Davies J.E., Sarkar S., Rubinsztein D.C. Trehalose reduces aggregate formation and delays pathology in a transgenic mouse model of oculopharyngeal muscular dystrophy. Hum Mol Genet. 2006;15:23–31. doi: 10.1093/hmg/ddi422. [DOI] [PubMed] [Google Scholar]
- 23.Trollet C., Anvar S.Y., Venema A., Hargreaves I.P., Foster K., Vignaud A., Ferry A., Negroni E., Hourde C., Baraibar M.A., 't Hoen P.A.C., Davies J.E., Rubinsztein D.C., Heales S.J., Mouly V., van der Maarel S.M., Butler-Browne G., Raz V., Dickson G. Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum Mol Genet. 2010;19:2191–2207. doi: 10.1093/hmg/ddq098. [DOI] [PubMed] [Google Scholar]
- 24.Blumen S.C., Brais B., Korczyn A.D., Medinsky S., Chapman J., Asherov A., Nisipeanu P., Codere F., Bouchard J.P., Fardeau M., Tome F.M., Rouleau G.A. Homozygotes for oculopharyngeal muscular dystrophy have a severe form of the disease. Ann Neurol. 1999;46:115–118. [PubMed] [Google Scholar]
- 25.Hino H., Araki K., Uyama E., Takeya M., Araki M., Yoshinobu K., Miike K., Kawazoe Y., Maeda Y., Uchino M., Yamamura K. Myopathy phenotype in transgenic mice expressing mutated PABPN1 as a model of oculopharyngeal muscular dystrophy. Hum Mol Genet. 2004;13:181–190. doi: 10.1093/hmg/ddh017. [DOI] [PubMed] [Google Scholar]
- 26.Apponi L.H., Leung S.W., Williams K.R., Valentini S.R., Corbett A.H., Pavlath G.K. Loss of nuclear poly(A)-binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum Mol Genet. 2010;19:1058–1065. doi: 10.1093/hmg/ddp569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim Y.J., Noguchi S., Hayashi Y.K., Tsukahara T., Shimizu T., Arahata K. The product of an oculopharyngeal muscular dystrophy gene, poly(A)-binding protein 2, interacts with SKIP and stimulates muscle-specific gene expression. Hum Mol Genet. 2001;10:1129–1139. doi: 10.1093/hmg/10.11.1129. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q., Bag J. Ectopic expression of a polyalanine expansion mutant of poly(A)-binding protein N1 in muscle cells in culture inhibits myogenesis. Biochem Biophys Res Commun. 2006;340:815–822. doi: 10.1016/j.bbrc.2005.12.078. [DOI] [PubMed] [Google Scholar]
- 29.Anvar S.Y., 't Hoen P., Venema A., van der Sluijs B., van Engelen B., Snoeck M., Vissing J., Trollet C., Dickson G., Chartier A., Simonelig M., van Ommen G.J., van der Maarel S., Raz V. Deregulation of the ubiquitin-proteasome system is the predominant molecular pathology in OPMD animal models and patients. Skelet Muscle. 2011;1(1):15. doi: 10.1186/2044-5040-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Needham M., Egerton M., Millest A., Evans S., Popplewell M., Cerillo G., McPheat J., Monk A., Jack A., Johnstone D., Hollis M. Further development of the locus control region/murine erythroleukemia expression system: high level expression and characterization of recombinant human calcitonin receptor. Protein Expr Purif. 1995;6:124–131. doi: 10.1006/prep.1995.1015. [DOI] [PubMed] [Google Scholar]
- 31.Tam J.L., Triantaphyllopoulos K., Todd H., Raguz S., de Wit T., Morgan J.E., Partridge T.A., Makrinou E., Grosveld F., Antoniou M. The human desmin locus: gene organization and LCR-mediated transcriptional control. Genomics. 2006;87:733–746. doi: 10.1016/j.ygeno.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 32.Chubet R.G., Brizzard B.L. Vectors for expression and secretion of FLAG epitope-tagged proteins in mammalian cells. Biotechniques. 1996;20:136–141. doi: 10.2144/96201pf01. [DOI] [PubMed] [Google Scholar]
- 33.Morgan J.E., Beauchamp J.R., Pagel C.N., Peckham M., Ataliotis P., Jat P.S., Noble M.D., Farmer K., Partridge T.A. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev Biol. 1994;162:486–498. doi: 10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- 34.Carmeli E., Coleman R., Reznick A.Z. The biochemistry of aging muscle. Exp Gerontol. 2002;37:477–489. doi: 10.1016/s0531-5565(01)00220-0. [DOI] [PubMed] [Google Scholar]
- 35.Goeman J.J., van de Geer S.A., de Kort F., van Houwelingen H.C. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20:93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- 36.Raz V., Carlotti F., Vermolen B.J., van der P.E., Sloos W.C., Knaan-Shanzer S., de Vries A.A., Hoeben R.C., Young I.T., Tanke H.J., Garini Y., Dirks R.W. Changes in lamina structure are followed by spatial reorganization of heterochromatic regions in caspase-8-activated human mesenchymal stem cells. J Cell Sci. 2006;119:4247–4256. doi: 10.1242/jcs.03180. [DOI] [PubMed] [Google Scholar]
- 37.Verheesen P., de Kluijver A., van Koningsbruggen S., de Brij M., de Haard H.J., van Ommen G.J., van der Maarel S.M., Verrips C.T. Prevention of oculopharyngeal muscular dystrophy-associated aggregation of nuclear polyA-binding protein with a single-domain intracellular antibody. Hum Mol Genet. 2006;15:105–111. doi: 10.1093/hmg/ddi432. [DOI] [PubMed] [Google Scholar]
- 38.Abu-Baker A., Messaed C., Laganiere J., Gaspar C., Brais B., Rouleau G.A. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum Mol Genet. 2003;12:2609–2623. doi: 10.1093/hmg/ddg293. [DOI] [PubMed] [Google Scholar]
- 39.Bao Y.P., Sarkar S., Uyama E., Rubinsztein D.C. Congo red, doxycycline, and HSP70 overexpression reduce aggregate formation and cell death in cell models of oculopharyngeal muscular dystrophy. J Med Genet. 2004;41:47–51. doi: 10.1136/jmg.2003.014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corbeil-Girard L.P., Klein A.F., Sasseville A.M., Lavoie H., Dicaire M.J., Saint-Denis A., Page M., Duranceau A., Codere F., Bouchard J.P., Karpati G., Rouleau G.A., Massie B., Langelier Y., Brais B. PABPN1 overexpression leads to upregulation of genes encoding nuclear proteins that are sequestered in oculopharyngeal muscular dystrophy nuclear inclusions. Neurobiol Dis. 2005;18:551–567. doi: 10.1016/j.nbd.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 41.Klein A.F., Ebihara M., Alexander C., Dicaire M.J., Sasseville A.M., Langelier Y., Rouleau G.A., Brais B. PABPN1 polyalanine tract deletion and long expansions modify its aggregation pattern and expression. Exp Cell Res. 2008;314:1652–1666. doi: 10.1016/j.yexcr.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Messaed C., Dion P.A., Abu-Baker A., Rochefort D., Laganiere J., Brais B., Rouleau G.A. Soluble expanded PABPN1 promotes cell death in oculopharyngeal muscular dystrophy. Neurobiol Dis. 2007;26:546–557. doi: 10.1016/j.nbd.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Antoniou M., Geraghty F., Hurst J., Grosveld F. Efficient 3′-end formation of human beta-globin mRNA in vivo requires sequences within the last intron but occurs independently of the splicing reaction. Nucleic Acids Res. 1998;26:721–729. doi: 10.1093/nar/26.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collis P., Antoniou M., Grosveld F. Definition of the minimal requirements within the human beta-globin gene and the dominant control region for high level expression. EMBO J. 1990;9:233–240. doi: 10.1002/j.1460-2075.1990.tb08100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Custodio N., Carmo-Fonseca M., Geraghty F., Pereira H.S., Grosveld F., Antoniou M. Inefficient processing impairs release of RNA from the site of transcription. EMBO J. 1999;18:2855–2866. doi: 10.1093/emboj/18.10.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Millevoi S., Geraghty F., Idowu B., Tam J.L., Antoniou M., Vagner S. A novel function for the U2AF 65 splicing factor in promoting pre-mRNA 3′-end processing. EMBO Rep. 2002;3:869–874. doi: 10.1093/embo-reports/kvf173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davies J.E., Sarkar S., Rubinsztein D.C. Wild-type PABPN1 is anti-apoptotic and reduces toxicity of the oculopharyngeal muscular dystrophy mutation. Hum Mol Genet. 2008;17:1097–1108. doi: 10.1093/hmg/ddm382. [DOI] [PubMed] [Google Scholar]
- 48.Fan X., Dion P., Laganiere J., Brais B., Rouleau G.A. Oligomerization of polyalanine expanded PABPN1 facilitates nuclear protein aggregation that is associated with cell death. Hum Mol Genet. 2001;10:2341–2351. doi: 10.1093/hmg/10.21.2341. [DOI] [PubMed] [Google Scholar]
- 49.Hjerpe R., Aillet F., Lopitz-Otsoa F., Lang V., England P., Rodriguez M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009;10:1250–1258. doi: 10.1038/embor.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemay J.F., Lemieux C., St-Andre O., Bachand F. Crossing the borders: poly(A)-binding proteins working on both sides of the fence. RNA Biol. 2010;7:291–295. doi: 10.4161/rna.7.3.11649. [DOI] [PubMed] [Google Scholar]
- 51.Ferrington D.A., Husom A.D., Thompson L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005;19:644–646. doi: 10.1096/fj.04-2578fje. [DOI] [PubMed] [Google Scholar]
- 52.Kuhn U., Wahle E. Structure and function of poly(A) binding proteins. Biochim Biophys Acta. 2004;1678:67–84. doi: 10.1016/j.bbaexp.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 53.Kühn U., Gündel M., Knoth A., Kerwitz Y., Rüdel S., Wahle E. Poly(A) tail length is controlled by the nuclear poly(A)-binding protein regulating the interaction between poly(A) polymerase and the cleavage and polyadenylation specificity factor. J Biol Chem. 2009;284:22803–22814. doi: 10.1074/jbc.M109.018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Junghans R.P. Dystrophia myotonia: why focus on foci? Eur J Hum Genet. 2009;17:543–553. doi: 10.1038/ejhg.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Mezer M., Wojciechowska M., Napierala M., Sobczak K., Krzyzosiak W.J. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011;39:3852–3863. doi: 10.1093/nar/gkq1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fold-change of fusion markers between IM2 myoblasts and myotubes. Histograms show the fold change in expression of MLP4, MEF2C, TropC, MyoD, and MyoG between unfused (myoblasts) and fused myotubes of IM2 cells. ΔCT was normalized to cyclophilin B housekeeping gene, and ΔΔCT was normalized to unfused cells. SDs represent two biological experiments. The fold change of the well-known MyoD and MyoG differentiation markers was approximately 10- to 100-fold lower than that obtained for MLP4, MEF2C, and TropC.
WT-PABPN1-FLAG expression levels in WTA and WTD clones. qPCR of WT hPABPN1 in stable IM2 clones, WTA, and WTD. Expression levels were normalized to mDES.
Detection of apoptosis in D7E myotubes. Myotubes of WTA, D7E, or IM2 parental were subjected to TUNEL assay (Roche) (A) or immunofluorescence with anti-cleaved caspase 3 (B). A: The nuclear localization of fluorerescein-dUTP, which marks apoptotic cells, was studied in the merge image with bright field. Nuclear TUNEL staining was detected only after DNaseI treatment was served as a positive control. B: The anti-cleaved caspase-3 antibody (R&D Systems, Minneapolis, MN) was visualized with Alexa 594, and PABPN1-FLAG was visualized with Alexa 488. Nuclei were counterstained with DAPI. Together, these experiments reveal activation of apoptosis in myotubes expressing WT or expPABPN1. For the TUNEL staining, 4-day fused myotubes were labeled with UTP-fluorescein (Cell Death Detection Kit; Roche, Indianapolis, IN) for 15 minutes. Cells were washed with PBS, fixed with 2% formaldehyde, and imaged with a 485- to 535-nm filter. DNaseI-treated cells (according to the manufacturer's protocol) were used as a positive control.