

Abstract
Polo-like kinase 1 (Plk1) is widely established as one of the most promising targets in oncology. Although the protein kinase domain of Plk1 is highly conserved, the polo-box domain (PBD) of Plk1 provides a much more compelling site to specifically inhibit the localization and target binding of Plk1. We recently identified, via fluorescence polarization assay, the natural product derivative, Poloxin, as the first small-molecule inhibitor specifically targeting the function of the Plk1 PBD. In this study, we characterized its mitotic phenotype and its function in vitro and in vivo. Poloxin induces centrosome fragmentation and abnormal spindle and chromosome misalignment, which activate the spindle assembly checkpoint and prolong mitosis. Notably, centrosomal fragmentation induced by Poloxin is partially attributable to dysfunctional Kizuna, a key substrate of Plk1 at centrosomes. Moreover, Poloxin strongly inhibits proliferation of a panel of cancer cells by inducing mitotic arrest, followed by a surge of apoptosis. More important, we report, for the first time to our knowledge, that the PBD inhibitor, Poloxin, significantly suppresses tumor growth of cancer cell lines in xenograft mouse models by lowering the proliferation rate and triggering apoptosis in treated tumor tissues. The data highlight that targeting the PBD by Poloxin is a powerful approach for selectively inhibiting Plk1 function in vitro and in vivo.
Polo-like kinases (Plks) are a family of Ser-Thr kinases regulating a variety of functions during the cell cycle and proliferation, from yeast to mammals.1,2 In mammalian cells, three closely related members (ie, Plk1, Plk2/Snk, and Plk3/Prk) and one distantly related kinase (ie, Plk4/Sak) have been identified, with apparently different expression patterns and physiological functions. Recently, mouse Plk5 has also been identified and human Plk5 is an interesting member lacking the protein kinase domain,3 which is implicated in specific roles in neuron differentiation and glioblastoma suppression.4 Plk1, the most thoroughly characterized member among the mammalian Plks, has multiple important functions during mitosis.1,5 In line with this multitude of proposed functions, Plk1 localizes to diverse mitotic structures, including centrosomes, kinetochores, the central spindle, and the midbody.1,6–8 Structurally, in addition to the conserved protein kinase domain, the members of the Plk family share, at the C-terminus, a conserved region termed the polo-box domain (PBD), which is essential for substrate targeting and Plk1 localization. The PBD, comprising two polo-box motifs, constitutes a binding region with maximal affinity to phosphopeptides containing a consensus sequence S-pS/pT-P/X.9,10
Plk1 is highly expressed in a broad spectrum of cancer types, and its expression often correlates with poor prognosis, suggesting its involvement in oncogenesis and its potential as a therapeutic target.11 Overexpression of Plk1 in human tumor cells, but not in normal nondividing cells, makes it an attractive and selective target for molecular intervention. Over the years, efforts have been made to identify Plk1 inhibitors, yielding several potent compounds that competitively inhibit the catalytic activity of Plk1.11 The protein kinase domain of Plk1 is closely related to several members of the superfamily of protein kinases, whereas the PBD is unique and a specific signature of the Plk kinase family. Moreover, although Plk1 is strongly associated with oncogenic transformation,11,12 Plk2 and Plk3 are critical for checkpoint-mediated cell cycle arrest to ensure genetic stability and prevent oncogenic transformation.13–15 Thus, the ideal inhibitor should selectively target the PBD of Plk1 but not that of Plk2 and Plk3.
Based on a fluorescence polarization assay, we have recently identified the natural product thymoquinone (TQ) and its synthetic derivative Poloxin as the first small-molecule inhibitors targeting the PBD of Plk1.16 We have shown that Poloxin exhibits a high specificity toward the PBD of Plk1, reduces both centrosomal and kinetochoral localization of Plk1, induces chromosome congression defects, arrests cells in prometaphase, and triggers further apoptosis in HeLa cells. The data demonstrate that inhibition of the PBD is sufficient to interfere with some of the mitotic functions of Plk1, including chromosome congression and mitotic progression. Herein, we analyzed the Poloxin-induced phenotype in more detail, its efficacy in various cancer cell lines, and its effect on proliferation using xenograft mouse models.
Materials and Methods
Cell Culture and Synchronization
All cell lines were grown according to the supplier's suggestions (DSMZ, Braunschweig, Germany). HeLa 776-6 and HeLa P25 cells were established as previously described.17–19 Briefly, HeLa cells transfected with plasmids phH1/shRNA/cyclin B1 or Plk1 were selected with medium containing G418 for 6 weeks. Cell clones with various levels of cyclin B1 or Plk1 were obtained. Cell synchronization to the G1-S boundary was performed by double thymidine block, as previously described.17
Western Blot Analysis and Indirect Immunofluorescence Staining
These experiments were performed as previously described.17,19,20 The following antibodies were used for Western blot analysis: mouse monoclonal antibodies against caspase-3, caspase-9, and Cdc25C (Santa Cruz Biotechnology, Heidelberg, Germany); rabbit polyclonal antibodies against poly(ADP)ribose polymerase (Cell Signaling, Beverly, MA); mouse monoclonal antibodies against β-actin (Sigma-Aldrich, Taufkirchen, Germany); and secondary anti-mouse or anti-rabbit antibodies (Santa Cruz Biotechnology). The following primary antibodies were used for staining: rat polyclonal anti-α-tubulin (Biozol, Eching, Germany), rabbit polyclonal antibodies against pericentrin (Cell Signaling), mouse monoclonal anti-phospho-histone H3 (p-HH3, or Ser10; Millipore, Schwalbach, Germany), mouse monoclonal anti-Plk1 (Santa Cruz Biotechnology), rabbit polyclonal-anti-Plk1 (Millipore), mouse monoclonal antibodies against BubR1 (BD Biosciences, Heidelberg), human anti-centromere antibody (ImmunoVision, Springdale, AR), and mouse anti-γ-tubulin (Abcam, Cambridge, PA). Fluorescein isothiocyanate–conjugated donkey anti-mouse, Cy3-conjugated goat anti-rabbit, or anti-rat and Cy5-conjugated goat anti-mouse (Jackson Immunoresearch, West Grove, PA) were used as secondary antibodies.
The assays for cell cycle analysis, cell viability assay, annexin staining, and activity of caspase-3/7 were performed as previously described.17,19
In Vivo Experiments, Western Blot Analysis, and IHC with Tumor Tissue
Viable MDA-MB-231 or HeLa cells (1 × 106) were resuspended in 300 μL of 0.9% NaCl and s.c. injected into both flanks of nude mice (MDA-MB-231: n = 8 mice in each group, total N = 16; HeLa: n = 7 mice in each group, total N = 14). Approximately 3 weeks after inoculation, mice were treated with Poloxin (40 mg/kg) or TQ (20 mg/kg) by intratumoral injection on Mondays, Wednesdays, and Fridays for 5 to 6 weeks. The tumor area was calculated by multiplication of the greatest diameter with the perpendicular diameter every 2 to 3 days. Measurements of all tumors within the group were represented by the mean value. U-tests and Student's t-tests were performed for statistical evaluation among MDA-MB-231 groups and between HeLa groups, respectively. All mice were properly treated in accordance with the guidelines of the local animal committee.
Cellular extracts from tumor tissues were prepared by using standard protocol. Briefly, tumor pieces were digested in lysis buffer [50 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EDTA, 2.5 mmol/L EGTA, 10% glycerol, 0.1% Tween 20, 1 mmol/L dithiothreitol, 10 mmol/L β-glycerol-phosphate, 1 mmol/L NaF, and 0.1 mmol/L Na3VO4 with Complete Protease Inhibitor Cocktail; Roche, Mannheim, Germany] and then homogenized. After a 30-minute incubation on ice, the lysates were centrifuged with 10,000 × g at 4°C for 20 minutes. Cellular extracts were obtained by a further 20-minute incubation on ice and centrifugation. Sections of formalin-fixed, paraffin-embedded tissues were used for immunohistochemical (IHC) analysis. Slides were pretreated in a microwave oven in 10 mmol/L citrate buffer to improve antigen retrieval. Monoclonal mouse anti-human Ki-67 antibodies (Dako, Glostrup, Denmark), polyclonal rabbit anti-p-HH3 (Ser10) antibodies (Millipore), and polyclonal rabbit anti-cleaved caspase-3 antibodies (Cell Signaling) were used for staining. Sections were stained using alkaline phosphatase anti-alkaline phosphatase or avidin-biotin peroxidase complex techniques.
Results
Poloxin Induces Defects in Centrosome Integrity and Chromosome Alignment During Mitosis
Poloxin induces mitotic arrest and prolongs the mitotic duration (see Supplemental Figure S1, A and B, at http://ajp.amjpathol.org), accompanied by Plk1's mislocalization at kinetochores and centrosomes with reduced γ-tubulin (see Supplemental Figure S2, A and B, at http://ajp.amjpathol.org), as previously reported,16 corroborating that Poloxin specifically targets the PBD of Plk1 in cells. To further explore the reasons for mitotic arrest, we analyzed the mitotic phenotype in more detail in Poloxin-treated HeLa cells by immunofluorescence microscopy. HeLa cells, synchronized by double thymidine block and released into medium with either 25 μmol/L Poloxin or dimethyl sulfoxide (DMSO) for 10 hours, were stained for α-tubulin, pericentrin, and DNA. Strikingly, compared with DMSO-treated cells (Figure 1A), Poloxin-treated mitotic cells exhibited frequently fragmented and disassociated centrosomes (pericentrin in Figure 1A), associated with aberrant mitotic spindles (α-tubulin in Figure 1A), which could be induced either indirectly, by perturbing microtubule nucleation due to fragmented centrosomes, or directly, by interfering with tubulin-stabilizing proteins, such as traditionally controlled tumor protein21,22 in Poloxin-treated cells. In addition, consistent with our previous results, defects in chromosome alignment were easily observable (ie, unaligned chromosomes surrounding the metaphase plate; DAPI in Figure 1A). Further analysis revealed that almost 50% of the 25 μmol/L Poloxin-treated cells displayed abnormal mitotic spindles (Figure 1B), 37% showed defects in chromosome alignment (Figure 1C), and almost 60% exhibited centrosomal fragmentation (Figure 1D) during mitosis. In addition, we performed the same experiment in colon cancer HCT116 p53−/− cells, which enter mitosis despite cellular stress. Fragmented centrosomes in mitotic cells were 7% for DMSO-treated and 31% for 25 μmol/L Poloxin-treated HCT116 p53−/− cells, respectively (see Supplemental Figure S2, C and D, at http://ajp.amjpathol.org). The data suggest that Poloxin-treated cells face severe problems with proper spindle formation, chromosome alignment, and centrosome integrity during mitosis.
Figure 1.
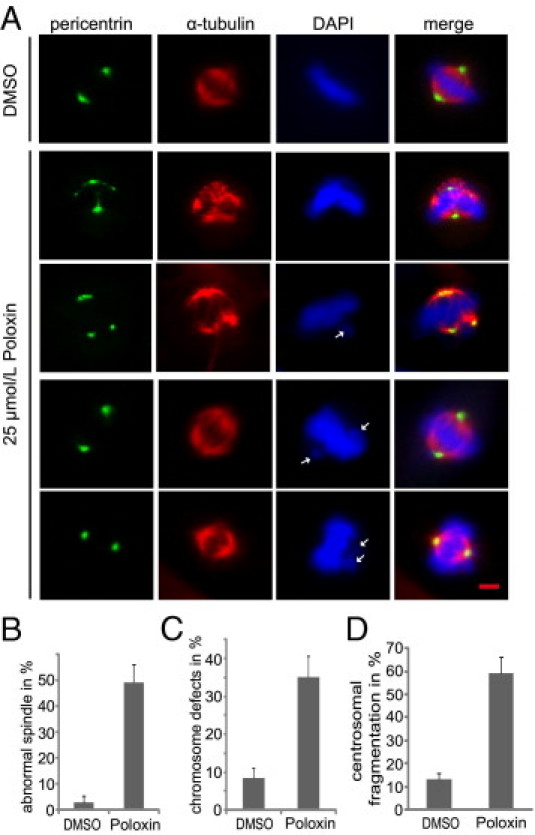
Poloxin induces defects in centrosome integrity, spindle formation, and chromosome alignment in mitosis. A: HeLa cells were synchronized with double thymidine block and released into fresh medium with either DMSO or 25 μmol/L Poloxin for 10 hours. Cells were fixed and stained for α-tubulin, pericentrin, and DNA. The morphological features of centrosomes, spindles, and chromosomes were examined by fluorescence microscopy. Examples are displayed for centrosome fragmentation with aberrant mitotic spindles (second and third rows, pericentrin and α-tubulin) and for misaligned chromosomes (third through fifth rows, DAPI). White arrows: Misaligned chromosome. Cells treated with DMSO were taken as control (first row). Scale bar = 5 μm. Quantification of abnormal spindles (B), chromosome misalignment (C), and centrosomal fragmentation (D) in approximately 300 mitotic HeLa cells treated with DMSO or 25 μmol/L Poloxin. The results are presented as mean ± SD.
Fragmented Centrosomes Induced by Poloxin Are Associated with Dysfunction of Kiz
Centrosomal fragmentation induced by Poloxin was attracting our attention. Phosphorylation of Kizuna (Kiz) at T379 is thought important for stabilizing mitotic centrosomes.23 To answer whether Kiz was affected by Poloxin, a rescue experiment was performed, as illustrated in Figure 2A: HeLa cells were cotransfected with human Myc-tagged wild-type Kiz, its nonphosphorylatable mutant Kiz T379A, or phosphomimetic Kiz T379E and pBabe-puro constructs. After selection with puromycin, cells were synchronized and released into fresh medium containing either DMSO or Poloxin. Cells were then stained for immunofluorescence microscopy. Approximately 40% of treated mitotic cells showed centrosomal fragmentation with 15 μmol/L Poloxin treatment (Figure 2, B and C). Interestingly, although the phosphomimetic Kiz T379E was able to rescue approximately 36% of the fragmentation (Figure 2, B and C), wild-type Kiz and alanine-mutant Kiz T379A hardly affected the phenotype (Figure 2, B and C). The expression levels of Myc-tagged wild-type Kiz and its variants in HeLa cells were comparable using a Western blot analysis (Figure 2D). HeLa cells, treated as described in Figure 2A but without Poloxin, were also stained for control (see Supplemental Figure S3, A, B, and C, at http://ajp.amjpathol.org).
Figure 2.
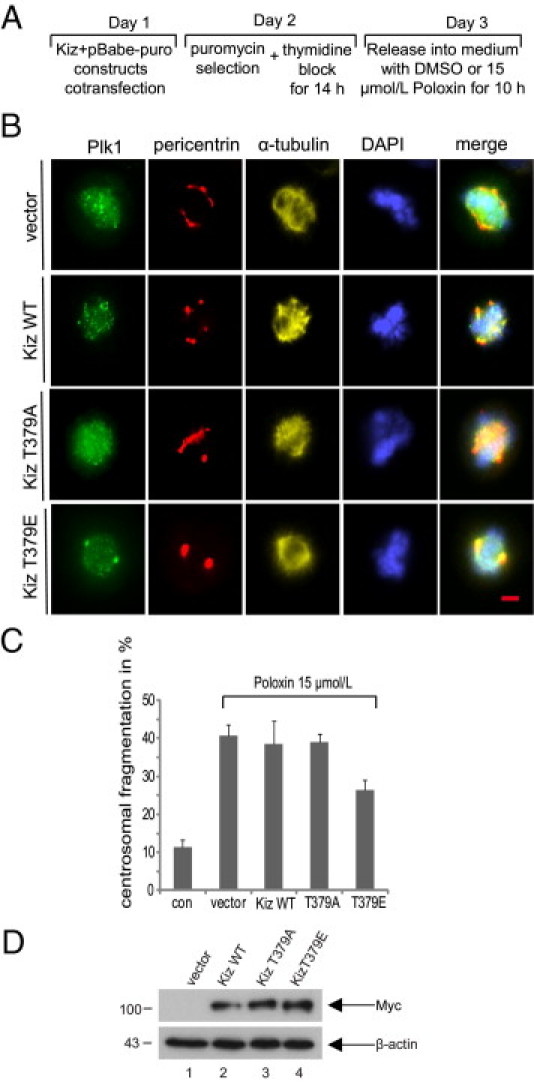
Centrosomal fragmentation induced by Poloxin is partially rescued by Kiz T379E. A: Working schedule. To obtain sufficient mitotic cells for analysis, HeLa cells, transfected, selected, and synchronized, were treated for 10 hours with a lower concentration of 15 μmol/L Poloxin, because 25 μmol/L Poloxin detaches HeLa cells, treated with transfection, selection, and synchronization, from the cell culture slides, along with increasing the mitotic cell population. B: HeLa cells were treated as described in A and stained for Plk1, pericentrin, α-tubulin, and DNA. Cells treated with DMSO were taken as control (top row). Scale bar = 5 μm. C: Evaluation of centrosomal fragmentation in approximately 300 mitotic cells from each group in B. The results are presented as mean ± SD. con, control. D: Expression levels of Myc-tagged wild-type Kiz and its variants in HeLa cells.
Poloxin Activates the Spindle Assembly Checkpoint
Inactivation of Plk1 causes a prometaphase arrest by activating the spindle assembly checkpoint.24–26 To explore whether Poloxin is capable of activating the checkpoint, Poloxin-treated HeLa cells were stained for α-tubulin, DNA, and BubR1, an important regulator of the spindle assembly checkpoint. Plk1 phosphorylates BubR1, which regulates the stability of kinetochore-microtubule interaction.27 Yet, Plk1 is not required for BubR1's localization at kinetochores in HeLa cells,27–29 although Plx1 is responsible for the loading of xBubR1 onto kinetochores in Xenopus laevis.28 In DMSO-treated cells, BubR1 localized at kinetochores before chromosome alignment in prometaphase, and it disappeared when chromosomes properly aligned at the equatorial plate in metaphase (Figure 3A). On the other hand, most of the Poloxin-treated cells were retained during prometaphase, with strong BubR1 staining at kinetochores of not yet aligned chromosomes (Figure 3A). However, strong signals of BubR1 were still bound to kinetochores in those Poloxin-treated cells, which managed to reach metaphase regardless of aberrant spindles (Figure 3A). BubR1 possibly senses misattachments or reduced tensions and thereby activates the spindle assembly checkpoint in Poloxin-treated cells. To corroborate these results, HeLa cells were synchronized by thymidine treatment and released into fresh medium with 25 μmol/L of Poloxin for 12 hours. The shake-off cells were then incubated with medium containing Poloxin or in combination with 2 μmol/L of the Aurora A/B inhibitor ZM 447439 (Tocris Bioscience, Ellisville, MO) for a further 2 hours. Poloxin-treated cells were still kept in mitosis, as evidenced by high levels of mitotic protein Plk1, cyclin B1, and p-HH3 (Figure 3B). However, in the presence of ZM 447439, the Plk1 and cyclin B1 levels were strongly reduced and the p-HH3 signal almost disappeared (Figure 3B), implying that cells escaped the prometaphase arrest and exit from mitosis after inactivating the spindle assembly checkpoint with ZM 447439. BI 2536, an inhibitor of the protein kinase domain of Plk1, was used as a positive control (Figure 3B) because it also activates the spindle assembly checkpoint and arrests cells in prometaphase, and cells exit from mitosis on treatment with the Aurora A/B inhibitor hesperidin.26 The data suggest that the prometaphase delay induced by Poloxin is ascribed to activation of the spindle assembly checkpoint.
Figure 3.
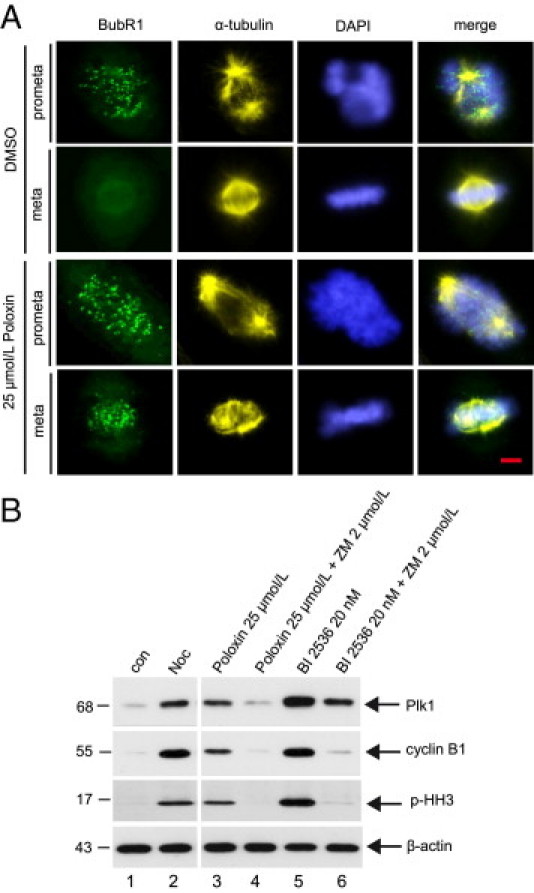
Poloxin activates the mitotic checkpoint. A: HeLa cells were synchronized by thymidine treatment and released into medium containing DMSO or 25 μmol/L Poloxin for 10 hours. Cells were stained for BubR1, α-tubulin, and DNA. Scale bar = 5 μm. B: HeLa cells were synchronized by thymidine treatment and released into medium containing 25 μmol/L Poloxin or 20 nmol/L BI 2536. Twelve hours later, the shake-off cells were further incubated with Poloxin alone, Poloxin plus 2 μmol/L ZM 447439, BI 2536 alone, or BI 2536 plus ZM 447439 for a further 2 hours, as indicated. Cellular extracts were then prepared for Western blot analyses with antibodies against Plk1, cyclin B1, and p-HH3. Nontreated (con) or nocodazole-treated (Noc) cells were taken as mitotic-negative and mitotic-positive controls, respectively. β-Actin served as the loading control.
Massive Apoptosis Caused by Poloxin
Cancer cells with depletion/inhibition of Plk1 undergo apoptosis, mostly caused by catastrophic mitotic defects.30–33 To corroborate that inhibiting Plk1 by Poloxin triggers cancer cells to apoptosis, asynchronous HeLa cells were treated with increasing amounts of Poloxin for 24 hours and cell cycle profiles were analyzed by fluorescence-activated cell sorting. As shown in Figure 4A, Poloxin arrested cells in the G2/M phase with a distinctive sub-G1 peak in a concentration-dependent manner, indicative of apoptosis induction after prolonged mitosis. Apoptosis induction by Poloxin was further underlined by annexin staining (Figure 4B). In addition to HeLa cells, we also measured the sub-G1 peak and performed annexin staining in MDA-MB-231, SW 480, MCF7, and A549 cells treated with Poloxin, and comparable results were obtained (data not shown). The induction of apoptosis was further confirmed by using Western blot analyses by showing cleaved poly(ADP)ribose polymerase and reduced procaspase-3 and procaspase-9 (Figure 4C), consistent with the observation that the activities of caspase-3/7 were increased in the treated cellular extracts (Figure 4D). Furthermore, Poloxin induced the cleavage of poly(ADP)ribose polymerase in connection with increased Emi1 and decreased p-Cdc25C (see Supplemental Figure S3D at http://ajp.amjpathol.org), two specific targets of Plk1, implying that apoptosis induction is associated with the function of Poloxin by targeting Plk1 within cells.
Figure 4.
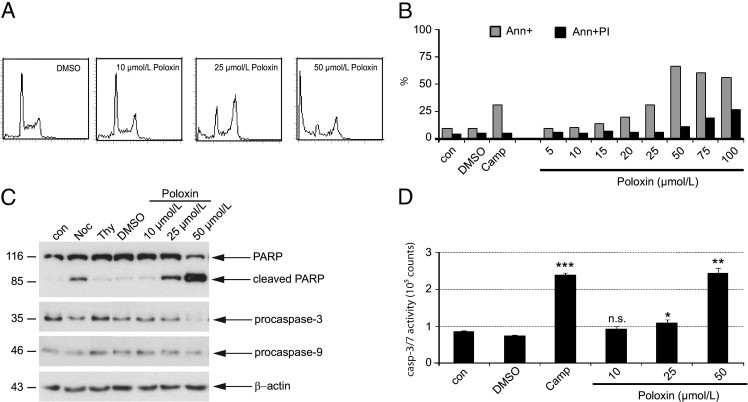
Poloxin induces apoptosis. A: The sub-G1 population is increased in Poloxin-treated HeLa cells. Cells were treated with Poloxin with indicated concentrations for 24 hours and analyzed by fluorescence-activated cell sorting. B: Cells were treated with increasing concentrations of Poloxin for 24 hours and stained for annexin (Ann) and propidium iodide (PI). Cells treated with DMSO or camptothecin (Camp, 10 μmol/L) were taken as negative and positive controls, respectively. C: HeLa cells were treated as indicated for 24 hours. Cellular extracts were prepared for Western blot analyses with corresponding antibodies. β-Actin served as the loading control. Cellular extracts from nontreated cells (con) or cells treated with DMSO, nocodazole (Noc), or thymidine (Thy) were taken as controls. Two lanes (the same amounts of DMSO as that in 10 and 25 μmol/L of Poloxin) between Thy and DMSO were omitted, and the equal amount of DMSO in 50 μmol/L of Poloxin remained as the DMSO control. PARP, poly(ADP)ribose polymerase. D: HeLa cells were treated as in B, and cellular extracts were prepared for active caspase-3/7 assays using the Caspase-Glo Assay (Promega, Mannheim, Germany). Cellular extracts from control (con) and DMSO- or Camp-treated cells were taken as negative and positive controls, respectively. The results are presented as the mean ± SD (n = 3) and analyzed by the Student's t-test. *P < 0.05, **P < 0.01, and ***P < 0.001. n.s., no significance.
Poloxin Inhibits Proliferation of Various Cell Types
To study its inhibitory effect on proliferation, cancer cells from various origins were treated with increasing doses of Poloxin for different time periods. Poloxin inhibited proliferation of metastatic breast cancer MDA-MB-231 cells in a dose-dependent manner through 72 hours (Figure 5A). However, a similar inhibitory effect was also observable in human normal retinal epithelial cells (hTERT-RPE1 in Figure 5B). These dose and time kinetics were also performed in various cancer and normal cell lines, and EC50 values of all cell lines tested were illustrated in Table 1. We could not find a distinctively different response to Poloxin between cancer and normal cell lines, at least in the cell culture system, suggesting that Plk1 is required for all proliferating cells.
Figure 5.
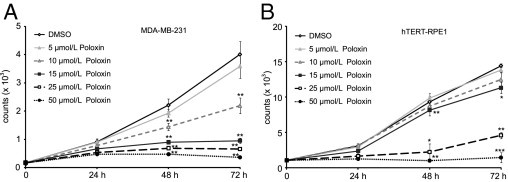
Poloxin inhibits proliferation. A: MDA-MB-231 cells were treated with increasing concentrations of Poloxin and harvested at indicated time points. Cell viability assays were performed by using a CellTiter-Blue assay (Promega). Cells treated with DMSO served as the control. B: Cell viability assays were also performed with human normal retinal epithelial cells (hTERT-RPE1). Cells treated with DMSO served as the control. The results are presented as the mean ± SD (n = 3) and analyzed by the Student's t-test. *P < 0.05, **P < 0.01, and ***P < 0.001.
Table 1.
Inhibitory Effect of Poloxin on Proliferation in Various Cell Lines
| Cells/cell lines | Tissue type | EC50 (μmol/L) |
|---|---|---|
| Tumor | ||
| MCF7 | Breast | 35 |
| MDA-MB-231 | Breast | 15 |
| Detroit 562 | Pharynx | 15 |
| T47D | Breast | 25 |
| A549 | Lung | 20 |
| SW 480 | Colon | 22 |
| HCT 116 | Colon | 25 |
| HeLa | Cervix | 25 |
| HeLa P25⁎ | Cervix | 22 |
| HeLa 776–6† | Cervix | 22 |
| PC-3 | Prostate | 20 |
| Normal | ||
| hTERT-RPE1 | Retinal epithelial | 20 |
| MTSV-1 | Mammary epithelial | 20 |
| Fibroblasts | Foreskin fibroblastic | 25 |
| HUVECs | Umbilical vein endothelial | 20 |
HUVEC, human umbilical vein endothelial cell.
Stably Plk1-depleted HeLa.
Stably cyclin B1–depleted HeLa.
Poloxin Inhibits Tumor Growth in Vivo
We next investigated the ability of Poloxin to inhibit tumor growth in nude mice xenografted with MDA-MB-231 or HeLa cells. We could observe a significant reduction of tumor volume after approximately 6 weeks of treatment with Poloxin or TQ in MDA-MB-231 xenograft mice compared with vehicle DMSO treatment (Figure 6A). Suppression of tumor growth by TQ was previously reported.34 Moreover, tumor regression was also observed in HeLa xenograft mice treated with Poloxin (Figure 6B). Poloxin treatment was well tolerated, as judged by body weight (see Supplemental Figure S4A at http://ajp.amjpathol.org). Furthermore, by using Western blot analysis and IHC staining, we showed that tumor regression of MDA-MB-231 cells after Poloxin or TQ treatment was accompanied by a decrease in proliferation rate, as evidenced by Plk1 levels (see Supplemental Figure S4, B and C, at http://ajp.amjpathol.org), Ki-67 and p-HH3 staining, and an increase in apoptosis, demonstrated by active caspase-3 staining, compared with DMSO-treated tumor tissues (Figure 6C).
Figure 6.
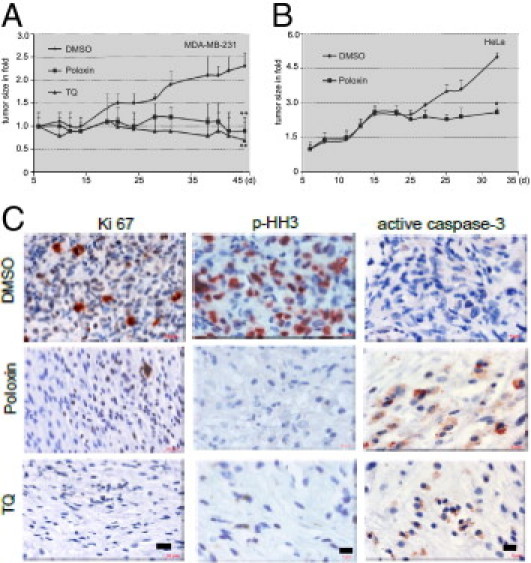
Poloxin suppresses tumor growth. A and B: Nude mice bearing established xenografts of MDA-MB-231 (A, n = 8 mice in each group, N = 16 mice per group) or HeLa cells (B, n = 7 mice in each group, N = 14 mice per group) were intratumorally treated with DMSO, Poloxin (40 mg/kg), or TQ (20 mg/kg) on Mondays, Wednesdays, and Fridays. Tumor size is displayed. Data are presented as the mean ± SD and statistically analyzed by the U-test (A) and the Student's t-test (B), respectively, because of different numbers of mice in each trial. *P < 0.05, **P < 0.001. C: IHC staining. MDA-MB-231 tumor tissues from xenograft mice treated with DMSO, Poloxin, or TQ for 6 weeks were stained for proliferation marker Ki-67 (left), mitotic marker p-HH3 (middle), or apoptosis marker active caspase-3 (right). Scale bars: 20 μm (Ki-67); 10 μm (p-HH3 and active caspase-3).
Discussion
Anti-mitotic agents targeting tubulin are widely used, with effectiveness in treating cancer, but they affect both dividing and nondividing cells, inducing unwanted adverse effects.35 Therefore, the development of a new generation of anti-mitotic therapy that targets proteins with specific functions in mitosis is much desired. Plk1, the key regulator of mitosis, has been established as one of such promising candidates. In fact, several interesting Plk1 inhibitors, most of them against the protein kinase domain of Plk1, are being tested in clinical trials, as recently summarized.5,36–38 In a more selective manner to the widely conserved kinase domain, the PBD of Plk1 poses a compelling site to block the Plk1 function. The first identified small-molecule compound, Poloxin, shows its high specificity by aiming at the PBD of Plk1,16 which is directly followed by another inspiring report that purpurogallin (PPG), a benzotropolone-containing natural compound derived from nutgalls, also blocks the PBD of Plk1 with selectivity.39 The data demonstrate that inhibition of the PBD is sufficient to specifically interfere with the multiple functions of Plk1.
Herein, we further characterize the phenotype and effect induced by Poloxin. Poloxin-treated cells display centrosome fragmentation, an aberrant mitotic spindle, and chromosome misalignment (Figure 1), which activate the mitotic checkpoint (Figure 3), further leading to prolonged mitosis (see Supplemental Figure S1 at http://ajp.amjpathol.org), followed by strong induction of apoptosis (Figure 4). Moreover, centrosome fragmentation induced by Poloxin can be attributed, at least in part, to dysfunction of a centrosomal protein, Kiz (Figure 2). More important, Poloxin strongly inhibits proliferation of a panel of proliferating cells (Figure 5, Table 1). More interestingly, Poloxin significantly suppresses tumor growth in xenograft mice (Figure 6, A and B). We found a low proliferation rate and an increased induction of apoptosis in tumor tissues after 5 to 6 weeks of treatment with Poloxin (Figure 6C; see also Supplemental Figure S4, B and C, at http://ajp.amjpathol.org). The data highlight that Poloxin works in vitro and in vivo by specifically interfering with the functions of Plk1, leading to mitotic prolongation and apoptosis induction.
It is well established that the function of Plk1 is required for centrosome maturation, separation, and spindle pole integrity.24,40,41 We have observed a distinctive centrosomal fragmentation with aberrant mitotic spindles in cells treated with Poloxin (Figure 1, A and D, and Figure 2, B and C). Notably, centrosomes were unfocused and distanced in cells treated with another PBD inhibitor, PPG.39 Moreover, cells treated with a pan-PBD inhibitor, poloxipan, also displayed fragmented centrosomes.42 However, enforced PBD expression did not impair centrosome maturation/separation.43,44 It will be interesting to clarify whether overexpression of the PBD also induces centrosome fragmentation.
We have closely looked into possible mechanisms for centrosomal fragmentation induced by Poloxin. It has been convincingly reported that Plk1 associates with Kiz, an important centrosomal substrate for Plk1, in a PBD-dependent manner and regulates its function for centrosome integrity by phosphorylating its residue, T379.23 Blocking this regulation or depletion of Kiz causes fragmentation and dissociation of the pericentriolar material from centrioles at prometaphase, which will be not able to endure the forces that converge on centrosomes during spindle formation.23 Based on these data, we reasoned that Kiz could be one of the centrosomal key molecules affected by Poloxin. This notion is underlined by the results that Kiz T379E, the phosphomimetic form of Kiz, is capable of partially rescuing the Poloxin-induced centrosomal fragmentation (Figure 2), indicating that Plk1-mediated Kiz function is disrupted by Poloxin. Because Plk1 is involved in centrosome stability and centriole cohesion during mitosis,45 it will be important to clarify the molecular mechanisms of centrosome fragmentation and to explore further critical molecules, such as aster-associated protein (ASAP),46 that are responsible for spindle pole integrity and could be affected by Poloxin. Nevertheless, the data suggest that Poloxin is specifically targeting the Plk1 function at centrosomes in cells.
In addition to HeLa cells, Poloxin strongly inhibits the proliferation of a panel of human cancer cell lines from diverse organ derivations (Table 1). The EC50 ranges from 15 to 35 μmol/L. The proliferation of exponentially normal growing cells, such as human retinal primary epithelial cells, human umbilical vein endothelial cells, mammary epithelial (MTSV-1) cells, and fibroblasts, is also suppressed by Poloxin; EC50 values range from 20 to 30 μmol/L, implying a comparable sensitivity of nontransformed cycling cells to Plk1 inhibition. This phenomenon has already been observed with BI 2536 treatment.47 Furthermore, apoptosis was strongly induced in normal rat kidney cells and NIH/3T3 cells on treatment with the PBD inhibitor, PPG.39 Therefore, we suppose that Plk1 is required for the proliferation of both normal/nontransformed cells and tumor cells. Thus, the Plk1 inhibitors, targeting either the protein kinase domain, such as BI 2536, or the PBD domain of Plk1, such as Poloxin and PPG, affect both cancer cells and nontransformed but proliferating cells with a comparable efficacy, at least in cell culture system. Yet, this does not necessarily represent situations in vivo because these nontransformed cells are artificially kept in cycling by additionally supplementing various growth factors. In addition, we are aware that these results from the small-molecule compounds are not consistent with previous data31,48 that knockdown of Plk1 by small-interfering RNA inhibits proliferation more dramatically in cancer cells than in normal cells. Different time courses could possibly result in this discrepancy: small-molecule compounds, such as BI 2536 or Poloxin, work immediately within cells on addition, whereas small-interfering RNA requires some time to deplete Plk1. During this period, normal cells with intact checkpoints could have enough time to respond to the challenges induced by depleting Plk1 by activating corresponding rescue pathways, such as halting the cell cycle, and to survive the challenges. This is not the case for tumor cells. Alternatively, depletion of Plk1 by small-interfering RNA is not able to totally knock out Plk1 and the remaining Plk1 may not be sufficient for survival of Plk1-addicted cancer cells, but for normal cells. This is in accordance with a recent report49 that sensitivity of normal cells to Plk1 depletion is dependent on the depletion level. However, an interesting review by Lens et al50 suggests that it is unlikely to have Plk1-addicted cells, and the contribution of Plk1 to tumor formation might be largely due to the induction of chromosome instability. Thus, the efficacy of Plk1 inhibitors most probably primarily depends on their antimitotic effect in tumor cells, whereas depleting Plk1 generates a form of cellular stress in primary cells and causes cells to arrest during G2. Yet, it still cannot explain why the small-molecule compounds, such as BI 2536 and Poloxin, exert comparable efficacy in both normal proliferating cells and tumor cells. Further studies (in particular, evidence from primary cells and tumor tissues in vivo) are required to precisely answer this question. Moreover, although Poloxin shows a higher specificity toward the PBD of Plk1 than that of Plk2 and Plk3 in vitro, it is important to define whether it interferes with the functions of Plk2 and Plk3 in vivo. It will also be critical to delineate the relationship between Poloxin and Plk5, the only PBD protein. All these data will shed light on the application strategy of small-molecule compounds targeting Plk1.
Taken together, in this study, we have further characterized the PBD inhibitor Poloxin and demonstrated, for the first time to our knowledge, its effectiveness in xenograft models. We believe that inhibitors specifically targeting the PBD, such as Poloxin and PPG, hold much promise for developing a new generation of molecular anti-mitotic agents, although many challenges are posed by their clinical development.
Acknowledgments
We thank Drs. Tadashi Yamamoto and Miho Ohsugi (Institute of Medical Science, University of Tokyo) for offering the Myc- and HA-tagged wild-type Kiz plasmids and their variants, Kiz T379A and Kiz T379E, for this study; and Drs. Kenneth W. Kinzler and Bert Vogelstein (Ludwig Center at Johns Hopkins, Howard Hughes Medical Institute, Baltimore, MD) for the HCT116 p53−/− cell line.
Footnotes
Supported by grants from Deutsche Krebshilfe (107594 and 108553), BANSS Stiftung and LOEWE präbionik grants (BOSS 4 to M.H.) W.R. and T.B. were supported by the Department of Molecular Biology (director,Dr. Axel Ullrich) at the Max Planck Institute of Biochemistry, Martinsried.
T.B. and K.S. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.06.031.
Current address of W.R.: Life Sciences Division, Lawrence Berkeley National Lab, Berkeley, California.
Contributor Information
Juping Yuan, Email: yuan@em.uni-frankfurt.de.
Klaus Strebhardt, Email: strebhardt@em.uni-frankfurt.de.
Supplementary data
Poloxin retains cells in mitosis. A: Cell cycle analysis. Top: HeLa cells were synchronized by double thymidine block and released into fresh medium containing either DMSO or 25 μmol/L Poloxin for the indicated time points, and cell cycle distributions were measured. Bottom: Quantification of subphases (sub) of the cell cycle. B: p-HH3 staining. Left: HeLa cells, treated as in A for 12 hours, were stained for p-HH3, a mitotic maker. Representatives are presented for DMSO and 25 μmol/L Poloxin-treated cells. Scale bar = 10 μm. Right: Evaluation of positive p-HH3 in DMSO and Poloxin-treated cells. The results are presented as the mean ± SD.
Subcellular localization of Plk1, human anti-centromere antibody (ACA; a kinetochore/centromere marker), and γ-tubulin on treatment of Poloxin. HeLa cells were synchronized with thymidine treatment and released into fresh medium containing either DMSO or 25 μmol/L Poloxin for 10 hours. Cells were fixed and stained for Plk1, ACA, and DNA (A) or for Plk1, γ-tubulin, and DNA (B). Scale bars: 5 μm (A and B). Poloxin induces centrosomal fragmentation in HCT116 p53−/− cells. C: HCT116 p53−/− cells were synchronized with thymidine block and released into fresh medium with either DMSO or 25 μmol/L Poloxin for 10 hours. Cells were fixed and stained for α-tubulin, pericentrin, and DNA. Examples are displayed for centrosome fragmentation with aberrant mitotic spindles (second and third rows, pericentrin and α-tubulin). Cells treated with DMSO were taken as the control, and one representative is shown (first row). Scale bar = 5 μm. D: Quantification of cells with fragmented centrosomes in approximately 200 mitotic HCT116 p53−/− cells treated with DMSO or 25 μmol/L Poloxin. The results are presented as the mean ± SD.
HeLa cells transfected with different Kiz constructs exhibited almost normal centrosomes. A: Working schedule. B: HeLa cells were treated as illustrated in A, fixed, and stained for γ-tubulin, α-tubulin, and DNA. This set of experiments served as the control for Figure 2. Scale bar = 5 μm. C: Expression levels of Myc-tagged wild-type Kiz and its variants in HeLa cells. D: Apoptosis induction is associated with Poloxin's function by targeting Plk1. HeLa cells were treated as described in Figure 3B. Cellular lysates were prepared for Western blot analyses with antibodies against poly(ADP)ribose polymerase (PARP), Cdc25C, and Emi1. β-Actin served as the loading control (con), which is also used in Figure 3B, because the same lysates were used. Noc, nocodazole.
Poloxin suppresses tumor growth. Nude mice bearing established xenografts of MDA-MB-231 (n = 8 mice in each group, N = 16 mice per group) (A) or HeLa cells (n = 7 mice in each group, N = 14 mice per group) (B) were intratumorally treated with the vehicle control DMSO, Poloxin (40 mg/kg), or TQ (20 mg/kg) on Mondays, Wednesdays, and Fridays. Tumor volume and body weight were measured every 2 to 3 days. A: Body weight during treatment time. B: Decreased Plk1 levels in Poloxin- and TQ-treated tumor tissues. Cellular extracts were prepared from MDA-MB-231 xenografts treated with DMSO (samples D1 to D10), Poloxin (samples P1 to P10), or TQ (samples TQ1 to TQ3) for Western blot analysis with Plk1 antibodies. β-Actin served as the loading control. C: Quantification of Plk1 expression levels of tumor tissues in B, relative to corresponding loading control β-actin. Data were presented as the mean ± SD and analyzed by the Student's t-test. **P < 0.01.
References
- 1.Barr F.A., Sillje H.H., Nigg E.A. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 2.Archambault V., Glover D.M. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol. 2009;10:265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 3.Andrysik Z., Bernstein W.Z., Deng L., Myer D.L., Li Y.Q., Tischfield J.A., Stambrook P.J., Bahassi E.M. The novel mouse Polo-like kinase 5 responds to DNA damage and localizes in the nucleolus. Nucleic Acids Res. 2010;38:2931–2943. doi: 10.1093/nar/gkq011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Cárcer G., Escobar B., Higuero A.M., Garcia L., Ansón A., Pérez G., Mollejo M., Manning G., Meléndez B., Abad-Rodríguez J., Malumbres M. Plk5, a polo box domain-only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol Cell Biol. 2011;31:1225–1239. doi: 10.1128/MCB.00607-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 6.Golsteyn R.M., Mundt K.E., Fry A.M., Nigg E.A. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol. 1995;129:1617–1628. doi: 10.1083/jcb.129.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee K.S., Yuan Y.L., Kuriyama R., Erikson R.L. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol Cell Biol. 1995;15:7143–7151. doi: 10.1128/mcb.15.12.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnaud L., Pines J., Nigg E.A. GFP tagging reveals human Polo-like kinase 1 at the kinetochore/centromere region of mitotic chromosomes. Chromosoma. 1998;107:424–429. doi: 10.1007/s004120050326. [DOI] [PubMed] [Google Scholar]
- 9.Elia A.E., Rellos P., Haire L.F., Chao J.W., Ivins F.J., Hoepker K., Mohammad D., Cantley L.C., Smerdon S.J., Yaffe M.B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 10.Yun S.M., Moulaei T., Lim D., Bang J.K., Park J.E., Shenoy S.R., Liu F., Kang Y.H., Liao C., Soung N.K., Lee S., Yoon D.Y., Lim Y., Lee D.H., Otaka A., Appella E., McMahon J.B., Nicklaus M.C., Burke T.R., Jr, Yaffe M.B., Wlodawer A., Lee K.S. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strebhardt K., Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 12.Smith M.R., Wilson M.L., Hamanaka R., Chase D., Kung H., Longo D.L., Ferris D.K. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun. 1997;234:397–405. doi: 10.1006/bbrc.1997.6633. [DOI] [PubMed] [Google Scholar]
- 13.van de Weerdt B.C., Medema R.H. Polo-like kinases: a team in control of the division. Cell Cycle. 2006;5:853–864. doi: 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- 14.Xie S., Wu H., Wang Q., Cogswell J.P., Husain I., Conn C., Stambrook P., Jhanwar-Uniyal M., Dai W. Plk3 functionally links DNA damage to cell cycle arrest and apoptosis at least in part via the p53 pathway. J Biol Chem. 2001;276:43305–43312. doi: 10.1074/jbc.M106050200. [DOI] [PubMed] [Google Scholar]
- 15.Smith P., Syed N., Crook T. Epigenetic inactivation implies a tumor suppressor function in hematologic malignancies for Polo-like kinase 2 but not Polo-like kinase 3. Cell Cycle. 2006;5:1262–1264. doi: 10.4161/cc.5.12.2813. [DOI] [PubMed] [Google Scholar]
- 16.Reindl W., Yuan J., Kramer A., Strebhardt K., Berg T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol. 2008;15:459–466. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Kreis N.N., Sommer K., Sanhaji M., Krämer A., Matthess Y., Kaufmann M., Strebhardt K., Yuan J. Long-term downregulation of Polo-like kinase 1 increases the cyclin-dependent kinase inhibitor p21(WAF1/CIP1) Cell Cycle. 2009;8:460–472. doi: 10.4161/cc.8.3.7651. [DOI] [PubMed] [Google Scholar]
- 18.Yuan J., Kramer A., Matthess Y., Yan R., Spankuch B., Gatje R., Knecht R., Kaufmann M., Strebhardt K. Stable gene silencing of cyclin B1 in tumor cells increases susceptibility to taxol and leads to growth arrest in vivo. Oncogene. 2006;25:1753–1762. doi: 10.1038/sj.onc.1209202. [DOI] [PubMed] [Google Scholar]
- 19.Kreis N.N., Sanhaji M., Krämer A., Sommer K., Rödel F., Strebhardt K., Yuan J. Restoration of the tumor suppressor p53 by downregulating cyclin B1 in human papillomavirus 16/18-infected cancer cells. Oncogene. 2010;29:5591–5603. doi: 10.1038/onc.2010.290. [DOI] [PubMed] [Google Scholar]
- 20.Sanhaji M., Friel C.T., Kreis N.N., Kramer A., Martin C., Howard J., Strebhardt K., Yuan J. Functional and spatial regulation of mitotic centromere-associated kinesin by cyclin-dependent kinase 1. Mol Cell Biol. 2010;30:2594–2607. doi: 10.1128/MCB.00098-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yarm F.R. Plk phosphorylation regulates the microtubule-stabilizing protein TCTP. Mol Cell Biol. 2002;22:6209–6221. doi: 10.1128/MCB.22.17.6209-6221.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng Y., Hodge D.R., Palmieri G., Chase D.L., Longo D.L., Ferris D.K. Association of polo-like kinase with alpha-, beta- and gamma-tubulins in a stable complex. Biochem J. 1999;339(Pt 2):435–442. [PMC free article] [PubMed] [Google Scholar]
- 23.Oshimori N., Ohsugi M., Yamamoto T. The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat Cell Biol. 2006;8:1095–1101. doi: 10.1038/ncb1474. [DOI] [PubMed] [Google Scholar]
- 24.van Vugt M.A., van de Weerdt B.C., Vader G., Janssen H., Calafat J., Klompmaker R., Wolthuis R.M., Medema R.H. Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for anaphase promoting complex/Cdc20 activation and initiation of cytokinesis. J Biol Chem. 2004;279:36841–36854. doi: 10.1074/jbc.M313681200. [DOI] [PubMed] [Google Scholar]
- 25.Santamaria A., Neef R., Eberspacher U., Eis K., Husemann M., Mumberg D., Prechtl S., Schulze V., Siemeister G., Wortmann L., Barr F.A., Nigg E.A. Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate functions of Plk1 in early and late stages of mitosis. Mol Biol Cell. 2007;18:4024–4036. doi: 10.1091/mbc.E07-05-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenart P., Petronczki M., Steegmaier M., Di F.B., Lipp J.J., Hoffmann M., Rettig W.J., Kraut N., Peters J.M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 27.Elowe S., Hummer S., Uldschmid A., Li X., Nigg E.A. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 2007;21:2205–2219. doi: 10.1101/gad.436007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong O.K., Fang G. Plx1 is the 3F3/2 kinase responsible for targeting spindle checkpoint proteins to kinetochores. J Cell Biol. 2005;170:709–719. doi: 10.1083/jcb.200502163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahonen L.J., Kallio M.J., Daum J.R., Bolton M., Manke I.A., Yaffe M.B., Stukenberg P.T., Gorbsky G.J. Polo-like kinase 1 creates the tension-sensing 3F3/2 phosphoepitope and modulates the association of spindle-checkpoint proteins at kinetochores. Curr Biol. 2005;15:1078–1089. doi: 10.1016/j.cub.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 30.Liu X., Erikson R.L. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci U S A. 2003;100:5789–5794. doi: 10.1073/pnas.1031523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X., Lei M., Erikson R.L. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol. 2006;26:2093–2108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yim H., Erikson R.L. Polo-like kinase 1 depletion induces DNA damage in early S prior to caspase activation. Mol Cell Biol. 2009;29:2609–2621. doi: 10.1128/MCB.01277-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan J., Kramer A., Eckerdt F., Kaufmann M., Strebhardt K. Efficient internalization of the polo-box of polo-like kinase 1 fused to an Antennapedia peptide results in inhibition of cancer cell proliferation. Cancer Res. 2002;62:4186–4190. [PubMed] [Google Scholar]
- 34.Gali-Muhtasib H., Ocker M., Kuester D., Krueger S., El-Hajj Z., Diestel A., Evert M., El-Najjar N., Peters B., Jurjus A., Roessner A., Schneider-Stock R. Thymoquinone reduces mouse colon tumor cell invasion and inhibits tumor growth in murine colon cancer models. J Cell Mol Med. 2008;12:330–342. doi: 10.1111/j.1582-4934.2007.00095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson J.R., Patrick D.R., Dar M.M., Huang P.S. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer. 2007;7:107–117. doi: 10.1038/nrc2049. [DOI] [PubMed] [Google Scholar]
- 36.Schmit T.L., Ledesma M.C., Ahmad N. Modulating polo-like kinase 1 as a means for cancer chemoprevention. Pharm Res. 2010;27:989–998. doi: 10.1007/s11095-010-0051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Degenhardt Y., Lampkin T. Targeting Polo-like kinase in cancer therapy. Clin Cancer Res. 2010;16:384–389. doi: 10.1158/1078-0432.CCR-09-1380. [DOI] [PubMed] [Google Scholar]
- 38.Schoffski P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist. 2009;14:559–570. doi: 10.1634/theoncologist.2009-0010. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe N., Sekine T., Takagi M., Iwasaki J., Imamoto N., Kawasaki H., Osada H. Deficiency in chromosome congression by the inhibition of Plk1 polo box domain-dependent recognition. J Biol Chem. 2009;284:2344–2353. doi: 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- 40.Lane H.A., Nigg E.A. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol. 1996;135:1701–1713. doi: 10.1083/jcb.135.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sumara I., Gimenez-Abian J.F., Gerlich D., Hirota T., Kraft C., de la Torre C., Ellenberg J., Peters J.M. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol. 2004;14:1712–1722. doi: 10.1016/j.cub.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 42.Reindl W., Yuan J., Krämer A., Strebhardt K., Berg T. A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. Chembiochem. 2009;10:1145–1148. doi: 10.1002/cbic.200900059. [DOI] [PubMed] [Google Scholar]
- 43.Seong Y.S., Kamijo K., Lee J.S., Fernandez E., Kuriyama R., Miki T., Lee K.S. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J Biol Chem. 2002;277:32282–32293. doi: 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- 44.Hanisch A., Wehner A., Nigg E.A., Sillje H.H. Different Plk1 functions show distinct dependencies on Polo-Box domain-mediated targeting. Mol Biol Cell. 2006;17:448–459. doi: 10.1091/mbc.E05-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X., Yang Y., Duan Q., Jiang N., Huang Y., Darzynkiewicz Z., Dai W. sSgo1, a major splice variant of Sgo1, functions in centriole cohesion where it is regulated by Plk1. Dev Cell. 2008;14:331–341. doi: 10.1016/j.devcel.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eot-Houllier G., Venoux M., Vidal-Eychenié S., Hoang M.T., Giorgi D., Rouquier S. Plk1 regulates both ASAP localization and its role in spindle pole integrity. J Biol Chem. 2010;285:29556–29568. doi: 10.1074/jbc.M110.144220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steegmaier M., Hoffmann M., Baum A., Lenart P., Petronczki M., Krssak M., Gurtler U., Garin-Chesa P., Lieb S., Quant J., Grauert M., Adolf G.R., Kraut N., Peters J.M., Rettig W.J. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 48.Spankuch-Schmitt B., Bereiter-Hahn J., Kaufmann M., Strebhardt K. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Inst. 2002;94:1863–1877. doi: 10.1093/jnci/94.24.1863. [DOI] [PubMed] [Google Scholar]
- 49.Lei M., Erikson R.L. Plk1 depletion in nontransformed diploid cells activates the DNA-damage checkpoint. Oncogene. 2008;27:3935–3943. doi: 10.1038/onc.2008.36. [DOI] [PubMed] [Google Scholar]
- 50.Lens S.M., Voest E.E., Medema R.H. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer. 2010;10:825–841. doi: 10.1038/nrc2964. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Poloxin retains cells in mitosis. A: Cell cycle analysis. Top: HeLa cells were synchronized by double thymidine block and released into fresh medium containing either DMSO or 25 μmol/L Poloxin for the indicated time points, and cell cycle distributions were measured. Bottom: Quantification of subphases (sub) of the cell cycle. B: p-HH3 staining. Left: HeLa cells, treated as in A for 12 hours, were stained for p-HH3, a mitotic maker. Representatives are presented for DMSO and 25 μmol/L Poloxin-treated cells. Scale bar = 10 μm. Right: Evaluation of positive p-HH3 in DMSO and Poloxin-treated cells. The results are presented as the mean ± SD.
Subcellular localization of Plk1, human anti-centromere antibody (ACA; a kinetochore/centromere marker), and γ-tubulin on treatment of Poloxin. HeLa cells were synchronized with thymidine treatment and released into fresh medium containing either DMSO or 25 μmol/L Poloxin for 10 hours. Cells were fixed and stained for Plk1, ACA, and DNA (A) or for Plk1, γ-tubulin, and DNA (B). Scale bars: 5 μm (A and B). Poloxin induces centrosomal fragmentation in HCT116 p53−/− cells. C: HCT116 p53−/− cells were synchronized with thymidine block and released into fresh medium with either DMSO or 25 μmol/L Poloxin for 10 hours. Cells were fixed and stained for α-tubulin, pericentrin, and DNA. Examples are displayed for centrosome fragmentation with aberrant mitotic spindles (second and third rows, pericentrin and α-tubulin). Cells treated with DMSO were taken as the control, and one representative is shown (first row). Scale bar = 5 μm. D: Quantification of cells with fragmented centrosomes in approximately 200 mitotic HCT116 p53−/− cells treated with DMSO or 25 μmol/L Poloxin. The results are presented as the mean ± SD.
HeLa cells transfected with different Kiz constructs exhibited almost normal centrosomes. A: Working schedule. B: HeLa cells were treated as illustrated in A, fixed, and stained for γ-tubulin, α-tubulin, and DNA. This set of experiments served as the control for Figure 2. Scale bar = 5 μm. C: Expression levels of Myc-tagged wild-type Kiz and its variants in HeLa cells. D: Apoptosis induction is associated with Poloxin's function by targeting Plk1. HeLa cells were treated as described in Figure 3B. Cellular lysates were prepared for Western blot analyses with antibodies against poly(ADP)ribose polymerase (PARP), Cdc25C, and Emi1. β-Actin served as the loading control (con), which is also used in Figure 3B, because the same lysates were used. Noc, nocodazole.
Poloxin suppresses tumor growth. Nude mice bearing established xenografts of MDA-MB-231 (n = 8 mice in each group, N = 16 mice per group) (A) or HeLa cells (n = 7 mice in each group, N = 14 mice per group) (B) were intratumorally treated with the vehicle control DMSO, Poloxin (40 mg/kg), or TQ (20 mg/kg) on Mondays, Wednesdays, and Fridays. Tumor volume and body weight were measured every 2 to 3 days. A: Body weight during treatment time. B: Decreased Plk1 levels in Poloxin- and TQ-treated tumor tissues. Cellular extracts were prepared from MDA-MB-231 xenografts treated with DMSO (samples D1 to D10), Poloxin (samples P1 to P10), or TQ (samples TQ1 to TQ3) for Western blot analysis with Plk1 antibodies. β-Actin served as the loading control. C: Quantification of Plk1 expression levels of tumor tissues in B, relative to corresponding loading control β-actin. Data were presented as the mean ± SD and analyzed by the Student's t-test. **P < 0.01.