

Abstract
The TP53 tumor suppressor gene is mutated in the majority of human cancers. Inactivation of p53 in a variety of animal models results in early-onset tumorigenesis, reflecting the importance of p53 as a gatekeeper tumor suppressor. We generated a mutant Tp53 allele in the rat using a target-selected mutagenesis approach. Here, we report that homozygosity for this allele results in complete loss of p53 function. Homozygous mutant rats predominantly develop sarcomas with an onset of 4 months of age with a high occurrence of pulmonary metastases. Heterozygous rats develop sarcomas starting at 8 months of age. Molecular analysis revealed that these tumors exhibit a loss-of-heterozygosity of the wild-type Tp53 allele. These unique features make this rat highly complementary to other rodent p53 knockout models and a versatile tool for investigating tumorigenesis processes as well as genotoxic studies.
The TP53 gene is arguably the most extensively studied tumor suppressor and is found mutated in the majority of human cancers.1,2 To study the in vivo role of p53 loss in mammalian tumorigenesis, transgenic mouse models have proven to be extremely useful. Loss of p53 in mice accelerates neoplasia formation reflecting strain predispositions, resulting in relatively uniform tumor spectra with thymic lymphoma as the dominant tumor type.3–6 In human lymphomas, TP53 is less frequently mutated.7,8 Although lymphomas have been found in patients with Li-Fraumeni syndrome, who are born with a defective p53 germ line allele, the tumor spectrum in these patients is more diverse and reveals a high frequency of sarcomas.9 This discrepancy can be explained by the fact that human TP53 mutations occur in heterozygous state and loss of the wild-type allele (loss of heterozygosity [LOH]) has to precede tumor development. Indeed, heterozygous mutant p53 mouse models, which require LOH before tumorigenesis can occur, develop a spectrum of tumors that is more similar to humans. However, this slows down tumor onset to approximately 9 months of age,10 increasing the costs of using these models for the development of therapeutic interventions.
As a mammalian genetic model organism, the rat is highly complementary to the mouse model and is well-suited for studying human disease,11 including cancer.12–14 Although genetic tools to manipulate the rat genome have been scarce for a long time, recent technical developments have enabled a variety of gene targeting methods in rat model systems, including zinc-finger nuclease (ZFN)-mediated gene targeting15 and homologous recombination (HR) in embryonic stem (ES) cells.16 In fact, Tp53 was the first and to date the only gene that was knocked out in the rat using ES cell-based HR.16 Although the development of hemangiosarcomas in these rats was reported in a brief communication while finishing this study,17 no thorough phenotypic analysis has been reported for a P53-deficient rat line until now.
An alternative method for generating rat mutants is N-ethyl-N-nitrosourea (ENU)-driven target-selected mutagenesis.18,19 This technique is based on treating male animals with the supermutagen ENU, which very efficiently introduces random point mutations in the DNA of spermatogenial stem cells. By crossing the ENU-treated males with untreated females, a F1 library is generated in which each individual carries unique heterozygous mutations.20 The DNA of the F1 library can then be screened for mutations in genes of interest that affect protein function, like the introduction of a premature translation stop. Using this technique a range of rat knockout models have been successfully generated,21 including animals deficient for tumor suppressors like Apc,12 Bcra2,13 and Msh6.14
Here we report the first in-depth characterization of p53 deficiency in the rat. Homozygous mutant rats completely lack TP53 protein and display a decrease in survival as a result of tumorigenesis. However, unlike the p53 knockout mouse models that primarily develop lymphomas, the rat predominantly develops sarcomas, indicating species-specificity in tumor spectrum and demonstrating the importance of the availability and use of complementary mammalian mutant models. Heterozygous mutant rats exhibit a delay in tumor onset when compared with homozygous mutant rats, although the tumor spectrum is not significantly different. Taken together, the Tp53C273X rat can be a unique and valuable tool for studying early onset p53-deficient tumor development.
Materials and Methods
Animals and Primary Rat Embryonic Fibroblasts Isolation
All experiments were approved by the Animal Care Committee of the Royal Dutch Academy of Sciences according to the Dutch legal ethical guidelines. Experiments were designed to minimize the number of required animals and their suffering. Animals were housed under standard conditions in groups of two to three per cage per sex under controlled experimental conditions (12-hour light/dark cycle, 21 ± 1°C, 60% relative humidity, food and water ad libitum). ENU treatment of male rats was done as described.22 Genes of interest were screened using PCR amplification followed by capillary sequencing as described.18 For rat embryonic fibroblast (REF) isolation heterozygous carriers were mated and at E13.5 embryos were isolated. After washing the embryo thoroughly, the head and visceral organs were removed and used for DNA isolation and genotyping. The embryos were minced and treated with trypsin to get a single cell suspension. REFs were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum.
Histological Analysis
Tissues were sampled and fixed in 4% neutral buffered formaldehyde and fixed overnight. After routine processing and paraffin embedding, sections of 4–5 μm thickness were cut and mounted on glass slides for H&E staining. For immunohistological stainings, 4 μm paraffin tissue sections were mounted on silan-coated glass microscope slides, dried overnight at 55°C, and deparaffinized in 2 × 5 minutes xylene, 2 × 3 minutes 100% ethanol, 96% or 80% (MyoD-1), 70% ethanol, and rehydrated in water. If needed, slides were then pretreated, blocked with 1:10 normal horse or goat serum in PBS for 15 minutes, and 30 minutes 1% H2O2 in methanol before incubation with the primary antibodies, Factor VIIIra (Dako, Heverlee, Belgium), MyoD-1 (Dako), NSE (Lab Vision Products, Fremont, CA), Synaptophysin (Dako), CD3 (Cell Marque Corporation, Rocklin, CA), CD79a (Dako) or Vimentin (Fremont, CA). Slides were three times rinsed in PBS/Tween and incubated for 30 minutes with ABC/PO. After rinsing (three times, PBS/Tween), DAB (500 mg/L in Tris/HCl buffer, pH 7.8 + 1 mL 35% H2O2; for MyoD-1: 400 mg/L in Tris/Maleate buffer, pH 7.6 + 0.2 mL 30% H2O2) was added to visualize immunoreactivity.
LOH Analyses
DNA was isolated from snap-frozen tumor and healthy tissue of heterozygous mutant animals. Tp53 was amplified with gene-specific primers encompassing the ENU-induced knockout allele. The resulting amplicon was resequenced and LOH was determined by genotyping the knockout mutation.
Results
Tp53C273X Results in a Complete Lack of p53
In an ENU-driven target-selected mutagenesis screen in the outbred Wistar background, we isolated an F1 animal carrying a nonsense mutation in the sixth exon of rat Tp53 (Figure 1A). The mutation truncates the protein at the DNA binding domain, eliminating functionally essential domains including the nuclear localization domain and the homo-oligomerization domain. Even when the truncated protein would be stable, it is likely that the mutation will result in a complete loss of p53 function. To test the molecular phenotype of Tp53C273X, primary REFs were isolated and treated with ultraviolet radiation to introduce double strand breaks in the DNA that will result in p53 stabilization and translocation to the nucleus.23 Whereas wild-type REFs reveal p53 stabilization and translocation to the nucleus as expected, no full-length or truncated p53 could be detected in homozygous mutant REFs (Figure 1), indicating that Tp53C273X/C273X results in loss of p53 protein. Furthermore, Western blot analysis of doxorubicin-treated REF lysates confirms complete lack of p53 in homozygous mutant cells (Figure 1C). Notably, no truncated protein was detected in homozygous mutant fibroblasts, probably due to nonsense-mediated decay of the mRNA, which has also been observed in other ENU-induced knockout models.14
Figure 1.
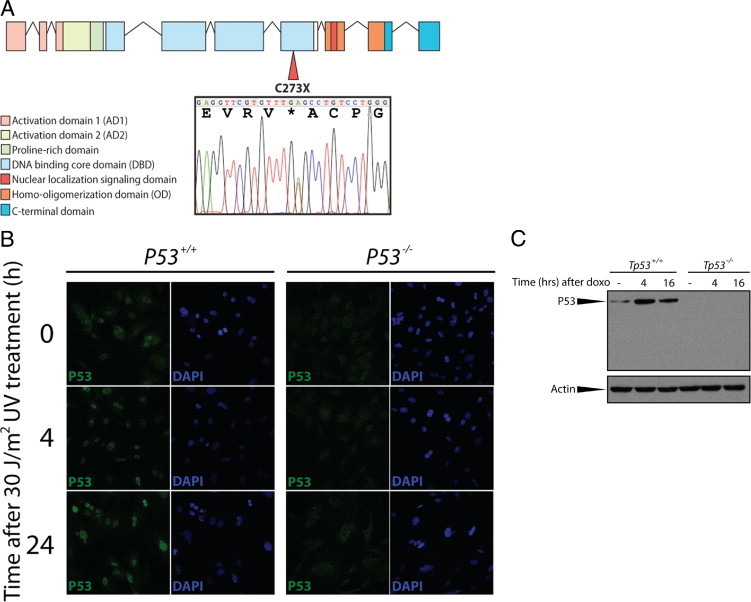
A: Schematic representation of the genomic DNA encoding rat P53. An N-ethyl-N-nitrosourea (ENU)-induced mutation in the sixth exon of the gene results in a premature stop codon, truncating the protein within the DNA binding domain. The sequence trace indicates the T to A transition in a heterozygous rat resulting in a premature stop codon. Asterisk indicates the stopcodon that was introduced by the ENU-induced mutagenesis. B:Tp53C273X results in the lack of p53 protein and function in primary rat embryonic fibroblasts (REFs). Whereas wild-type REFs show p53 stabilization and translocation to the nucleus on UV radiation, no p53 protein is observed in homozygous mutant REFs. C:Tp53C273X results in a complete lack of p53 protein. Wild-type and homozygous mutant REFs were treated with 0.5 μmol/L doxorubicin and lysed at the indicated time points. Western Blotting reveal the presence of p53 in wild type REFs, which is stabilized on induction of double strand breaks, whereas no protein can be detected in homozygous mutant cells. Notably, the epitope of the anti-p53 monoclonal antibody 1C12 is located N-terminally from the ENU-induced premature translational stop, demonstrating complete absence of potentially truncated p53 protein.
p53-Deficient Rats Display Early-Onset Tumorigenesis
Next, we bred Tp53+/C273X to homozygosity by crossing heterozygous carriers and monitored tumor development. As expected, homozygous mutant animals showed a decrease in survival as a result of early onset tumorigenesis. Remarkably, almost all Tp53C273X/C273X animals developed grossly visible tumors or suffered marked conditional decline around the same time at 4 months of age (Figure 2A). From the 11 homozygous mutant animals that were monitored, 10 could be histologically examined (Figure 2B and Table 1). Hemangiosarcomas, or pulmonary metastases thereof, were the most frequently observed neoplasms (7 of 10 animals). Interestingly, one animal with a hemangiosarcoma also carried a multicentric lymphoma. Hemangiosarcomas were generally composed of plump, spindle-shaped cells growing in bundles and solid sheets, with frequent blood-filled intercellular spaces (Figure 3A). These hemangiosarcomas stained positive for factor VIIIra by immunohistochemistry, confirming an endothelial origin of these neoplastic cells (Figure 3B). In two animals, tentative primary hemangiosarcomas were found attached to the wall of a visceral cavity (thoracic wall and adrenal region, respectively). In these cases the tumor cells formed multilayered cords; differential diagnoses of transitional cell carcinoma were deemed less likely based on negative keratin, and positive vimentin and factor VIIIra immunohistochemistry. The rhabdomyosarcoma was grossly found on the left kidney. The remaining five animals showed multiple pulmonary metastasis histologically consistent of hemangiosarcoma, typically located inside, or centered around blood vessels, indicating extensive hematogenic spread (Figure 3C), but no primary neoplasm was located. Enlarged spleen and liver in the rat with the hemangiosarcoma at the thoracic wall were the result of diffuse lymphoma infiltration (Figure 3D), a second neoplasm in this animal. Development of lymphoma was restricted to this one homozygous mutant rat and extensive CD79a immunoreactivity indicated this was of B-cell origin (Figure 3E). Apart from liver and spleen, lymphoma cells focally invaded the hemangiosarcoma (Figure 3E). We did not observe involvement of the thymus, as has been observed in p53 knockout mice.3–6 Further tumors were detected in two of the remaining animals: one carried an undifferentiated sarcoma, which showed immunostaining for MyoD-1, a skeletal muscle marker; one animal presented with a large abdominal tumor in the region of the left adrenal. The tumor cells were relatively uniform and organized along thin fibrovascular septa, as typically observed in masses from endocrine origin (Figure 3F). Some neoplastic cells showed immunoreactivity against neuron specific enolase, synaptophysin, or cromogranin A, indicating an adrenocortical origin (Figure 3G).
Figure 2.
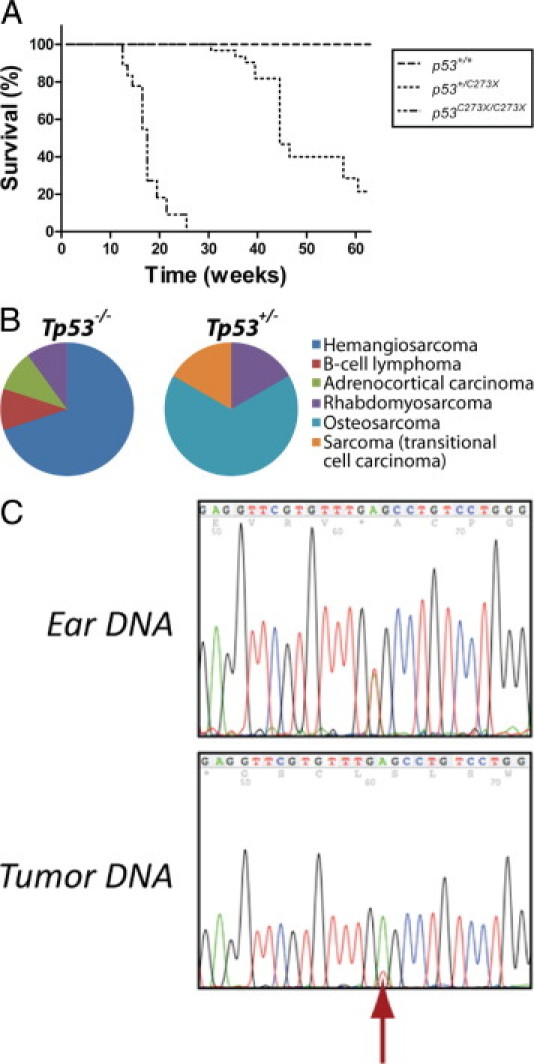
Tp53C273X mutant rats demonstrate a decrease in survival as a result of spontaneous tumor development. A: Homozygous mutant animals (n = 11) have sudden mortality at the age of 4 months, whereas a prolonged survival is observed in heterozygous carriers (n = 8) that reveals a median survival of approximately 9 months. No wild-type littermates (n = 4) became moribund during this study. B: Pie charts of the tumor types identified in homozygous (Tp53−/−) and heterozygous (Tp53+/−) mutant animals. C: Tumors in heterozygous (Tp53+/−) mutant rats display loss of heterozygosity of the wild-type Tp53 allele (red arrow). Sequencing analysis of DNA isolated from the ear revealed that the animals were heterozygous carriers of the Tp53C273X allele; however, loss of the wild-type allele was observed in tumor tissue.
Table 1.
Tumor Spectrum in Mutant Tp53 Rats
| Rat ID | Genotype | Sex | Age (weeks) | Tumor diagnose | IHC⁎ | LOH | Metastases |
|---|---|---|---|---|---|---|---|
| 60 | −/− | Male | 18 | Hemangiosarcoma | Factor VIIIra+++ | — | Lung |
| B-cell lymphoma | CD3+; CD79a+++ | — | Liver | ||||
| 61 | −/− | Male | 18 | Hemangiosarcoma† | Factor VIIIra+++ | — | Lung |
| 62 | −/− | Male | 18 | Hemangiosarcoma† | Factor VIIIra++ | — | Lung |
| 66 | −/− | Male | 20 | Adrenocortical carcinoma | NSE+; Factor VIIIra-; Synaptophysin-; Chromogranin A- | — | Lung |
| 76 | −/− | Male | 26 | Hemangiosarcoma† | ND | — | Lung |
| 77 | −/− | Male | 17 | ND‡ | — | — | |
| 140 | −/− | Female | 22 | No tumor detected | — | — | — |
| 164 | −/− | Male | 13 | Hemangiosarcoma | Factor VIIIra+++; Pankeratin-; Vimentin+++ | — | — |
| 198 | −/− | Male | 13 | Rhabdomyosarcoma; undifferentiated sarcoma | ND | — | — |
| 199 | −/− | Male | 14 | Hemangiosarcoma | ND | — | — |
| 223 | −/− | Female | 15 | Hemangiosarcoma | ND | — | Lung |
| 9 | +/− | Female | 36 | Rhabdomyosarcoma | MyoD1++; Factor VIIIra+ | Yes | Lung |
| 20 | +/− | Female | 45 | Osteosarcoma | ND | Yes | ND |
| 63 | +/− | Male | 50 | Sarcoma; transitional cell carcinoma in situ | ND | Yes | — |
| 74 | +/− | Male | 45 | No tumor detected | — | — | — |
| 75 | +/− | Male | 40 | ND‡ | — | ND | — |
| 82 | +/− | Male | 45 | Osteosarcoma | ND | Yes | Lung |
| 97 | +/− | Male | 47 | Osteosarcoma | ND | Yes | Lung |
| 100 | +/− | Female | 45 | Osteosarcoma | ND | Yes | — |
| 126 | +/− | Female | 31 | No tumor detected | — | — | — |
ND, not determined; LOH, loss of heterozygosity.
Immunohistochemistry used to determine tumor type. +++ indicates frequent expression of indicated marker, ++ indicates moderate expression, + means rare marker expression, and — means no marker expression found.
Primary tumor could not be identified; however multiple pulmonary metastases were found and used for IHC analyses.
Animal was found dead in the cage and in a decomposing state.
Figure 3.
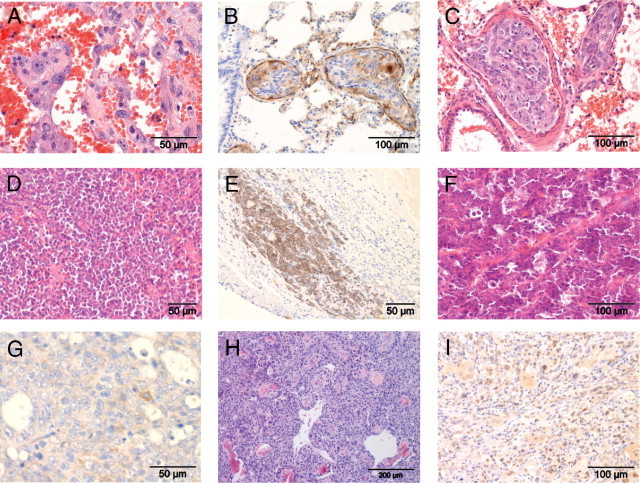
Tp53C273X predominantly develop sarcomas. A: Representative image of clusters of plump, pleomorphic tumor cells separated by blood-filled spaces, morphologically consistent with hemangiosarcoma. B: Marked Factor VIIIra immunoreactivity in intravascular pulmonary metastasis of hemangiosarcoma. Note positivity in pre-existing, non-neoplastic endothelium (internal positive control). C: Tumor emboli in pulmonary blood vessels consist of cells highly pleomorphic cells with indistinct cell borders and variably sized nuclei. D: Monotonous population of neoplastic lymphocytes obscuring the normal architecture of the spleen. E: Infiltration of neoplastic lymphocytes into a hemangiosarcoma from the same animal. CD79a immunoreactivity indicates B-cell origin. F: Abdominal mass from the region of the left adrenal. Small polygonal, relatively uniform cells aligned along fine fibrovascular stroma as is typical for endocrine tissue. G: Only rare activity for neuron-specific enolase was observed in the neoplasm shown in F, consistent with adrenocortical origin. H: Hind limb neoplasm in heterozygous p53 mutant rat. Neoplastic spindle cell concentrically arranged around islands of partly mineralized bone, consistent with osteosarcoma. I: Rhabdomyosarcoma showing multinucleate cells with rows of nuclei and cytoplasmic and nuclear MyoD1 immunoreactivity.
Animals Heterozygous for Tp53C273X Show Slower Onset and Different Spectrum
Animals heterozygous for Tp53C273X also displayed a decrease in tumor-free survival compared with wild-type littermates; however, it was considerably prolonged compared with homozygous mutant littermates (Figure 2A). This gene dosage effect has also been observed in mutant mouse models and can be attributed to the time needed to lose the wild-type allele before tumorigenesis is initiated.5,6 Indeed, LOH was confirmed in the tumors of all heterozygous animals (Figure 2C and Table 1). From eight heterozygous animals that were monitored, seven could be histologically examined. Of these, one animal presented with hind limb paralysis, but no tumor was macroscopically detected. Six Tp53+/− animals carried sarcomas, all located on the hind limb and in one case, extending into the abdomen. Four of these could be further classified as osteosarcomas based on their histological appearance of pleomorphic spindle cells with moderate amounts of pale eosinophilic intercellular material with occasional mineralization (osteoid), sometimes containing osteocytes within lacunae, consistent with osteosarcoma (Figure 3H). Of the remaining two, one tumor was classified as rhabdomyosarcoma based on weak but consistent MyoD-1 (a skeletal muscle marker) immunostaining (Figure 3I), and inconsistent Factor VIIIra staining. The final tumor could not be further differentiated. Interestingly, none of these neoplasms in heterozygotes resembled the tumors detected in homozygotes and pulmonary metastatic spread was observed in only three animals (50%; Table 1). This underlines a more aggressive behavior in homozygotes and provides additional explanation for the reduced survival of homozygote tumor carriers compared with heterozygous animals. At the time of finishing this article, three heterozygous carriers were still alive (age >12 months). Of the four wild-type littermates, none became moribund within 12 months, indicating that the early-onset tumorigenesis was a result of p53 deficiency.
Discussion
The ENU-induced Tp53C273X mutation results in a complete loss-of-function phenotype in the rat. At the molecular level we found that no p53 protein could be detected in homozygous mutant REFs, even after ultraviolet-induced DNA damage, in contrast to wild-type REFs. Consistent with multiple murine p53 knockout models3–6 and the high prevalence of p53 mutations found in human tumors,1 p53-deficiency results in an increased tumorigenesis in rats.
Whereas existing p53-deficient mouse models predominantly develop lymphomas, our present results indicate that homozygous mutant p53 rats predominantly develop metastasizing sarcomas, indicating species-specific phenotypes in different rodent models. Although the number of animals we followed in this study is relatively low (11 homozygous, 8 heterozygous, and 4 wild-type animals), the tumor spectrum was very consistent between all animals, as demonstrated by histological and immunohistochemical analyses.
Significant phenotypic differences between species were also observed in the APC mutant rat,12 the BRCA213 and MSH6 knockout rats14 when compared with similar mouse models. In general, differences in tumor spectra are observed, like a predominant development of colonic tumors in the APC mutant rat in contrast to a predominant development of small intestinal tumors in the Apcmin mouse.12 In addition, the mutant rat models seem to exhibit increased tolerance to genetically-induced tumorigenesis because APC mutants as well as MSH6 knockout rats show a delayed onset of tumor onset and prolonged survival compared with similar mouse models.12,14 Even more surprising, whereas BCRA2 mouse knockout mutants are not viable, BCRA2 knockout rats are viable, although a considerable decrease in survival was observed as a result of increased tumorigenesis.13
Our present observations indicate a similar rate of tumor onset in the rat when compared with p53 knockout mouse models (tumor onset of 3 to 4 months of age in mice versus 4 months in rats), and perhaps a slightly increased rate in heterozygotes. However, the observed differences in tumor spectra underline the necessity of interspecies phenotypic comparisons to better understand the in vivo role of p53-deficiency in tumorigenesis. Interestingly, requirement of loss of heterozygosity suggests that the resulting tumor spectrum in heterozygotes is mostly dependent on the stochastic distribution of spontaneous somatic mutations.5 Our present results suggest a predominance of osteosarcoma in heterozygote p53+/C273X rats, whereas early spontaneous tumors in Han Wistar rats are most frequently lymphomas.24 In contrast, only a single lymphoma was observed in the present study, and the predominant occurrence of sarcomas in both homo-, and heterozygotes suggests an additional factors may determine the tissue predisposition of Tp53 C273X –induced tumors.
In summary, our results demonstrate that the Tp53C273X/C273X rat is highly complementary to existing mouse models for studying human cancer biology. The predominant development of (hemangio)sarcomas in homozygous mutant rats with a high occurrence of lung metastases and a relatively early onset of 4 months makes this rat model a unique and especially useful tool for testing therapeutic interventions.
Footnotes
Supported by grants of the Cancer Genomics Center (CGC) program of the Netherlands Genomics Initiative (NGI) and the award “Exploiting natural and induced genetic variation in the laboratory rat” to E.C. from the European Heads of Research Councils and European Science Foundation EURYI (European Young Investigator) Award scheme.
R.v.B. and R.V.K. contributed equally to this work.
References
- 1.Chang F., Syrjanen S., Tervahauta A., Syrjanen K. Tumourigenesis associated with the p53 tumour suppressor gene. Br J Cancer. 1993;68:653–661. doi: 10.1038/bjc.1993.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hollstein M., Sidransky D., Vogelstein B., Harris C.C. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 3.Donehower L.A., Harvey M., Slagle B.L., McArthur M.J., Montgomery C.A., Jr, Butel J.S., Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 4.Harvey M., McArthur M.J., Montgomery C.A., Jr, Butel J.S., Bradley A., Donehower L.A. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet. 1993;5:225–229. doi: 10.1038/ng1193-225. [DOI] [PubMed] [Google Scholar]
- 5.Jacks T., Remington L., Williams B.O., Schmitt E.M., Halachmi S., Bronson R.T., Weinberg R.A. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 6.Purdie C.A., Harrison D.J., Peter A., Dobbie L., White S., Howie S.E., Salter D.M., Bird C.C., Wyllie A.H., Hooper M.L. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 1994;9:603–609. [PubMed] [Google Scholar]
- 7.Krug U., Ganser A., Koeffler H.P. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene. 2002;21:3475–3495. doi: 10.1038/sj.onc.1205322. [DOI] [PubMed] [Google Scholar]
- 8.Preudhomme C., Vanrumbeke M., Detourmignies L., Facon T., Lepelley P., Soussi T., Fenaux P. Very low incidence of p53 antibodies in adult non-Hodgkin's lymphoma and multiple myeloma. Br J Haematol. 1998;100:184–186. doi: 10.1046/j.1365-2141.1998.00516.x. [DOI] [PubMed] [Google Scholar]
- 9.Malkin D., Li F.P., Strong L.C., Fraumeni J.F., Jr, Nelson C.E., Kim D.H., Kassel J., Gryka M.A., Bischoff F.Z., Tainsky M.A. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 10.Clarke A.R. Murine models of neoplasia: functional analysis of the tumour suppressor genes Rb-1 and p53. Cancer Metastasis Rev. 1995;14:125–148. doi: 10.1007/BF00665796. [DOI] [PubMed] [Google Scholar]
- 11.Jacob H.J. Functional genomics and rat models. Genome Res. 1999;9:1013–1016. doi: 10.1101/gr.9.11.1013. [DOI] [PubMed] [Google Scholar]
- 12.Amos-Landgraf J.M., Kwong L.N., Kendziorski C.M., Reichelderfer M., Torrealba J., Weichert J., Haag J.D., Chen K.S., Waller J.L., Gould M.N., Dove W.F. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc Natl Acad Sci USA. 2007;104:4036–4041. doi: 10.1073/pnas.0611690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cotroneo M.S., Haag J.D., Zan Y., Lopez C.C., Thuwajit P., Petukhova G.V., Camerini-Otero R.D., Gendron-Fitzpatrick A., Griep A.E., Murphy C.J., Dubielzig R.R., Gould M.N. Characterizing a rat Brca2 knockout model. Oncogene. 2007;26:1626–1635. doi: 10.1038/sj.onc.1209960. [DOI] [PubMed] [Google Scholar]
- 14.van Boxtel R., Toonen P.W., van Roekel H.S., Verheul M., Smits B.M., Korving J., de Bruin A., Cuppen E. Lack of DNA mismatch repair protein MSH6 in the rat results in hereditary non-polyposis colorectal cancer-like tumorigenesis. Carcinogenesis. 2008;29:1290–1297. doi: 10.1093/carcin/bgn094. [DOI] [PubMed] [Google Scholar]
- 15.Geurts A.M., Cost G.J., Freyvert Y., Zeitler B., Miller J.C., Choi V.M., Jenkins S.S., Wood A., Cui X., Meng X., Vincent A., Lam S., Michalkiewicz M., Schilling R., Foeckler J., Kalloway S., Weiler H., Menoret S., Anegon I., Davis G.D., Zhang L., Rebar E.J., Gregory P.D., Urnov F.D., Jacob H.J., Buelow R. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325:433. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong C., Li P., Wu N.L., Yan Y., Ying Q.L. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature. 2010;467:211–213. doi: 10.1038/nature09368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang G., Tong C., Kumbhani D.S., Ashton C., Yan H., Ying Q.L. Beyond knockout rats: new insights into finer genome manipulation in rats. Cell Cycle. 2011;10:1059–1066. doi: 10.4161/cc.10.7.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smits B.M., Mudde J.B., van de Belt J., Verheul M., Olivier J., Homberg J., Guryev V., Cools A.R., Ellenbroek B.A., Plasterk R.H., Cuppen E. Generation of gene knockouts and mutant models in the laboratory rat by ENU-driven target-selected mutagenesis. Pharmacogenet Genomics. 2006;16:159–169. doi: 10.1097/01.fpc.0000184960.82903.8f. [DOI] [PubMed] [Google Scholar]
- 19.Zan Y., Haag J.D., Chen K.S., Shepel L.A., Wigington D., Wang Y.R., Hu R., Lopez-Guajardo C.C., Brose H.L., Porter K.I., Leonard R.A., Hitt A.A., Schommer S.L., Elegbede A.F., Gould M.N. Production of knockout rats using ENU mutagenesis and a yeast-based screening assay. Nature Biotechnol. 2003;21:645–651. doi: 10.1038/nbt830. [DOI] [PubMed] [Google Scholar]
- 20.van Boxtel R., Gould M.N., Cuppen E., Smits B.M. ENU mutagenesis to generate genetically modified rat models. Methods Mol Biol. 2010;597:151–167. doi: 10.1007/978-1-60327-389-3_11. [DOI] [PubMed] [Google Scholar]
- 21.van Boxtel R., Cuppen E. Rat traps: filling the toolbox for manipulating the rat genome. Genome Biol. 2010;11:217. doi: 10.1186/gb-2010-11-9-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Boxtel R., Toonen P.W., Verheul M., van Roekel H.S., Nijman I.J., Guryev V., Cuppen E. Improved generation of rat gene knockouts by target-selected mutagenesis in mismatch repair-deficient animals. BMC Genomics. 2008;9:460. doi: 10.1186/1471-2164-9-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riley T., Sontag E., Chen P., Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 24.Son W.C., Bell D., Taylor I., Mowat V. Profile of early occurring spontaneous tumors in Han Wistar rats. Toxicol Pathol. 2010;38:292–296. doi: 10.1177/0192623309359794. [DOI] [PubMed] [Google Scholar]