

Abstract
The phenotype of hereditary apolipoprotein A-I amyloidosis is heterogeneous with some patients developing extensive visceral amyloid deposits and end-stage renal failure as young adults and others having only laryngeal and/or skin amyloid, which may be of little clinical consequence. Clinical management and prognosis of patients with systemic amyloidosis depend entirely on correct identification of the fibril protein, such that light chain amyloidosis (AL, previously referred to as “primary”), the most frequently diagnosed type, is treated with chemotherapy, which has absolutely no role in hereditary apolipoprotein A-I amyloidosis. We report five novel apolipoprotein A-I variants, four of which were amyloidogenic and one of which was incidental in a patient with systemic AL amyloidosis. Interestingly, only one of four patients with apolipoprotein A-I amyloidosis had a family history of similar disease. Laser microdissection and tandem mass spectrometry–based proteomics were used to confirm the amyloid fibril protein and, for the first time in apolipoprotein A-I amyloidosis, demonstrated that only mutated protein as opposed to wild-type apolipoprotein A-I was deposited as amyloid. The clinical spectrum and outcome of hereditary apolipoprotein A-I amyloidosis are reviewed in detail and support the need for sequencing of the apolipoprotein A-I gene among patients with apparent localized amyloidosis in whom IHC is nondiagnostic of the fibril protein, even in the absence of a family history of disease.
Amyloidosis is a rare disorder characterized by extracellular deposition of fibrillar protein that results in a progressive disruption of structure and function of affected tissues and organs. Amyloidosis is a remarkably heterogeneous disease; it can be acquired or hereditary and systemic or localized.
The most common form of systemic amyloidosis is AL (light chain, previously referred to as “primary”), in which the fibrils are derived from monoclonal immunoglobulin light chains and consist of the whole or part of the variable (VL) domain. The prognosis of systemic AL amyloidosis is generally worse than in other amyloid types, although there is marked heterogeneity in the extent of organ involvement and rate of disease progression. Treatment of AL amyloidosis is with chemotherapy for which there is no role in other amyloid types.
Hereditary systemic amyloidosis is a rare autosomal dominant condition caused by deposition of variant proteins as amyloid fibrils. The term “hereditary nonneuropathic systemic amyloidosis” (Online Mendelian Inheritance of Man no. 105200) was coined by Ostertag1 in 1932 following the discovery of two families with dominantly inherited renal amyloidosis. Mutations in the genes encoding apolipoprotein A-I (apoAI),2–12 apolipoprotein A-II (apoAII),13 fibrinogen Aα-chain,14–17 and lysozyme18 have since been identified as the cause of hereditary renal amyloidosis in different kindreds (Online Mendelian Inheritance of Man no. 105200). The clinical amyloidosis syndromes that accompany the various mutations in these different genes are diverse with respect to age at onset, mode of presentation, pattern of organ distribution, rate of progression, and prognosis. Indeed, certain apoAI variants are associated with neuropathy19,20 such that the nomenclature, which includes the term “nonneuropathic,” is confusing and probably no longer appropriate.
Patients with hereditary apoAI amyloidosis typically present with hypertension, proteinuria, and renal impairment6 and frequently develop extensive visceral deposits that affect the liver, spleen, and kidneys, with occasional involvement of the heart, nerves, larynx, and gastrointestinal tract. Although the condition may lead to end-stage renal disease and other organ failure, the natural history of the disease is often slow, contrasting systemic AL amyloidosis in which the median patient survival without therapy is approximately 6 to 15 months from diagnosis.21,22
Here we report five novel apoAI variants, four of which were associated with hereditary systemic amyloidosis and one of which was nonamyloidogenic, and an incidental finding discovered in a patient with systemic AL amyloidosis. The importance of correct identification of the amyloid fibril protein is highlighted, and the novel use of mass spectrometry to identify apoAI amyloidosis is reported. We review the UK National Amyloidosis Centre experience and published literature regarding the clinical spectrum and outcome of hereditary apoAI amyloidosis.
Materials and Methods
Five unrelated patients were referred to the UK National Amyloidosis Centre after discovery of amyloid deposits and underwent detailed investigations to elucidate the cause. Informed consent was obtained from all patients, and clinical care was in accordance with the Declaration of Helsinki.
Patients
A 35-year-old English man presented with hoarseness (patient 1). Vocal cord nodules were discovered at laryngoscopy and excised. A 51-year-old English woman presented with a soft tissue mass growing on her palate (patient 2). A 29-year-old Polish woman presented with edema (patient 3). A 77-year-old English woman presented with hypertension and was noted to have proteinuria and renal impairment (patient 4). A 56-year-old English man presented with proteinuria and progressive renal impairment (patient 5).
Histology and IHC
Biopsy specimens of vocal cord and testicles from patient 1, palatal mass from patient 2, rectum from patient 3, and kidney from patients 4 and 5 were stained at the UK National Amyloidosis Centre. Sections, 6-μm thick, were stained for amyloid deposits with Congo red and viewed under crossed polarized light.23 Immunohistochemistry (IHC) staining of the amyloid deposits was performed using monospecific antibodies reactive with serum amyloid A protein, lysozyme, apoAI, fibrinogen Aα-chain, transthyretin, and κ and λ immunoglobulin light chains, as previously described.24 Specificity of staining was confirmed by prior absorption of the antiserum with pure antigen in each case, and positive and negative controls were included in each run.
Direct DNA Sequencing
Genomic DNA from all five patients was extracted from whole blood treated with EDTA.25 Exons 3 (c.5352 to c.5711) and 4 (c.6116 to 6859) of the APOAI gene (National Center for Biotechnology Information accession no. NG_012021) were amplified by PCR performed with Ready-To-Go tubes (Amersham Pharmacia Biotech, Piscataway, NJ). PCR amplification of exons 3 and 4 was performed with the following oligonucleotides: exon 3 forward 5′-GGCAGAGGCAGCAGGTTTCTCAC-3′ (c.5352−5374) and reverse 5′-CCAGACTGGCCGAGTCCTCACCTA-3′ (c.5688−5711) and exon 4 forward 5′-CACTGCACCTCCGCGGACA-3′ (c.6116−6134) and reverse 5′-CTTCCCGGTGCTCAGAATAAACGTT-3′ (c.6835−6859). The total volume of PCR reaction was 25 μL, and it consisted of the following: 2 μL of DNA (at a concentration of 100 ng/μL), 1 μL of each primer (at a concentration of 10 μmol/L), and 21 μL of water. PCR cycling conditions were as followed: initial denaturation at 96°C for 5 minutes; five cycles of 96°C for 30 seconds and 72°C for 30 seconds; 35 cycles at 96°C for 30 seconds, 72°C for 30 seconds, and 72°C for 1 minute; and final elongation step at 72°C for 7 minutes. Negative and positive controls were included.
The PCR products were purified with a QIAquick PCR purification kit (Oiagen, Velno, the Netherlands) according to the manufacturer's protocol and sequenced with the ABI BigDye Terminator v 3.1 Ready Reaction Cycle Sequencing kit (Applied Biosystems, Foster City, CA). The primers 5′-GATCTCAGCCCACAGCTGGCC-3′ (c.5417−5437) and 5′-AGGGCTCACCCCTGATAGGCTG-3′ (c.6144−6166) were used for sequencing of exons 3 and 4, respectively. APOAI gene sequences were analyzed on the ABI 3130xl Genetic Analyzer, using Sequencing Analysis Software, version 5.4 (Applied Biosystems, Carlsbad, CA).
Laser Microdissection and Tandem Mass Spectrometry–Based Proteomics
The proteome of amyloid deposits was analyzed by combining microdissection and mass spectrometry–based proteomics. Sections (10-μm thick) of formalin-fixed, paraffin-embedded tissues were placed on DIRECTOR slides (Expression Pathology, Rockville, MD). Sections were air dried and then melted, deparaffinized, and stained in hematoxylin followed by Congo red.
Congo red–stained sections were examined for the presence of amyloid deposits under fluorescence (B/G/R filter cube; Leica, Wetzler, Germany). Positive areas were microdissected into 0.5-mL microcentrifuge tube caps containing 10 mmol TRIS/1 mmol EDTA/0.002% Zwittergent 3−16 (Calbiochem, San Diego, CA) using a Leica DM6000B Microdissection System (Leica). For each case, two to four different microdissections were collected, and each microdissection contained 50,000 to 60,000 μm2 of the tissue section. Collected tissues were heated at 98°C for 90 minutes with occasional vortexing. After 60 minutes of sonication in a waterbath, samples were digested overnight at 37°C with 1 μg of trypsin (Promega, Madison, WI). The trypsin-generated digests were reduced with dithiothreitol and separated by nanoflow liquid chromatography–electrospray tandem mass spectrometry using a ThermoFinnigan LTQ Orbitrap Hybrid Mass Spectrometer (Thermo Electron, Bremen, Germany) coupled to an Eksigent nanoLC-2D HPLC system (Eksigent, Dublin, CA). A 0.25-μL trap (Optimize Technologies, Oregon City, OR) packed with Michrom Magic C-8 was plumbed into a 10-port valve. A 75 μm × 15 cm C-18 column was used for the separation using an organic gradient from 6% to 86% in 55 minutes at 400 nL/min. The Thermo-Fisher Scientific (Waltham, MA) MS/MS raw data files were searched using three different algorithms (Mascot, Sequest, and X!Tandem) and the results assigned peptide and protein probability scores. The results were then combined and displayed using Scaffold (Proteome Software, Portland, OR). All searches were conducted with variable modifications and restricted to full trypsin-generated peptides, allowing for two missed cleavages. Peptide mass search tolerances were set to 10 ppm and fragment mass tolerance to 1.00 Da. The search algorithms used to identify peptides interrogate the human SwissProt database, which does not contain the mutated sequences identified in this study. To show that the peptides containing the mutation were part of amyloid deposits, these sequences were added to the human SwissProt database using an in-house script, and a second-round data analysis was performed. Peptide identifications below the 90% confidence level were not considered in our analysis.
Radiolabeled SAP Scintigraphy
All five patients underwent whole body anterior and posterior scintigraphic imaging 24 hours after administration of 123I-labeled serum amyloid P component (SAP) using a GE Infinia Hawkeye (GE Healthcare, Chalfont St Giles, UK) gamma camera, as previously described.26 The labeled SAP study results were interpreted by a panel of physicians with experience of more than 10,000 SAP scans.
Results
Clinical Findings
Patient 1 presented with a hoarse voice and no other significant history. He underwent direct laryngoscopy and excision of multiple nodules on the vocal cords and aryepiglottic folds, which were discovered to be amyloid on histologic analysis. On direct questioning there was no history of hemoptysis, dyspnea, rashes, petechiae, weight loss, anorexia, or edema. There was no history of paraesthesia or postural dizziness. The only medical history was of hypogonadism and azoospermia for which he had undergone a testicular biopsy 8 to 10 years previously. He did not take regular medication, and there was no family history of similar illness, cardiac disease, or renal disease. Clinical examination was unremarkable. Baseline investigations showed normal blood cell count, clotting profile, renal, and liver function. There was no evidence of a plasma cell dyscrasia by sensitive nephelometric serum free light chain assay,27 conventional electrophoresis and immunofixation of serum or urine, or bone marrow examination. There was no proteinuria, and ECG and echocardiography did not reveal evidence of cardiac amyloid infiltration.
Patient 2 presented with a 4-year history of a gradually enlarging mass on the palate within an area of scarring from a childhood injury. There had been no bleeding or pain associated with the lump, and the patient remained otherwise well. She experienced an occasional dry mouth and dry eyes and very rare tingling in her fingertips, with no other features to suggest a peripheral neuropathy or carpal tunnel syndrome. There was no history of weight loss or autonomic symptoms. There was no family history of amyloidosis, renal disease, or cardiac disease. Clinical examination was unremarkable. She had a medical history that included celiac disease, dermatitis herpetiformis, and multinodular goiter for which she had undergone a partial thyroidectomy at the age of 29 years. Shortly before her presentation with the lump in the palate, she had developed a recurrent clinically overt goiter. There was no evidence of a plasma cell dyscrasia by serum free light chain assay, conventional electrophoresis and immunofixation of serum or urine, or bone marrow examination. There was no proteinuria, and ECG, echocardiogram, and N-terminal pro-B–type natriuretic peptide were not suggestive of cardiac amyloid infiltration.
Patient 3 presented with edema and was discovered to have subnephrotic range proteinuria (1.68 g/24 h) and stage 3 chronic kidney disease (CKD). A kidney biopsy was performed in 2008 and revealed amyloidosis, thought on initial IHC in her local hospital to be AA type. The results of investigations to identify an underlying inflammatory condition were all negative. Treatment with colchicine was commenced empirically, and she was referred to the UK National Amyloidosis Centre. There was no history of chronic inflammatory disease in her or her family and no family history of kidney disease. There was no history to suggest peripheral or autonomic neuropathy and no dyspnea. Baseline investigations revealed normal blood cell count, clotting profile, and liver function. There was no evidence of a plasma cell dyscrasia. An echocardiogram demonstrated a 12-mm interventricular wall thickness but normal diastolic dysfunction and good left ventricular systolic function, but there was mild thickening of her heart valves; overall, there was no definite evidence of cardiac amyloidosis, but it could not be completely excluded. There was insufficient tissue remaining in her renal biopsy specimen to definitively confirm the amyloid type, and there was doubt about the diagnosis of AA amyloidosis; therefore, a rectal biopsy was requested.
Patient 4 presented with hypertension and was discovered to be nephrotic (7.9 g/24 h; serum albumin, 24 g/L) with CKD stage 3 on a background of type 2 diabetes mellitus and ischemic heart disease. A renal biopsy specimen revealed amyloid deposits in the glomeruli and interstitium. At presentation, she was experiencing paraesthesia in the feet and minimal dyspnea on exertion. There was no evidence of a plasma cell dyscrasia. Cardiac investigations did not suggest amyloid cardiomyopathy. The patient's sister was subsequently discovered to have amyloid deposits on a renal biopsy specimen, undertaken for proteinuria.
Patient 5 presented with edema and was discovered to have proteinuria, advanced CKD, and mildly obstructive derangement of liver function tests. A renal biopsy specimen revealed amyloid deposits. There was mild breathlessness on exertion but no clinical evidence of peripheral or autonomic neuropathy. Nephelometric serum free light chain assay revealed a marked κ excess (κ, 393 mg/L; λ, 30 mg/L). There was no paraprotein by serum or urine immunoelectrophoresis, and bone marrow examination revealed less than 5% clonal plasma cells. Cardiac investigations did not show any evidence of cardiac involvement by amyloid. The patient received 15 days of high-dose dexamethasone for presumed AL amyloidosis, which substantially suppressed the κ serum free light chain concentration. Despite this, however, there was a progressive decline in renal function and the patient commenced hemodialysis.
Histology and IHC
Extensive amyloid deposits were identified in the vocal cord and testes of patient 1, the palate of patient 2, the rectum of patient 3, and the kidneys of patients 4 and 5 by their pathognomonic green birefringence when stained with Congo red and viewed under crossed polarized light (Figure 1A–B). The amyloid from patients 1 through 4 stained specifically with antibodies to apoAI (Figure 1C), and staining was completely abolished by prior absorption of the antiserum with an excess of pure human apoAI. There was no other staining with antibodies to κ light chains and no staining with antibodies against other known amyloid fibril proteins, including lysozyme, transthyretin, serum amyloid A protein, or λ light chains. The amyloid from patient 5 stained with antibodies against both κ light chains and apoAI such that IHC results were inconclusive.
Figure 1.
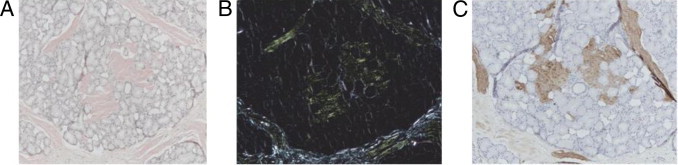
Palate biopsy specimen from a patient with apoAI amyloidosis. Staining with Congo red viewed under bright field light (A) showed amorphous eosinophilic deposits, which showed apple green birefringence when viewed under crossed polarized light (B) diagnostic of amyloid. The amyloid deposits stained with an antibody against apoAI (C), and staining was completely abolished by prior incubation of the antiserum with pure apoAI, thus confirming apoAI as the amyloid fibril protein.
APOAI Gene Mutations
Genetic analysis of exons 3 and 4 of the APOAI gene revealed five novel mutations (Figure 2). Four were single nucleotide substitutions, c.595G>C, c.284T>A, c.172G>A, and c.178T>G, resulting in amino acid change: alanine to proline at position 175, A175P in patient 1; phenylalanine to tyrosine at position 71, F71Y in patient 2; glutamic acid to lysine at position 34, E34K in patient 3; and serine to alanine at position 36, S36A in patient 5, respectively. The other novel variant p. His155MetfsX46, discovered in patient 4, resulted from a frameshift mutation caused by a deletion of cytosine at position c.535. The new reading frame created by this single nucleotide deletion results in premature termination, and consequently the mutated protein was 44 nucleotides shorter than wild-type apoAI. The remainder of the APOAI gene in all five patients was identical to the published sequence.28
Figure 2.
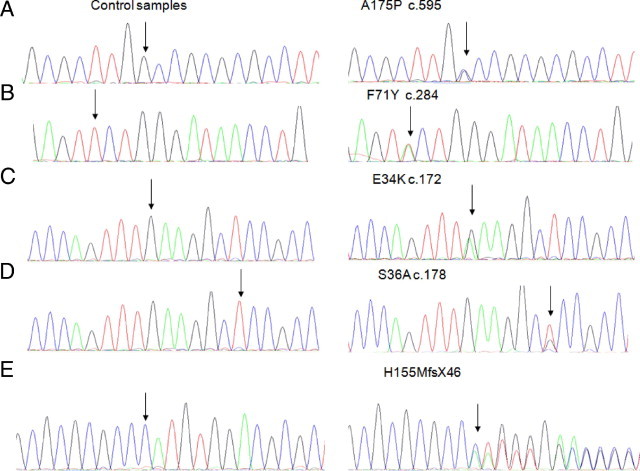
Partial DNA sequences of exons 3 and 4 of the APOAI gene. Right column shows five novel mutations indicated by arrows; four single nucleotide substitutions: A175P (A), F71Y (B), E34K (C), S36A (D), and a frameshift mutation H155MfsX46 (E). Left column shows the corresponding wild-type sequences.
Amyloid Typing by Laser Microdissection and Tandem Mass Spectrometry–Based Proteomics
Laser microdissection and tandem mass spectrometry showed that, in patients 1 through 4, apoAI was one of the most abundant proteins present in the amyloid deposits (Figure 3A). In contrast, there were no apoAI peptides present in the amyloid deposits of patient 5, which were composed of κ light chains. In addition to apoAI, other identified proteins included serum proteins, such as vitronectin and albumin, along with proteins that are known to be associated with all types of amyloidosis, such as apolipoprotein E and serum amyloid P component.
Figure 3.
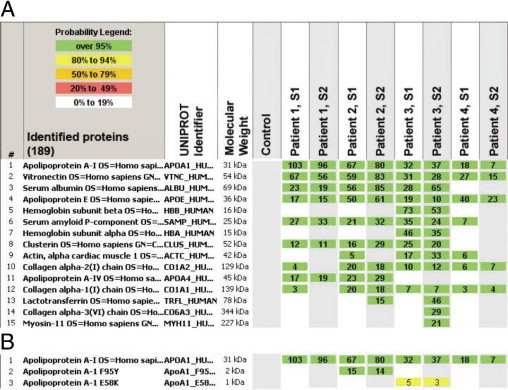
Results of mass spectrometry–based proteomic analysis of amyloid deposits (A). The 15 most abundant proteins identified among four patients are listed. At least two different samples (S1 and S2) were run for each patient. The columns show the protein name, the UnitProt identifier (protein accession code in the UniProt database, http://www.uniprot.org), the molecular weight of the protein, the results of the blank control sample, and two samples from each of the four patients. The numbers indicate number of total peptide spectra identified for each protein. ApoAI is the most abundant protein in this sample set, confirming apoAI as the amyloid fibril protein. Identification of mutated protein in amyloid deposits by mass spectrometry–based proteomics (B). After addition of the mutated sequences to the human SwissProt database using an in-house script and a second-round data analysis, the presence of mutated protein (a tryptic peptide carrying the altered amino acid sequence) within the amyloid deposits of patients 2 and 3 was demonstrated. The nomenclature for the location of the mutated amino acid includes the 24 amino acid signal peptide; thus, F95Y and E58K in the figure correspond to the F71Y and E34K protein variants, respectively.
Identification of Mutated Protein in Amyloid Deposits by Tandem Mass Spectrometry–Based Proteomics
To show the presence of mutated proteins in the amyloid deposits, we searched the mass spectrometry raw data files using the human SwissProt database supplemented with the APOAI gene variants identified in this study. The mutated protein (a tryptic peptide carrying the altered amino acid sequence) was present in the amyloid deposits of patients 2 and 3 (Figure 3B). Detailed examination of the apoAI protein coverage showed that certain tryptic peptides, which would have been present had protein encoded by the normal allele been incorporated into amyloid deposits, were absent, indicating that only the protein encoded by the abnormal allele was amyloidogenic (Figure 4). Interestingly, the mutation in patient 1 affected an area of the protein that cannot be detected by the mass spectrometry–based proteomics technology used in the study due to the presence of numerous trypsin cutting sites in this area (Figure 4). In patient 4, the frameshift mutation in APOAI generates a novel C terminus amino acid sequence, which, theoretically, could be detected by mass spectrometry–based proteomics. Although this novel sequence was included in our supplemented database, we did not identify peptides representing the novel sequence, suggesting that this part of the protein was unstable and was most likely cleaved, generating a truncated apoAI protein that was deposited as amyloid.
Figure 4.
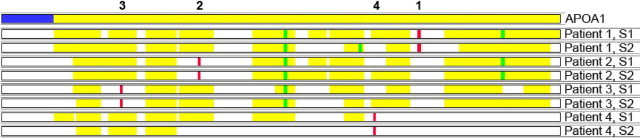
ApoAI protein coverage in apoAI amyloidosis. The top line represents the native apoAI protein (signal peptide in blue and secreted protein in yellow). The numbers above the top line indicate the individual patient's number throughout this article. Two samples (S1 and S2) from four patients are shown. The location of the amino acid residues affected by the mutations in each patient are represented by red bars and oxidized residues are represented by green bars. Most of the apoAI protein is deposited in patients 1, 2, and 3, but the tryptic peptides carrying the variant amino acid sequence are notably absent. In patient 4, where the mutation identified is predicted to cause a frameshift, only the N-terminal part of the protein before the mutation is present. The absence of the peptide carrying the mutated amino residue in each of these four patients indicates that the amyloid fibrils were composed solely of mutated protein and not wild-type apoAI. As discussed in the text, mutated peptides are not detected by the algorithms that only interrogate the data for wild-type sequences. If wild-type apoAI encoded by the normal allele had been incorporated into the amyloid deposits, these peptides would have been detected.
SAP Scintigraphy
SAP scintigraphy did not show any visceral amyloid deposits in patient 1 and showed only renal amyloid deposits in patients 4 and 5. Subclinical hepatic amyloid deposits were detected in patient 2. A large amyloid load affecting the spleen and liver with an obscured kidney signal was identified in patient 3. It should be noted that amyloid deposits in the skin, vocal cord, heart, nerves, and testes do not show up by this technique.
Discussion
Determining the fibril protein remains a challenge in many patients with amyloidosis. Indeed, AL amyloidosis, the most commonly diagnosed systemic form of amyloid, is not infrequently a diagnosis of exclusion, which is complicated by the fact that there is considerable overlap between the clinical features of the different amyloid types. Furthermore, it is important to recognize that the presence of a plasma cell dyscrasia, which occurs in approximately 3% of the population older than 50 years and approximately 5% of those older than 70 years,29 in a patient with amyloidosis does not prove AL type and may be incidental to their amyloidosis.30 The deposits in approximately 20% of patients with AL amyloidosis fail to stain with antibodies against κ and λ immunoglobulin light chains, presumably reflecting the fact that the fibrils consist of misfolded, variable light chain domains within which certain epitopes may be masked.31–33 Similarly, to confirm the amyloidogenicity of a novel mutation, even within a known amyloid fibril protein gene, one is required to demonstrate the protein composition of the amyloid fibrils. Until recently, this could only be achieved by IHC or direct protein sequencing. The latter is technically challenging and requires a substantial quantity of fresh tissue, which often cannot be obtained during life. Classification of amyloidosis by mass spectrometry–based proteomic analysis therefore represents a significant advance in amyloid diagnostics, enabling the fibril protein to be reliably identified from small quantities of formalin-fixed tissue.33
The demonstration, by IHC and/or mass spectrometry, that the amyloid fibrils in four patients were composed of apoAI in conjunction with a typical phenotype of laryngeal involvement in patient 1, liver amyloidosis in patient 2, liver and renal manifestations in patient 3, and renal amyloidosis in patient 4 indicates that these novel APOAI mutations were the cause of their amyloidosis. The IHC was supported by mass spectrometry–based proteomics, which confirmed, for the first time in apoAI amyloidosis, that the amyloid fibrils were composed uniquely of variant protein. Interestingly, only one of these four patients had a family history of amyloidosis. The absence of a family history is apparently unusual in apoAI amyloidosis and might suggest a de novo mutation; however, variable penetrance is well recognized in other types of hereditary systemic amyloidosis.34 We have not been able to obtain DNA from the parents of the three relevant patients to investigate this in more detail. Patient 5 presented a diagnostic challenge. In the presence of a κ light chain–secreting clonal dyscrasia and a novel APOA1 mutation, the IHC was nondiagnostic of the amyloid fibril protein. Only laser microdissection of amyloid fibrils from fixed tissue and mass spectrometry enabled definitive confirmation of monoclonal κ light chain–derived (AL) rather than apoAI amyloidosis in this patient.33 The mass spectrometry findings were later corroborated by SAP scintigraphic evidence of amyloid regression in response to successful suppression of κ light chain production with steroids, thereby excluding apoAI amyloidosis.
ApoAI (Mendelian Inheritance of Man no. 107680) is synthesized in the liver and small intestine and is encoded by a gene located on chromosome 11q23-q24. It is the major protein component of high-density lipoprotein in plasma, and it promotes efflux of cholesterol from cells. The APOAI gene sequence is composed of 8870 nucleotides (National Center for Biotechnology Information accession no. NG_012021) encoding a 267 amino acid primary transcript containing an 18-amino acid prepeptide and 6-amino acid propeptide connected to the amino terminus of the 243 amino acid mature apoAI.35 Although the precise mechanism for the amyloidogenicity of variant apoAI remains unclear, it has been shown that mutated apoAI undergoes proteolytic cleavage releasing the amyloidogenic N-terminal fragment from the full-length protein.5,7,9
There are now 19 mutations in the APOAI gene known to be association with hereditary systemic apoAI amyloidosis, including the four novel amyloidogenic variants described here (Table 1). The phenotype of apoAI amyloidosis may be fairly aggressive and present in teenagers with CKD, hepatomegaly, cardiomyopathy, and sometimes neuropathy19,44 or may be extremely indolent and clinically insignificant. Interestingly, most C-terminal variants seem to be associated with hoarseness due to laryngeal amyloid deposits (Table 1),7,9,10,20,40 although there is even substantial heterogeneity in the age at onset and clinical manifestations between patients with identical mutations. G26R, which is the most frequently reported amyloidogenic apoAI variant among Northern Europeans, is associated with amyloid deposition in the peripheral nerves, as well as the kidneys, liver, and gastrointestinal tract.36,45 Patient 4 in this series presented with symptoms of possible neuropathy and is awaiting confirmatory tests. Two other apoAI variants, L174S and L178H, are reportedly associated with varying degrees of neuropathy.20 Here we report the sixth apoAI variant to be associated with laryngeal amyloidosis, Ala175Pro. The nucleotide change in each of the previous kindreds occurred close to the carboxyl terminus (Leu170Pro,40 Arg173Pro,7 Leu174Ser,9,20 Leu178His10) apart from that in an Italian family with laryngeal, cardiac, and skin amyloidosis who carried the Leu90Pro apoAI variant.8 His155MetfsX46 is the third reported amyloidogenic frameshift mutation in the APOAI gene. The other two, Asn74fs and Ala154fs, were recently described by Rocken and coworkers.40 Interestingly, all three patients with frameshift mutations presented with renal manifestations.
Table 1.
Summary of APOAI Mutations Associated with Amyloidosis, Clinical Phenotype, and Outcome
| APOA1 variant | Ethnicity | Clinical presentation | Age at presentation, years/family history | Organ involvement by amyloid | SAP scintigraphic findings | Organs not involved to date | Clinical Outcome |
|---|---|---|---|---|---|---|---|
| Gly26Arg2,19,36,37 | British Scandinavian | HTN, renal impairment, macroscopic hematuria | 20–46/Y | Kidneys, liver, peripheral nerves, GI tract (peptic ulcers) | Large load: liver, spleen, kidneys | Heart | Slowly progressive CKD, dialysis 11–20 years after presentation. Excellent outcome with transplantation of kidney with or without liver. Death at 47 years of age with renal failure reported. Death after 1–18 years reported. |
| Glu34Lys (current study) | Polish | Renal impairment, subnephrotic proteinuria | 29/Unknown | Kidneys, liver | Large load: spleen and liver, small kidney | Heart, nerves | Slowly progressive renal failure |
| Trp50Arg4 | Jewish (Ashkenazi) | Macroscopic hematuria, proteinuria, renal impairment | 34/Y | Kidneys, liver, GI tract | Large load: liver, spleen, kidneys | Heart, nerves | Progressive renal failure, no reports of successful renal transplantation. Death at the age of 45 years (after 10 years), massive liver and renal amyloidosis. |
| Leu60Arg3 | British | HTN, renal impairment, macroscopic hematuria | 24/Y | Kidneys, liver, testes, heart | Large or small load: liver, spleen, kidneys | Nerves | Good with single organ (kidney, heart) transplantation. Regression after liver transplantation. Death between 35 and 65 years of age from “amyloidosis” |
| Leu64Pro11 | Italian | Proteinuria, renal impairment | 56–58/Unknown | Kidneys, liver | Large load: liver, spleen, kidneys | Heart, nerves | Progressive renal failure, death at 69 years of age with renal failure, good outcome with renal transplantation. |
| Leu60_Phe71delins 60Val_61Thr5,38 | Spanish | Hepatomegaly and abnormal LFT results | 35–45/Y | Liver | Large load: liver, spleen | Heart, kidneys, nerves | Progressively abnormal LFT results (usually slow), liver failure, no reports of successful OLT. Death at 48–66 years of age, after 1–20 years. |
| Glu70_Trp72del39 | British | HTN, renal impairment | 18–21/Y | Kidneys, liver, choroid | Large load: liver, spleen, kidneys | Heart, nerves | ESRD 5 years after presentation. Death at 32 years of age and middle-age reported. Excellent outcome with renal transplantation. (No clinical sequelae of liver and spleen involvement.) |
| Asn74Lysfs40 | German | Renal impairment, tumor of the right ovary | 48/Unknown | Kidneys, uterus, ovaries, pelvic lymph nodes, GI tract | Never performed | Not listed | Not mentioned |
| Phe71Tyr (current study) | British | Palatal mass | 51/Unknown | Liver, palate | Moderate load: liver | Heart, nerves, kidneys | Good outcome up to 52 years of age |
| Leu75Pro12,40–42 | Italian/German/Other | Hepatomegaly, renal impairment | 42–70/Y | Kidneys, liver, testes | Never performed | Heart, nerves, spleen | Gradually progressive liver amyloidosis, CKD, and TIN with death at 67–90 years of age, good outcome. |
| Leu90Pro8 | French/American | Skin lesions, cardiomyopathy (later), dysphonia | 37–51/Y | Skin, heart, larynx | Never performed L, S, A at autopsy | Nerves, kidneys | Progressive cardiac failure and death at 41–70 years of age, no reports of successful heart transplantation. |
| Lys107del43 | Scandinavian | Angina | 45/Unknown | Aortic intimal amyloid; not known (PM study) | Never performed | Nerves, Kidneys | Premature CAD, required CABG, and developed abdominal aortic aneurysm. Died at 68 years of age. |
| Ala154fsX4740 | German | Renal impairment | 58/Unknown | Kidneys | Never performed | Not listed | Not mentioned |
| Leu170Pro40 | German | Laryngeal amyloid deposits | 52/Unknown | Larynx | Never performed | Not listed | Not mentioned |
| His155MetfsX46 (current study) | British | HTN, nephrotic syndrome, renal impairment | 77/Unknown | Kidneys | Small load: kidneys | Heart, liver | CKD, short follow-up |
| Arg173Pro7 | American/British | Skin lesions, dysphonia, cardiomyopathy (later) | 20–45/Y | Kidneys, skin, heart, larynx | Small load with equivocal kidneys | Nerves, spleen | Progression of cardiomyopathy and renal failure, death at 52–63 years of age, good outcome with cardiac and renal transplantation |
| Leu174Ser9,20 | Italian/Dutch | Infertility, rash, cardiomyopathy, dysphonia | 42–45/Y | Skin, testes, heart, larynx, nerves | Small load: no visceral amyloid deposits | Nerves, liver, spleen | Progressive cardiomyopathy, death at 50–52 years of age, good outcome after cardiac transplantation |
| Ala175Pro (current study) | British | Dysphonia | 38/Unknown | Larynx, testes | Small load: no visceral amyloid deposits | Heart, nerves, liver, spleen, kidneys | Good outcome up to 43 years of age |
| Leu178His10,20 | French | Dysphonia, cardiomyopathy | 34/Y | Larynx, skin, heart, nerves | Never performed | Liver, spleen | Progressive cardiomyopathy, death at 39 years of age |
A, adrenal glands; CABG, coronary artery bypass graft; CAD, coronary artery disease; ESRD, end-stage renal disease; GI, gastrointestinal; HTN, hypertension; L, liver; LFT, liver function test; OLT, orthotopic liver transplantation; PM, post-mortem; S, spleen; TIN, tubulointerstitial nephritis.
In conclusion, we report here five novel apoAI variants, four of which cause hereditary systemic amyloidosis. Mass spectrometry–based proteomics identified apoAI as the main constituent of amyloid deposits in all four patients with pathogeneic APOAI mutations and was successful in identifying the amyloid fibril protein as monoclonal light chain (AL) rather than apoAI in one patient who had both a clonal dyscrasia and a novel APOAI mutation whose IHC was nondiagnostic of the fibril protein. Minimal formalin-fixed amyloidotic tissue is required for the procedure, making this an invaluable new diagnostic tool for amyloid typing. The substantial phenotypic heterogeneity among patients with identical apoAI variants implies that other genetic and environmental factors influence clinical manifestations and support the need for APOAI gene sequencing in patients with apparent localized amyloidosis, particularly involving the larynx or skin.
Acknowledgments
We acknowledge all of the physicians and surgeons who were involved in the clinical care of the patients reported in this study.
Footnotes
Supported by the UK Department of Health.
The licensed patent rights to perform protein extraction from paraffin-embedded tissue for the mass spectrometry–based amyloid typing test were granted by Expression Pathology, Inc.
References
- 1.Ostertag B. Demonstration einer eigenartigen familiaren paraamyloidose. Zentralbl Aug Pathol. 1932;56:253–254. [Google Scholar]
- 2.Jones L.A., Harding J.A., Cohen A.S., Skinner M. New USA family has apolipoprotein AI (Arg26) variant. In: Natvig J.B., Førre Ø., Husby G., Husebekk A., Skogen B., Sletten K., Westermark P., editors. Kluwer Academic Publishers; Dordrecht: 1991. pp. 385–388. [Google Scholar]
- 3.Soutar A.K., Hawkins P.N., Vigushin D.M., Tennent G.A., Booth S.E., Hutton T., Nguyen O., Totty N.F., Feest T.G., Hsuan J.J., Pepys M.B. Apolipoprotein AI mutation Arg-60 causes autosomal dominant amyloidosis. Proc Natl Acad Sci U S A. 1992;89:7389–7393. doi: 10.1073/pnas.89.16.7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Booth D.R., Tan S.Y., Booth S.E., Hsuan J.J., Totty N.F., Nguyen O., Hutton T., Vigushin D.M., Tennent G.A., Hutchinson W.L., Thomson N., Soutar A.K., Hawkins P.N., Pepys M.B. A new apolipoprotein AI variant: Trp50Arg, causes hereditary amyloidosis. Q J Med. 1995;88:695–702. [PubMed] [Google Scholar]
- 5.Booth D.R., Tan S.Y., Booth S.E., Tennent G.A., Hutchinson W.L., Hsuan J.J., Totty N.F., Nguyen O., Soutar A.K., Hawkins P.N., Bruguera M., Caballería J., Solé M., Campistol J.M., Pepys M.B. Hereditary hepatic and systemic amyloidosis caused by a new deletion/insertion mutation in the apolipoprotein AI gene. J Clin Invest. 1996;97:2714–2721. doi: 10.1172/JCI118725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Persey M.R., Booth D.R., Booth S.E., van Zyl-Smit R., Adams B.K., Fattaar A.B., Tennent G.A., Hawkins P.N., Pepys M.B. Hereditary nephropathic systemic amyloidosis caused by a novel variant apolipoprotein A-I. Kidney Int. 1998;53:276–281. doi: 10.1046/j.1523-1755.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- 7.Hamidi Asl K., Liepnieks J.J., Nakamura M., Parker F., Benson M.D. A novel apolipoprotein A-1 variant: Arg173Pro, associated with cardiac and cutaneous amyloidosis. Biochem Biophys Res Commun. 1999;257:584–588. doi: 10.1006/bbrc.1999.0518. [DOI] [PubMed] [Google Scholar]
- 8.Hamidi Asl L., Liepnieks J.J., Hamidi Asl K., Uemichi T., Moulin G., Desjoyaux E., Loire R., Delpech M., Grateau G., Benson M.D. Hereditary amyloid cardiomyopathy caused by a variant apolipoprotein A1. Am J Pathol. 1999;154:221–227. doi: 10.1016/S0002-9440(10)65268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Obici L., Bellotti V., Mangione P., Stoppini M., Arbustini E., Verga L., Zorzoli I., Anesi E., Zanotti G., Campana C., Viganò M., Merlini G. The new apolipoprotein A-I variant Leu174 → Ser causes hereditary cardiac amyloidosis, and the amyloid fibrils are constituted by the 93-residue N-terminal polypeptide. Am J Pathol. 1999;155:695–702. doi: 10.1016/S0002-9440(10)65167-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Sousa M.M., Vital C., Ostler D., Fernandes R., Pouget-Abadie J., Carles D., Saraiva M.J. Apolipoprotein AI and transthyretin as components of amyloid fibrils in a kindred with apoAI Leu178His amyloidosis. Am J Pathol. 2000;156:1911–1917. doi: 10.1016/S0002-9440(10)65064-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy C.L., Wang S., Weaver K., Gertz M.A., Weiss D.T., Solomon A. Renal apolipoprotein A-I amyloidosis associated with a novel mutant Leu64Pro. Am J Kidney Dis. 2004;44:1103–1109. doi: 10.1053/j.ajkd.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 12.Obici L., Palladini G., Giorgetti S., Bellotti V., Gregorini G., Arbustini E., Verga L., Marciano S., Donadei S., Perfetti V., Calabresi L., Bergonzi C., Scolari F., Merlini G. Liver biopsy discloses a new apolipoprotein A-I hereditary amyloidosis in several unrelated Italian families. Gastroenterology. 2004;126:1416–1422. doi: 10.1053/j.gastro.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Benson M.D., Liepnieks J.J., Yazaki M., Yamashita T., Hamidi Asl K., Guenther B., Kluve-Beckerman B. A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics. 2001;72:272–277. doi: 10.1006/geno.2000.6499. [DOI] [PubMed] [Google Scholar]
- 14.Benson M.D., Liepnieks J., Uemichi T., Wheeler G., Correa R. Hereditary renal amyloidosis associated with a mutant fibrinogen α-chain. Nat Genet. 1993;3:252–255. doi: 10.1038/ng0393-252. [DOI] [PubMed] [Google Scholar]
- 15.Uemichi T., Liepnieks J.J., Benson M.D. Hereditary renal amyloidosis with a novel variant fibrinogen. J Clin Invest. 1994;93:731–736. doi: 10.1172/JCI117027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uemichi T., Liepnieks J.J., Yamada T., Gertz M.A., Bang N., Benson M.D. A frame shift mutation in the fibrinogen A α-chain gene in a kindred with renal amyloidosis. Blood. 1996;87:4197–4203. [PubMed] [Google Scholar]
- 17.Hamidi Asl L., Liepnieks J.J., Uemichi T., Rebibou J.M., Justrabo E., Droz D., Mousson C., Chalopin J.M., Benson M.D., Delpech M., Grateau G. Renal amyloidosis with a frame shift mutation in fibrinogen a α-chain gene producing a novel amyloid protein. Blood. 1997;90:4799–4805. [PubMed] [Google Scholar]
- 18.Pepys M.B., Hawkins P.N., Booth D.R., Vigushin D.M., Tennent G.A., Soutar A.K., Totty N., Nguyen O., Blake C.C.F., Terry C.J., Feest T.G., Zalin A.M., Hsuan J.J. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993;362:553–557. doi: 10.1038/362553a0. [DOI] [PubMed] [Google Scholar]
- 19.Nichols W.C., Gregg R.E., Brewer H.B.J., Benson M.D. A mutation in apolipoprotein A-I in the Iowa type of familial amyloidotic polyneuropathy. Genomics. 1990;8:318–323. doi: 10.1016/0888-7543(90)90288-6. [DOI] [PubMed] [Google Scholar]
- 20.Hazenberg A.J., Dikkers F.G., Hawkins P.N., Bijzet J., Rowczenio D., Gilbertson J., Posthumus M.D., Leijsma M.K., Hazenberg B.P. Laryngeal presentation of systemic apolipoprotein A-I-derived amyloidosis. Laryngoscope. 2009;119:608–615. doi: 10.1002/lary.20106. [DOI] [PubMed] [Google Scholar]
- 21.Kyle R.A., Gertz M.A. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45–59. [PubMed] [Google Scholar]
- 22.Kyle R.A., Gertz M.A., Greipp P.R., Witzig T.E., Lust J.A., Lacy M.Q. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336:1202–1207. doi: 10.1056/NEJM199704243361702. [DOI] [PubMed] [Google Scholar]
- 23.Puchtler H., Sweat F., Levine M. On the binding of Congo red by amyloid. J Histochem Cytochem. 1962;10:355–364. [Google Scholar]
- 24.Tennent G.A., Cafferty K.D., Pepys M.B., Hawkins P.N. Congo red overlay immunohistochemistry aids classification of amyloid deposits. In: Kyle R.A., Gertz M.A., editors. Parthenon Publishing; Pearl River, New York: 1999. pp. 160–162. [Google Scholar]
- 25.Talmud P., Tybjaerg-Hansen A., Bhatnagar D., Mbewu A., Miller J.P., Durrington P., Humphries S. Rapid screening for specific mutations in patients with a clinical diagnosis of familial hypercholesterolaemia. Atherosclerosis. 1991;89:137–141. doi: 10.1016/0021-9150(91)90053-6. [DOI] [PubMed] [Google Scholar]
- 26.Hawkins P.N., Lavender J.P., Pepys M.B. Evaluation of systemic amyloidosis by scintigraphy with 123I-labeled serum amyloid P component. N Engl J Med. 1990;323:508–513. doi: 10.1056/NEJM199008233230803. [DOI] [PubMed] [Google Scholar]
- 27.Bradwell A.R., Carr-Smith H.D., Mead G.P., Tang L.X., Showell P.J., Drayson M.T., Drew R. Highly sensitive, automated immunoassay for immunoglobulin free light chains in serum and urine. Clin Chem. 2001;47:673–680. [PubMed] [Google Scholar]
- 28.Shoulders C.C., Kornblihtt A.R., Munro B.S., Baralle F.E. Gene structure of human apolipoprotein A1. Nucleic Acids Res. 1983;11:2827–2837. doi: 10.1093/nar/11.9.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kyle R.A., Therneau T.M., Rajkumar S.V., Larson D.R., Plevak M.F., Offord J.R., Dispenzieri A., Katzmann J.A., Melton L.J., III Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354:1362–1369. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 30.Lachmann H.J., Booth D.R., Booth S.E., Bybee A., Gilbertson J.A., Gillmore J.D., Pepys M.B., Hawkins P.N. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786–1791. doi: 10.1056/NEJMoa013354. [DOI] [PubMed] [Google Scholar]
- 31.Novak L., Cook W.J., Herrera G.A., Sanders P.W. AL-amyloidosis is underdiagnosed in renal biopsies. Nephrol Dial Transplant. 2004;19:3050–3053. doi: 10.1093/ndt/gfh503. [DOI] [PubMed] [Google Scholar]
- 32.Solomon A., Murphy C.L., Westermark P. Unreliability of immunohistochemistry for typing amyloid deposits. Arch Pathol Lab Med. 2008;132:14–15. doi: 10.5858/2008-132-14b-IR. [DOI] [PubMed] [Google Scholar]
- 33.Vrana J.A., Gamez J.D., Madden B.J., Theis J.D., Bergen H.R., III, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957–4959. doi: 10.1182/blood-2009-07-230722. [DOI] [PubMed] [Google Scholar]
- 34.Gillmore J.D., Lachmann H.J., Rowczenio D., Gilbertson J.A., Zeng C.H., Liu Z.H., Li L.S., Wechalekar A., Hawkins P.N. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A alpha-chain amyloidosis. J Am Soc Nephrol. 2009;20:444–451. doi: 10.1681/ASN.2008060614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brewer H.B., Jr., Fairwell T., LaRue A., Ronan R., Houser A., Bronzert T.J. The amino acid sequence of human APOA-I, an apolipoprotein isolated from high density lipoproteins. Biochem Biophys Res Commun. 1978;80:623–630. doi: 10.1016/0006-291x(78)91614-5. [DOI] [PubMed] [Google Scholar]
- 36.Nichols W.C., Dwulet F.E., Liepnieks J., Benson M.D. Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem Biophys Res Commun. 1988;156:762–768. doi: 10.1016/s0006-291x(88)80909-4. [DOI] [PubMed] [Google Scholar]
- 37.Vigushin D.M., Gough J., Allan D., Alguacil A., Penner B., Pettigrew N.M., Quinonez G., Bernstein K., Booth S.E., Booth D.R., Soutar A.K., Hawkins P.N., Pepys M.B. Familial nephropathic systemic amyloidosis caused by apolipoprotein AI variant Arg26. Q J Med. 1994;87:149–154. [PubMed] [Google Scholar]
- 38.Caballeria J., Bruguera M., Sole M., Campistol J.M., Rodes J. Hepatic familial amyloidosis caused by a new mutation in the apolipoprotein AI gene: clinical and pathological features. Am J Gastroenterol. 2001;96:1872–1876. doi: 10.1111/j.1572-0241.2001.03887.x. [DOI] [PubMed] [Google Scholar]
- 39.Persey M.R., Booth D.R., Booth S.E., van Zyl-Smit R., Hawkins P.N., Pepys M.B. A new deletion mutation of the apolipoprotein AI gene causing hereditary amyloidosis. Clin Sci. 1996;90(Suppl 34):33P. [Google Scholar]
- 40.Eriksson M., Schonland S., Yumlu S., Hegenbart U., von Hutten H., Gioeva Z., Lohse P., Buttner J., Schmidt H., Rocken C. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: identification of three novel mutations in the APOA1 gene. J Mol Diagn. 2009;11:257–262. doi: 10.2353/jmoldx.2009.080161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coriu D., Dispenzieri A., Stevens F.J., Murphy C.L., Wang S., Weiss D.T., Solomon A. Hepatic amyloidosis resulting from deposition of the apolipoprotein A-I variant Leu75Pro. Amyloid. 2003;10:215–223. doi: 10.3109/13506120309041738. [DOI] [PubMed] [Google Scholar]
- 42.Gregorini G., Izzi C., Obici L., Tardanico R., Rocken C., Viola B.F., Capistrano M., Donadei S., Biasi L., Scalvini T., Merlini G., Scolari F. Renal apolipoprotein A-I amyloidosis: a rare and usually ignored cause of hereditary tubulointerstitial nephritis. J Am Soc Nephrol. 2005;16:3680–3686. doi: 10.1681/ASN.2005040382. [DOI] [PubMed] [Google Scholar]
- 43.Amarzguioui M., Mucchiano G., Häggqvist B., Westermark P., Kavlie A., Sletten K., Prydz H. Extensive intimal apolipoprotein A1-derived amyloid deposits in a patient with an apolipoprotein A1 mutation. Biochem Biophys Res Commun. 1998;242:534–539. doi: 10.1006/bbrc.1997.8005. [DOI] [PubMed] [Google Scholar]
- 44.Van Allen M., Frohlich J., Davis J. Inherited predisposition to generalized amyloidosis. Neurology. 1969;19:10–25. doi: 10.1212/wnl.19.1.10. [DOI] [PubMed] [Google Scholar]
- 45.Joy T., Wang J., Hahn A., Hegele R.A. APOA1 related amyloidosis: a case report and literature review. Clin Biochem. 2003;36:641–645. doi: 10.1016/s0009-9120(03)00110-3. [DOI] [PubMed] [Google Scholar]