

Abstract
Adeno-associated virus (AAV)–mediated expression of wild-type or mutant P301L protein tau produces massive degeneration of pyramidal neurons without protein tau aggregation. We probed this novel model for genetic and structural factors and early parameters of pyramidal neurodegeneration. In yellow fluorescent protein–expressing transgenic mice, intracerebral injection of AAV-tauP301L revealed early damage to apical dendrites of CA1 pyramidal neurons, whereas their somata remained normal. Ultrastructurally, more and enlarged autophagic vacuoles were contained in degenerating dendrites and manifested as dark, discontinuous, vacuolated processes surrounded by activated astrocytes. Dendritic spines were lost in AAV-tauP301L–injected yellow fluorescent protein–expressing transgenic mice, and ultrastructurally, spines appeared dark and degenerating. In CX3CR1EGFP/EGFP-deficient mice, microglia were recruited early to neurons expressing human tau. The inflammatory response was accompanied by extravasation of plasma immunoglobulins. α2-Macroglobulin, but neither albumin nor transferrin, became lodged in the brain parenchyma. Large proteins, but not Evans blue, entered the brain of mice injected with AAV-tauP301L. Ultrastructurally, brain capillaries were constricted and surrounded by swollen astrocytes with extensions that contacted degenerating dendrites and axons. Together, these data corroborate the hypothesis that neuroinflammation participates essentially in tau-mediated neurodegeneration, and the model recapitulates early dendritic defects reminiscent of “dendritic amputation” in Alzheimer's disease.
Tauopathies include a wide variety of primary disorders including Pick's disease, progressive supranuclear palsy, corticobasal degeneration, and frontotemporal dementia, as well as the most frequent secondary tauopathy, Alzheimer's disease (AD). In AD, the intracellular inclusions in somata and processes consist of highly phosphorylated protein tau and develop concomitant with or subsequent to intracellular accumulations of amyloid peptides, presumably in multivesicular bodies. Subsequently, extracellular amyloid plaques develop together with neurofibrillary tangles and inflammation, which combined define the postmortem pathologic findings in AD. The relative timing and molecular relation between amyloid and tauopathies are still debated, whereas the link to kinases such as GSK3β is becoming accepted.1–5 Although aggregation of phosphorylated protein tau into filamentous inclusions in soma and neuropil is characteristic and diagnostic of all tauopathies, the neurotoxic phosphorylated tau species that damages synapses and neurons remains elusive. By analogy to amyloids, it is not the final tau deposits but the intermediate tau oligomers that were first suspected to cause disease; however, their cellular sites of action and the mechanisms whereby neurons succumb in tauopathy remain to be defined.
Progressive staging of AD is clinically based on symptoms, cognitive examination, and brain imaging. Postmortem pathologic staging of AD is based on tauopathy visualized using immunohistochemistry (IHC) with monoclonal antibody AT8, which is specific for phosphorylated protein tau.6 The follow-up study by Braak and Braak2 revealed that transient tauopathy in the dendritic segments located in the stratum lacunosum moleculare causes “dendritic amputation.” Of note, tau-related dendritic defects are an early, albeit transient, phenomenon in stages II and III, preceding the tauopathy in soma of pyramidal neurons in later stages of AD. The stratum lacunosum moleculare is the “connection hub” of the dendritic tree of CA1 pyramidal neurons with incoming myelinated axons of the temporoammonic path, which originates in the entorhinal cortex (medial and lateral layers II and III).7 Thereby, the stratum lacunosum moleculare confers the direct connection between the two brain regions that are the first to be affected by pathologic features of AD, and primarily by tauopathy.2,6,8
Adeno-associated virus (AAV)–mediated gene transfer of mutant amyloid precursor protein and of wild-type (WT) and mutant P301L protein tau in the hippocampus of WT mice replicates pathologic features of AD including intracellular and extracellular amyloid accumulation and phosphorylation of protein tau. Pyramidal neurodegeneration was evident only in mice injected with AAV-tau, without formation of large aggregates of protein tau or tangles.1 This model robustly recapitulates neurodegeneration in vivo, which is largely lacking in transgenic mouse models of amyloidopathy and tauopathy.4,5,9,10 We observed that protein tau, present in an apparent oligomeric state, effected cellular demise via non-apoptotic mechanisms, and a large panel of markers implicated inflammation as a major actor.1 The third major defect in AD-affected brain, neuroinflammation, was thereby implicated in the novel AAV-based model. In degenerating brain, inflammation is chronic and thought to contribute essentially to the disease process, although inflammatory reactions are proposed to provide protection.11–14 In this respect, amyloid is analyzed most extensively because of experimental and clinical studies of anti-inflammatory compounds and vaccination against amyloid in AD. Far less is known of the mechanisms by which protein tau contributes to neurodegeneration and neuroinflammation.
We analyzed the reciprocal relations of tauP301L-induced dendritic degeneration, gliosis, and vascular defects as contributing early to the subsequent neurodegeneration. Expression of mutant protein tau was initially evident in dendrites, causing degeneration of distal neuronal compartments, reminiscent of dendritic amputation in AD-affected brain.2 Degenerating neuronal axons and dendrites contained more and larger autophagic vacuoles and were surrounded by swollen astrocytic soma and processes. Moderate inflammatory reactions preceding the death of CA1 pyramidal neurons are proposed to increase the permeability of the blood-brain barrier (BBB), which was remarkably selective. The combined data are consistent with the hypothesis that tauP301L-mediated neurodegeneration is initiated by distal dendritic and axonal injury, closely associated temporally and spatially with early inflammatory responses and vascular defects. The combined effects eventually lead to annihilation of CA1 pyramidal neurons, which subsequently abrogate the neuroinflammatory reaction and plasma protein extravasation, corroborating their contribution to neurodegeneration.
Materials and Methods
Animals and Stereotaxic Injection
Adult WT FVB/N mice aged 3 to 4 months and of both sexes were used for most studies. To visualize neurodegeneration and microgliosis, we also studied yellow fluorescent protein (YFP)–expressing transgenic mice [B6.Cg-TgN(Thy1-YFP-H)2Jrs] and CX3CR1-deficient mice (B6.129P-Cx3cr1tm1Litt/J) (Jackson Laboratory, Bar Harbor, ME).15,16 Recombinant AAV vectors of hybrid serotype 1/2 to express either enhanced green fluorescent protein (EGFP) or mutant tauP301L under control of the human synapsin 1 gene promoter were used as described.1,17
Surgical procedures and unilateral intracerebral injection of viral particles into the left hemisphere were performed as described.1 In brief, stereotactic injection of 2 μL viral suspension containing 10E8 t.u. was at coordinates 1.94 mm posterior, 1.4 mm lateral, and 2.2 mm ventral relative to bregma.1,18
All experiments were performed by certified researchers conforming to regional, national, and European regulations concerning animal welfare and animal experimentation, and were authorized and supervised by the university animal welfare commission (Ethische Commissie Dierenwelzijn, Katholieke Universiteit Leuven).
Immunohistochemistry
At indicated times after infection, mice were anesthetized using pentobarbital (Nembutal) and perfused transcardially using 7 mL ice-cold saline solution for 2 minutes. Brains were removed rapidly and fixed overnight in 4% paraformaldehyde for subsequent IHC analysis on 40-μm free-floating coronal vibratome sections. Primary antibodies were either affinity-purified polyclonal antibodies or mouse monoclonal antibodies that were biotinylated or horseradish peroxidase–labeled (Table 1) to avert cross-reaction with mouse immunoglobulins (see Results). Immune reactions were developed using streptavidin–horseradish peroxidase complex for biotinylated monoclonal antibodies or via a three-step method for polyclonal antibodies or directly for horseradish peroxidase–labeled primary antibodies using diaminobenzidine as chromogen.1 Sections were counterstained using hematoxylin, dehydrated by passage through a graded series of alcohol and xylol, and mounted using DePeX mounting medium (Sigma-Aldrich Corp., St. Louis, MO) for microscopic analysis. For confocal analysis, after incubation with primary antibody, sections were processed using secondary antibodies coupled to Alexa Fluor 488 or 594 (Molecular Probes, Inc., Eugene, OR) counterstained with DAPI and mounted using Mowiol mounting medium (Polysciences, Inc., Warrington, PA) and DABCO mounting medium (Air Products and Chemicals, Inc., Allentown, PA).1
Table 1.
Antibodies Used in the Present Study
| Antibody | Type | Specificity or epitope | Label | Host | Concentration or dilution | Supplier |
|---|---|---|---|---|---|---|
| HT7 | Monoclonal | Human protein tau | Biotin | Mouse | IF, 0.55 μg/mL | Innogenetics |
| AT8 | Monoclonal | P-Ser202/P-Thr205 | Biotin | Mouse | IHC, 0.56 μg/mL | Innogenetics |
| MHCII | Polyclonal | Activated microglia | NA | Rat | IF, 0.5 μg/mL IHC, 0.05 μg/mL | Pharmingen |
| CD11b | Monoclonal | Mouse complement type 3 receptor | NA | Rat | IF, 1 μg/mL | Serotec |
| CD45 | Polyclonal | Mouse CD45 leukocyte common antigen, Ly-5 | NA | Rat | IF, 0.125 μg/mL IHC, 0.06 μg/mL | Pharmingen |
| A2 mol/L | Polyclonal | Mouse α2-macroglobulin | NA | Rabbit | IHC, 1/10,000 | Nordic |
| Immunoglobulins | Polyclonal | Mouse IgG, IgA, IgM | HRP | Goat | IHC, 2 μg/mL | Dako |
| IgM | Polyclonal | IgM μ-chain–specific | Biotin | Goat | IHC, 1 μg/mL | Vector |
| Albumin | Polyclonal | Mouse albumin | HRP | Goat | IHC, 0.05 μg/mL | Bethyl |
| Transferrin | Polyclonal | Mouse transferrin | HRP | Goat | IHC, 0.1 μg/mL | Bethyl |
| CD31 | Monoclonal | Mouse PECAM-1 | NA | Rat | IHC, 0.015 μg/mL | Pharmingen |
| PH2AXS139 | Polyclonal | Human phospho-histone H2A | NA | Rabbit | IHC, 0.5 μg/mL | Abcam |
Abcam, Inc., Cambridge, MA; Bethyl Laboratories, Inc., Montgomery, TX; Dako Corp., Carpinteria, CA; Immunogenetics, Inc., Buena, NJ; Nordic Immunologic Laboratories, Eindhoven, The Netherlands; BD Biosciences Pharmingen, Inc., San Diego, CA; Serotec, Inc., Raleigh, NC; Vector Laboratories, Inc., Burlingame, CA.
HRP, horseradish peroxidase; IF, immunofluorescence; NA, not available.
Evans Blue Dye Extravasation
To analyze integrity of the BBB, mice injected with AAV-tauP301L (10 days after infection) received an i.p. injection of 25 mg/kg Evans blue dye.19,20 At indicated times, mice were anesthetized and transcardially perfused, and the brain was removed as described. Hippocampi were dissected, weighed, and incubated for 72 hours in formamide in the dark at room temperature. After centrifugation at 10,000 × g for 10 minutes, the supernatant was collected, and absorbance was measured spectroscopically at 620 nm. Evans blue dye concentrations, calculated from standard curves, are given per unit brain weight. Another group of similarly treated AAV-tauP301L mice was euthanized via cervical dislocation to retain the Evans blue dye in blood vessels and tissue. The brains were processed for immunofluorescence and confocal microscopy on 40-μm vibratome sections and counterstained using DAPI.
Perls Prussian Blue Iron Staining
Perls staining for ferric iron was performed essentially as described previously.21 Vibratome sections of 40 μm were mounted on silanized glass slides and dried at 50°C. The sections were immersed in potassium ferrocyanide solution [1% K4(Fe)CN)63H2O in 0.11 mol/L HCl] for 60 minutes. After rinsing with PBS and 50 mmol/L Tris HCl buffer (pH 7.6), the reaction was enhanced via incubation with 0.5 mg/mL diaminobenzidine for 4 minutes at room temperature. The sections were counterstained using nuclear fast red, dehydrated, and mounted using DePeX.
Ultrastructural Analysis
Anesthetized mice were perfused transcardially using ice-cold saline solution.22 Brains were removed and stored in Karnovsky's solution. Vibratome sections 300 μm thick were incubated with 1% osmium tetroxide before dehydration via a graded series of ethanol and impregnation with Agar100 resin. Ultrathin sections (80 nm) were stained using uranyl acetate and lead citrate via standard procedures before analysis at transmission electron microscopy (CM100; Philips/FEI Corp., Eindhoven, The Netherlands). Toluidine blue staining was on thin sections (2 μm) immersed for 30 seconds in 0.1% toluidine blue, 0.1 mol/L Na2CO3, and air-dried before analysis at light microscopy.
Morphometric Study
Blood Vessels
Capillary profiles of transversely cut blood vessels 7.5 μm in diameter or smaller containing no nucleus of pericyte or endothelium were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). Four parameters were measured at 3400× magnification: area and inner diameter of the lumen, area and outer diameter of the lumen, area of endothelial cells, and area of astrocytes around capillary wall. Capillary wall area (in micrometers squared) was defined by subtracting the outer and inner areas of the lumen.
Synapses
Synaptic density was measured by counting postsynaptic density (PSD) at 10,500× magnification (expressed as number of synapses per 100 μm2). Analysis included 482 images containing 15,796 synapses (EGFP), 621 images with 17,779 synapses (tau 10 days after infection), and 696 images with 9189 synapses (tau 21 days after infection). The length of the active zone was measured at 10,500× magnification and expressed as average PSD length in micrometers. Analysis included 1547, 2079, and 2053 synapses on CA1 stratum radiatum dendritic spines from mice injected with AAV-EGFP (n = 3), AAV-tau 10 days after infection (n = 4), and AAV-tau 21 days after infection (n = 4), respectively. Digital image analysis was performed using dedicated software (ImageJ).
Statistical Analysis
Data are given as mean ± SEM, and were analyzed statistically using one way analysis of variance as specified. P < 0.05 was considered statistically significant.
Results
AAV-Tau Damages Dendrites and Axons of CA1 Neurons
AAV-mediated gene transfer of WT or mutant protein tau but not of mutant amyloid precursor protein or EGFP caused massive degeneration of CA1 pyramidal neurons in WT mouse hippocampus without appreciable tauopathy, defined as large fibrillar aggregates, threads, or tangles.1 Neurodegeneration was evident from 10 days after infection onward, and progressed rapidly, with elimination of almost all CA1 pyramidal neurons at 3 to 6 weeks after infection. The neurodegeneration was spatially and temporally closely associated with severe microglial inflammation.
To define the early aspects and essential parameters of neurodegeneration, the AAV-tauP301L vector was intracerebrally injected in transgenic mice that expressed YFP in their neurons, although we selected a substrain with a restricted expression pattern to enable observation of additional details such as dendritic spines.15 First, we confirmed that in YFP-expressing transgenic mice, the CA1 neurons were lost at 21 days after infection (Figure 1, A and B), as reported for WT mice.1 More remarkable, the fluorescent neurons enabled definition of damage to proximal dendrites in the stratum radiatum, and the associated CA1 cell bodies were still visibly intact (Figure 1A). Note that not all pyramidal neurons in this strain of YFP-expressing mice expressed the YFP transgene,15 which can be appreciated also in the contralateral hemisphere (Figure 1A, right panel).
Figure 1.
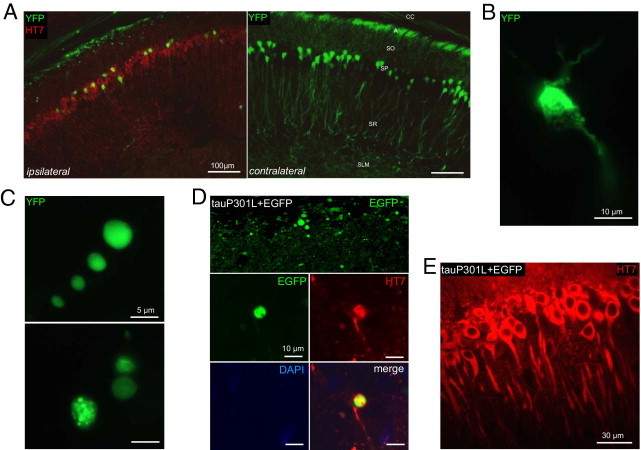
Tau damages dendrites and axons of CA1 neurons. A: YFP-expressing transgenic mice (n = 6) were analyzed to visualize degenerating neuritic processes. At 21 days after infection, AAV-tauP301L induced loss of apical and proximal dendrites and a significant decrease in the number of pyramidal neurons.1 Immunofluorescent staining was performed using HT7-bio and Strepta-Alexa 594 (red). A, alveus; SLM, stratum lacunosum moleculare; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. B: Degenerating neuron in the CA1 of mouse injected with AAV-tauP301L. Note shrinkage and fragmentation of cytoplasmic YFP signal. C: Examples of YFP-positive dilatations in the alveus in YFP-expressing mice injected with AAV-tauP301L. YFP signal appeared as punctate (upper panel) or smooth (lower panel). D: WT mice were co-injected with AAV-tauP301L (10E8 t.u.) and AAV-EGFP (10E8 t.u.) (n = 5) or injected only with AAV-EGFP (10E8 t.u.) (n = 4). Images were obtained in the CA1 from 40-μm thick sections using a microscope (Leica Microsystems GmbH, Wetzlar, Germany). Tau-induced degeneration was associated with deposition of EGFP in dilatations in alveus similarly as in YFP-expressing mice. E: Neurons co-injected with tauP301L and EGFP express tau (HT7 staining) in the somatodendritic compartment.
Of note, axons of CA1 pyramidal neurons projecting via the alveus demonstrated a marked punctate appearance of YFP-containing processes on the coronal sections, indicating that the axonal compartments of CA1 pyramidal neurons were compromised by expression of protein tau (Figure 1, A and C). Some axonal dilatations were not punctate but smooth, which we can refer to as multivesicular bodies and cytoplasmic protein aggregates, respectively (Figure 1C). Degenerating neurons were shrunken, with condensed and vacuolated cytoplasm (Figure 1B), although ultrastructurally they appeared as dark neurons (see Axonal, Dendritic, and Synaptic Pathologic Findings and Degeneration).
To confirm that protein tau caused the neuritic defects, we co-injected AAV-tauP301L and AAV-EGFP in WT mice compared with mice receiving only AAV-EGFP. Similar to YFP-expressing mice injected with AAV-tauP301L, axonal projections of CA1 pyramidal neurons in the alveus exhibited the same punctate pattern (Figure 1D). EGFP-positive dilated axons accumulated protein tau (Figure 1D), whereas CA1 pyramidal neurons contained protein tau in their apical dendrites (Figure 1E). Intracerebral injection of AAV-EGFP alone did not lead to any of these cellular defects, which indicated that they were mediated by expression of human mutant tau.
Axonal, Dendritic, and Synaptic Pathologic Findings and Degeneration
Protein tau is an axonal microtubule-associated protein that regulates microtubular dynamics and transport of proteins, organelles, and vesicles. Despite its reputation as an axonal microtubule-associated protein, protein tau was observed also in dendrites of AAV-tauP301L–injected WT mice (Figure 1, A and E; see AAV-Tau Damages Neuronal Processes and Causes Inflammation). In AD and other tauopathies, protein tau is redistributed from axonal to somatodendritic compartments, where it tends to aggregate into the typical fibrillary bundles. This finding corroborates the notion that most tauopathy in human patients is present as neuropil treads, implicating protein tau in dendrites and eventually in spines.23,24
Analysis of the ultrastructure of axons in AAV-tauP301L–injected WT mice demonstrated that many axons manifested vacuoles filled with aggregated electron-dense material (Figure 2, A and B). In addition, many electron-transparent circular structures represented degenerated axons that retained, to a variable extent, the original myelin sheath (Figure 2, C–F). These “axonal remnants” represent final stages of the evident axonal degeneration and resemble certain types of spongiform degeneration (Figure 2)25; however, it was not possible to define the actual state of cell bodies associated with these axonic remnants.
Figure 2.
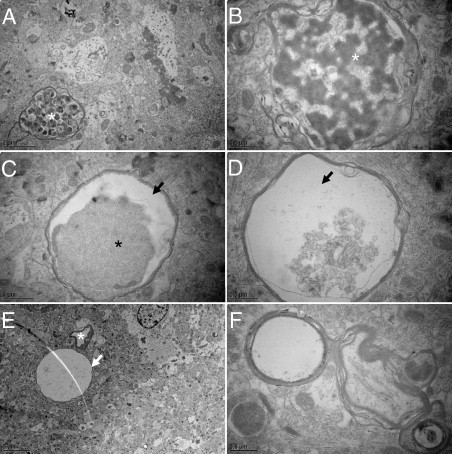
Axonal disease. Ultrastructural aspects of axonal degeneration representing various stages. A: Accumulation of autophagic vacuoles (asterisk). B: Example of digested axoplasm (asterisk). C and D: Accumulation and detachment (arrows) of dark axoplasm (asterisk). E: Extremely swollen degenerated axon is almost completely electron-lucent (arrow) next to a capillary (asterisk). F: Axon with split myelin sheath.
We continued to analyze the dendritic pathologic findings that have not been well-studied in human tauopathies.8,24 Nevertheless, the pathologic implication of tau in dendrites was observed long ago because dilated apical dendrites of CA1 pyramidal neurons filled with abnormal protein tau were described as a transient phenomenon in stages II to III in AD, leading eventually to dendritic amputation.2 The stratum radiatum was analyzed in WT mice injected with AAV-tauP301L at 10 and 21 days after infection compared with mice similarly injected with AAV-EGFP as controls. At 10 days after infection, toluidine blue staining of ultrathin sections revealed the relatively normal appearance of apical dendrites in AAV-tauP301L–injected WT mice (Figure 3, left and middle panels). In contrast, at 21 days after infection, many apical dendrites of CA1 neurons were disorganized or even lost (Figure 3, right panel).The observed dendritic degeneration was analyzed ultrastructurally in AAV-tauP301L–injected WT mice. At the onset of neurodegeneration, at 21 days after infection, dark discontinuous dendrites were observed that coexisted with dendrites of normal appearance (Figure 4A). However, many of these dendrites accumulated vacuoles (Figure 4B, inset), which were also observed in dark dendrites (Figure 4C), which suggests that most dendrites were undergoing a similar degenerative process but were observed in different stages at 21 days after infection. Dark dendrites demonstrated the most intense vacuolization and were surrounded by electron-lucent astrocytic processes (Figure 4D).
Figure 3.
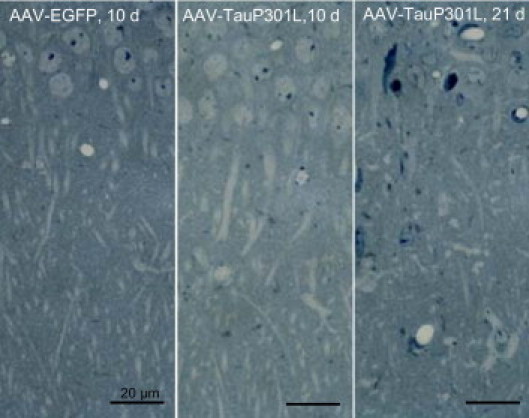
Dendritic changes on neurodegeneration. Thin sections of brain from AAV-EGFP-injected mice at 10 days after infection (n = 3), AAV-tauP301L–injected mice at 10 days (n = 4) and 21 days (n = 4), stained with toluidine blue, to compare projecting apical dendrites of CA1 pyramidal neurons into the stratum radiatum.
Figure 4.
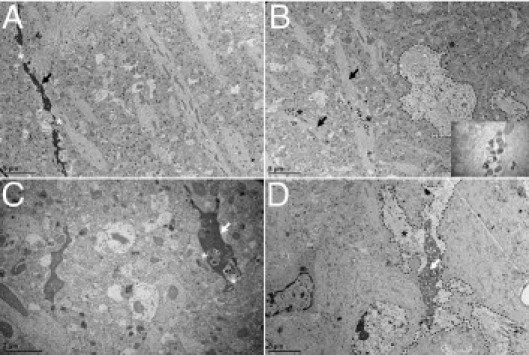
Ultrastructure of dendritic degeneration in CA1 stratum radiatum. Representative examples of the ultrastructure of the CA1 stratum radiatum reveal various stages of dendritic degeneration. A: Lack of continuity (asterisks) of dark degenerating dendrite (arrow). B: Healthy-looking dendrites (arrows) accumulating autophagic vacuoles (asterisks). Inset: Adjacent astrocyte is delineated (dashed line). C: Autophagic vacuoles (asterisks) in a dark dendrite (arrow). D: Dark perforated dendrite (arrow) surrounded by white astrocytic process (asterisk). Astrocyte is delineated (dashed line).
Loss of function of protein tau after increased phosphorylation is thought to be the prime cause for its release from microtubules and their destabilization. Subsequent missorting of protein tau to the somatodendritic compartments leads to the typical tauopathy. Nevertheless, impaired microtubule-dependent transport to synapses should be the first to be affected by the release of tau. This hypothesis was tested via ultrastructural analysis of synapses in the CA1 region in the AAV-tauP301L–mediated degeneration process. Many dark degenerating synapses were observed, with either their postsynaptic compartment (Figure 5A, upper and lower left panels) or both their presynaptic and postsynaptic compartments (Figure 5A, lower right panel) severely affected. Dark synapses, nevertheless, retained a rather normal gross structure, although they were decreased in size and mostly discontinuous with the dendrite (Figure 5A, upper right panel, and B).
Figure 5.
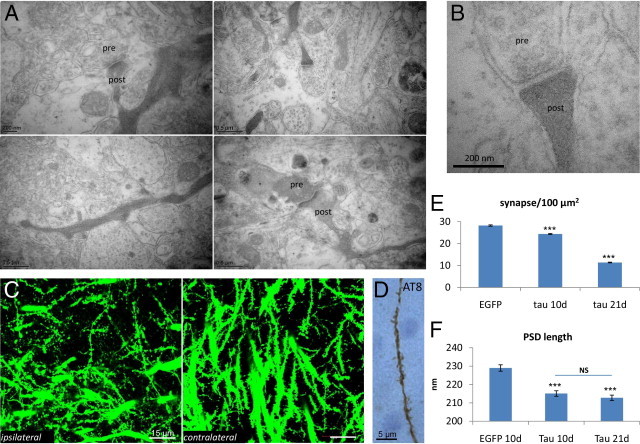
Synaptic degeneration. A: Representative examples of the ultrastructure of synapses in CA1 demonstrate dark synaptic degeneration. Presynaptic and postsynaptic compartments are denoted. B: Higher magnification of the upper right panel in A demonstrates degenerating dark synapse. C: Confocal microscopy of the CA1 stratum radiatum in brain from AAV-tauP301L–injected YFP-expressing transgenic mice (n = 6) at 21 days after infection to compare the extent of degenerating dendritic shafts and spines in injected and noninjected hippocampi. D: A single dendrite from CA1 stratum radiatum of AAV-tauP301L–injected WT mouse positive for AT8. Accumulation of phospho-tau along dendritic process and in postsynaptic spines is apparent. E: Quantification of synaptic density (PSD/100 μm2). Values are given as mean ± SEM. *P < 0.001 (one-way analysis of variance). AAV-EGFP–injected mice (n = 3) were compared with AAV-tauP301L–injected mice (n = 4) at 10 days after infection and with AAV-tauP301L–injected mice (n = 4) at 21 days after infection. Total number of synapses for EGFP-injected mice was 15,796 in area of 55,781.83 μm2, for tauP301L-injected mice at 10 days was 17,779 in area of 72,944.06 μm2, and for tauP301L-injected mice at 21 days was 9189 in area of 81,023.39 μm2. F: Quantification of PSD length is given in nanometers. Values are given as mean ± SEM. *P < 0.001 (one-way analysis of variance). Mice injected with AAV-EGFP at 10 days (n = 3) were compared with those injected with AAV-tauP301L at 10 days (n = 4) and AAV-tauP301L at 21 days (n = 4) after infection. Total number of nonperforated synapses for EGFP-injected mice was 1547, with total length of 354,257 μm; for tauP301L-injected mice at 10 days was 2079, with total length of 447,194 μm; and for tauP301L-injected mice at 21 days was 2053, with total length of 436,814 μm.
In AAV-tauP301L–injected YFP-expressing mice, the number of dendritic spines was significantly decreased (Figure 5C). This observation was confirmed at ultrastructural morphometric analysis of synapses in the CA1 stratum radiatum on ultrathin sections. The reduction in the number of synapses was already significant at 10 days after infection (14% reduction); however, a nearly 60% reduction was evident at 21 days after infection relative to AAV-EGFP–injected control mice (Figure 5E). Another important parameter of the postsynaptic compartments, ie, the average length of PSD, was already significantly decreased at 10 days after infection (6.1% reduction) but did not further decrease substantially in the remaining synapses at 21 days after infection (7.1% reduction) (Figure 5F).
IHC revealed that human protein tauP301L was located in the soma and along the apical dendrites of the pyramidal neurons at risk (Figure 1E) and occasionally also in dendritic spines (Figure 5D), although these were less numerous and likely reflected advanced stages of the degeneration process of dendrites and spines.
The combined data underline the conclusion that protein tau causes dendritic degeneration involving the postsynaptic compartment. Toluidine blue staining and ultrastructural analysis revealed large areas of empty space in the degenerating hemisphere injected with AAV-tau (Figure 6). These resemble “leftovers” or “ghosts” of degenerated dendrites and were present in the neuropil throughout the hippocampus and in deeper layers of the cortex, reminiscent of some specified aspects of spongiform degeneration.25
Figure 6.
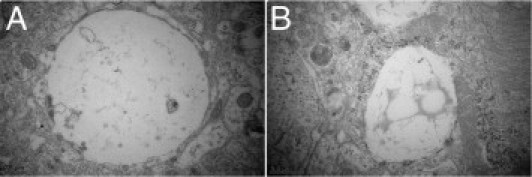
Spongiform degeneration. Examples of ultrastructure of neuropil illustrate vacuolization and spongiform appearance of CA1 stratum radiatum in hippocampus of AAV-tauP301L–injected WT mice at 21 days after infection. A shows a large empty vacuole. B shows a vacuole filled with flocculent material.
AAV-Tau Damages Neuronal Processes and Causes Inflammation
Details of the progression of the induced damage and of the associated inflammation were obtained by comparing AAV-tauP301L–injected WT mice at 10 and 21 days after infection. Inflammation was revealed using IHC for glial fibrillary acidic protein (GFAP) as a marker of astrogliosis and for major histocompatibility complex class II (MHCII) as a marker of active microgliosis, as described.1 Quantification of the respective signals in cortex and hippocampus of AAV-tauP301L–injected mice revealed that astrogliosis occurred early, whereas microgliosis coincided most closely with the onset of neurodegeneration (Figure 7A). Neurons that express protein tau in brain of AAV-tauP301L–injected YFP-expressing mice were in close contact with microglia that were clearly activated, as marked by MHCII (Figure 7B). In the same YFP-expressing mice, astroglia were also in close contact with degenerating neurons (Figure 7C).
Figure 7.
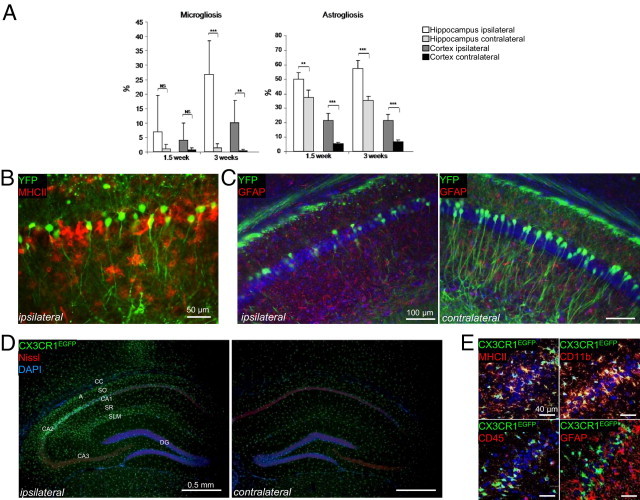
Tau expression causes neuronal loss intimately related to inflammation. A: Timeline of AAV-tauP301L–mediated inflammation. WT mice were intracerebrally injected with AAV-tauP301L and analyzed for GFAP and MHCII at 1½ weeks (n = 4) and 3 weeks (n = 6) after infection. Quantification of GFAP- and MHCII-positive signal in the ipsilateral (white bars) and contralateral (light gray bars) hippocampus and ipsilateral (gray bars) and contralateral (black bars) cortex. Values are given as mean ± SD. B: IHC for MHCII in YFP-expressing mice (n = 6) intracerebrally injected with AAV-tauP301L and analyzed at 21 days after infection. Degenerating neurons are in close proximity to activated microglial cells. Anti-MHCII antibody was followed by rabbit anti-rat biotinylated secondary antibody and Strepta-Alexa 594 (red). YFP-positive neurons are shown in green. C: IHC for GFAP in YFP-expressing mice (n = 6) intracerebrally injected with AAV-tauP301L and analyzed at 21 days after infection. Degenerating neurons are in close proximity to astroglial cells. Anti-GFAP antibody was followed by anti-rabbit biotinylated secondary antibody and Strepta-Alexa 594 (red). YFP-positive neurons are shown in green. Nuclei stained with DAPI are shown in blue. D: In AAV-tauP301L–injected CX3CR1EGFP/EGFP mice (n = 4), the fractalkine receptor–deficient EGFP-positive microglia are recruited to or by the degenerating neurons. Sections were stained for cytoplasmic Nissl substance (red) and nuclei (DAPI, blue). EGFP-positive microglia are shown in green. CA, cornu ammonis; DG, dentate gyrus; SLM, stratum lacunosum moleculare; SO, stratum oriens; SR, stratum radiatum. E: Confocal microscopy of glial markers in AAV-tauP301L–injected CX3CR1EGFP/EGFP mice (n = 4). Microglial markers MHCII and CD11b, unlike astroglial marker GFAP, co-localize with CX3CR1EGFP/EGFP-positive microglia. MHCII, CD11b, CD45, and GFAP were immunodetected using secondary antibody coupled to Alexa 594 (red). EGFP-positive microglia are shown in green, and nuclei stained with DAPI are shown in blue.
Having confirmed in this different mouse strain that microglial activation is closely associated with neuronal expression of protein tauP301L, as originally demonstrated in WT mice,1 we injected AAV-tauP301L intracerebrally in CX3CR1EGFP/EGFP mice.16 This knockin/knockout mouse strain was engineered via insertion of an active EGFP gene to disrupt the gene coding for the CX3CR1 fractalkine receptor, expressed specifically by microglia. The objective was dual: to test the eventual contribution of the microglial CX3CR1 receptor and to visualize via fluorescence microscopy all microglia activated and recruited by degenerating neurons.
Intracerebral injection of AAV-tau in CX3CR1EGFP/EGFP mice evoked a strong microglial reaction in the degenerating hippocampus (Figure 7D). The EGFP signal co-localized with CD11b and MHCII markers for activated microglia and not with astroglial GFAP (Figure 7E). Microglial activation was most intense in CA1/2, in which neurodegeneration was most intense, stopping abruptly and lacking in CA3, in which neurodegeneration was minimal (Figure 7D). Of note, microglial activation was unexpectedly intense in the stratum lacunosum moleculare, which comprises synapses of myelinated axons projecting from the entorhinal cortex to distal dendritic segments of CA1 pyramidal neurons, via the temporoammonic pathway.26
These observations considerably extend the proposed hypothetical relation of neurodegeneration and inflammation in the AAV-tau model.1 Neurodegeneration and microgliosis were also closely related in the inducible cdk5/p25 model of hippocampal sclerosis.27
AAV-Tau–Damaged Neuronal Processes Caused Inflammation and Neurovascular Defects
Next we defined the relative timing of the inflammatory responses with respect to pyramidal cell death. Brain ultrastructure was analyzed after intracerebral injection of AAV-tau at indicated times preceding evident neurodegeneration and focusing on neuronal processes and glia activation. This analysis revealed important neurovascular defects.
Dendrites of CA1 pyramidal neurons that expressed tau were surrounded by swollen footplates of astrocytes, with extended processes around the blood vessels. The structure of the blood vessels and their cellular linings seemed relatively normal (Figure 8, A and B), as in control animals injected with AAV-EGFP. The thickness of the wall of capillaries, defined as vessels with diameter less than 7.5 μm, was, however, significantly increased at 10 and 21 days after infection in the AAV-tauP301L–injected hemisphere relative to AAV-EGFP–injected mice at 10 days after infection (Figure 8C). Furthermore, many blood vessels within the CA1 region were surrounded by swollen astrocytes at 10 and 21 days after infection (Figure 8, A and D). These seemed to cause, or at least contribute to, the important constriction of these capillaries at the onset of neurodegeneration (Figure 8A).
Figure 8.
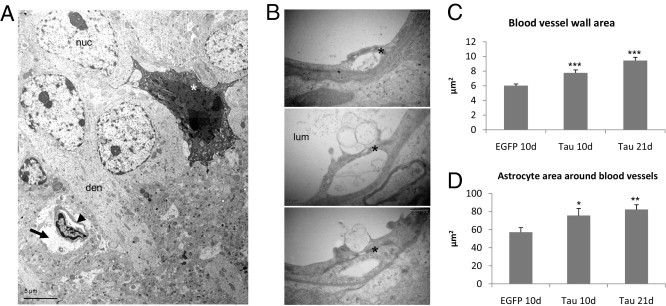
Inflammation is linked to vascular defects. A: Ultrastructure of degenerating CA1 region shows a dark neuron (asterisk) in proximity to a capillary (arrowhead), surrounded by a swollen astrocytic process (arrow) that likely is causing compression of the vessel. Den, apical dendrite; nuc, nucleus of CA1 pyramidal neuron. B: Ultrastructure of capillary wall in the brain of AAV-tauP301L–injected WT mice at 21 days after infection. Both the capillary wall linings and the tight junctions (asterisk) appeared normal and intact. Nevertheless, some membrane blebbing was evident at the luminal side (lum) of the endothelial cells in some capillaries. C: Quantification of the area occupied by the wall of capillaries in the brain of WT transgenic mice injected with AAV-EGFP at 10 days after infection (n = 3), with AAV-tauP301L at 10 days (n = 4), and at 21 days (n = 4). The number of capillaries analyzed for each condition was 75, 74, and 111, respectively. Data are given as mean ± SEM. *P < 0.05, **P < 0.01. D: Quantification of the astrocytic processes around blood vessels in the same mice as analyzed in C. *P < 0.05, **P < 0.01.
Early and Selective Permeability of the BBB
The cerebral circulation is separated from the neuropil by the BBB, which is formed by a complex and variable admixture of tight junctions between endothelial cells, by processes of pericytes at the basement membranes, and by astroglial footplates.28 Neurons, astroglia, and blood vessels form the neurovascular unit, and derailed communication between these cellular components can have severe repercussions on the proper functioning of the central nervous system.29
The foregoing results implicated that the BBB was compromised in the AAV-tauP301L–injected hemisphere in WT YFP-expressing transgenic mice and CX3CR1EGFP/EGFP mice. Previously, an unexpected high background reaction had been observed using IHC in a large panel of markers of neurodegeneration in the AAV-tauP301L model.1 The primary antibodies subsequently used in IHC were either affinity-purified polyclonal antibodies or mouse monoclonal antibodies labeled using biotin or peroxidase.1 Thereby, nonspecific reactions with endogenous mouse immunoglobulins in the brain tissue were circumvented.
We took advantage of earlier observations that indicated that the early inflammatory reaction was specifically restricted to the AAV-tauP301L–injected hemisphere. We first confirmed that in the brain of AAV-tauP301L–injected mice, but not in sham-operated or AAV-EGFP–injected mice, the immunoreactivity for IgG and IgM coincided with the neuronal degeneration (Figure 9A) and even appeared early after intracerebral injection of AAV-tauP301L (data not shown). We wondered whether extravasation of IgG and IgM was specific for immunoglobulins or whether other plasma proteins became lodged in the brain of AAV-tauP301L–injected WT mice. We were surprised to observe intense immunoreactivity for α2-macroglobulin (Figure 9B), the well-known high-molecular-weight proteinase inhibitor that has been genetically linked to AD.30 Conversely, levels of abundant plasma proteins albumin and transferrin were only marginally increased in the AAV-tauP301L–injected hemispheres (Figure 10, A and B). These observations demonstrated the high selectivity for passage of proteins across the BBB and suggested that active mechanisms operate in the AAV-tauP301L–injected hemisphere. An obvious candidate for this type of active transport is low-density lipoprotein receptor–related protein 1.31 However, IHC for this protein revealed no striking differences in intensity or distribution between ipsilateral and contralateral injected hemispheres in AAV-tauP301L–injected mice (data not shown).
Figure 9.
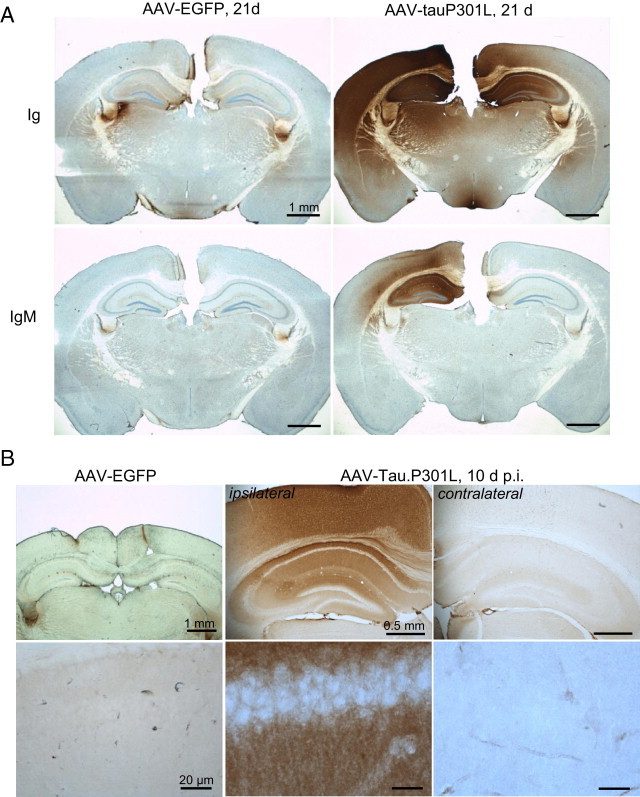
Selective extravasation of plasma proteins IgG, IgM, and α2-macroglobulin. A: IHC for mouse immunoglobulins (Ig) or mouse IgM on brain sections from AAV-tauP301L–injected WT mice (n = 6) at 21 days after infection compared with AAV-EGFP–injected WT mice (n = 4), as indicated. IHC was performed using specific horseradish peroxidase–labeled secondary antibodies only. B: Immunohistochemical detection of α2-macroglobulin in brain sections from AAV-tauP301L–injected WT mice at 10 days after infection (n = 4) compared with AAV-EGFP–injected WT mice (n = 4), as indicated. The lower panels are higher magnifications of pyramidal neurons in the CA1 region in the corresponding upper panels.
Figure 10.
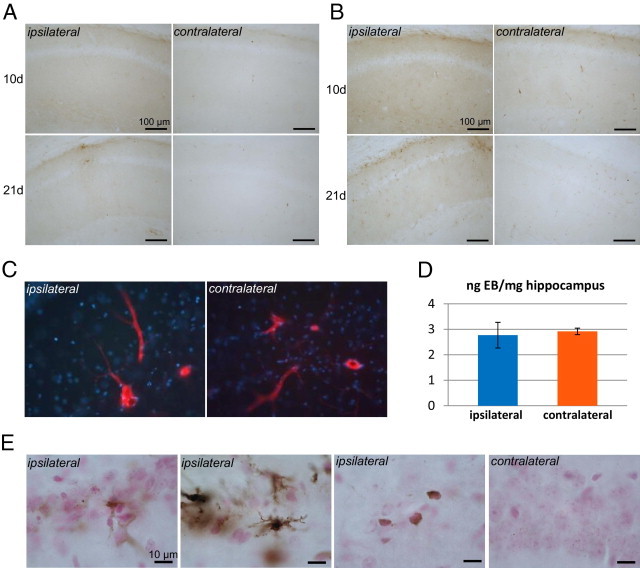
Selective extravasation. IHC for albumin (A) and transferrin (B) in sections of brain from AAV-tauP301L–injected mice at 10 days (n = 4) and 21 days (n = 6) after infection. C: WT mice were injected with AAV-tauP301L, and at 21 days after infection, 3 hours before euthanasia, were injected i.p. with 25 mg/kg Evans blue solution. The nonperfused brains were fixed in 4% paraformaldehyde and cut into 40-μm sections for microscopic analysis for Evans blue dye fluorescence. Note the positive reaction in blood vessels and the negative reaction in surrounding parenchyma. D: Mice (n = 3) were euthanized under anesthesia via perfusion with silane solution. The hippocampi were dissected and processed for Evans blue dye extraction (see Materials and Methods). Absorbance was measured at 620 nm, and Evans blue dye was quantified by comparison with a standard curve of various Evans blue dye concentrations. Data are expressed relative to the hippocampal weight in nanograms Evans blue dye per milligram tissue. E: Perls Prussian iron staining on sections of brain from AAV-tauP301L–injected mice (n = 3) at 21 days after infection. Nuclei were visualized using nuclear fast red. The stratum radiatum and stratum oriens are shown at the ipsilateral site (middle panels), and CA1 pyramidal regions ipsilateral versus contralateral (right and left panels).
Next assessed was the overall BBB integrity more classically using Evans blue dye, which on systemic injection binds to plasma albumin and does not penetrate the central nervous system in healthy animals. Two different approaches were used to detect eventual penetration of Evans blue dye in the brain parenchyma in AAV-tauP301L–injected WT mice: fluorescence microscopy of brain sections from nonperfused AAV-tauP301L–injected mice (Figure 10C) and homogenization, extraction, and spectroscopic quantification from the brain after perfusion of similarly treated animals (Figure 10D). Neither method revealed differences in tissue concentration of the Evans blue dye between ipsilateral and contralateral hippocampi at 10 days after infection (Figure 10, C and D). Thus, the BBB seemed to be intact for this low-molecular-weight compound.
Brains of AAV-tauP301L–injected mice were analyzed for hemosiderin, the degradation product of hemoglobin. Extravasation of red blood cells and hemoglobin is observed in pathologic brain conditions characterized by disrupted BBB, eg, microhemorrhage and trauma, and also in amyloid-damaged vessels in a subpopulation of patients with AD.32 Perls staining revealed small puncta of iron ions in the cell bodies of CA1 pyramidal neurons in all animals, ie, also in control mice and in the contralateral hemisphere (Figure 10E, right panel). No abnormal reactions were observed in the ipsilateral CA1 neurons (Figure 10E, left panel). Furthermore, no accumulation of iron was detected via Perls' reaction around blood vessels in the injected or noninjected hemispheres (Figure 10E). Occasionally, activated glial cells reacted more strongly for iron via Perls' reaction at the ipsilateral side (Figure 10E, second panel).
Together, the data convincingly indicate that the BBB was not grossly compromised in the brain of AAV-tauP301L–injected mice but that the leakiness was limited and specific. In particular, the eventual contribution of extravascular IgG or IgM and α2-macroglobulin remains to be explored.
Markers for Oxidative Stress and Vascular Damage
Increased phosphorylation of histone H2AX (S139), a marker of double-strand DNA breaks, was observed in AAV-tauP301L–injected WT mice (Figure 11A, upper panel). In control AAV-EGFP–injected mice, phosphorylated H2AX levels were also increased, which suggests that the AAV vectors themselves may act on the DNA repair pathway. Nevertheless, phosphorylated H2AX foci in the AAV-tauP301L–injected animals were far more intense than in the AAV-EGFP–injected mice, which suggests that oxidative stress contributed to tau-induced neurodegeneration. Nitrotyrosine, another marker of oxidative stress in pathologic conditions, was increased in glial cells (Figure 11A, lower panel).
Figure 11.
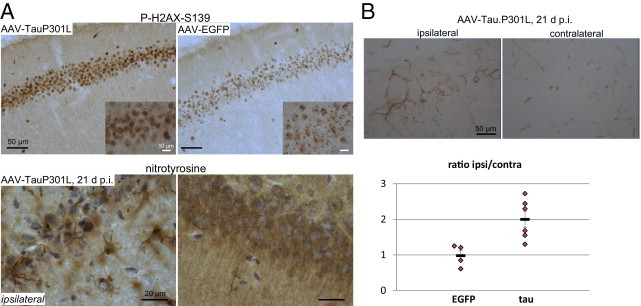
Oxidative stress and increased expression of PECAM-1. A: Upper panel: IHC for phosphorylated H2AX (S139) on sections of brain from AAV-tauP301L–injected mice (n = 4) at 10 days after infection compared with AAV-EGFP–injected mice (n = 4). Inset shows increased size and number of phosphorylated H2AX–positive foci in AAV-tauP301L–injected mice. Lower panel: IHC for nitrotyrosine on sections of brain from AAV-tauP301L–injected mice (n = 6) at 21 days after infection. Comparison of ipsilateral versus contralateral sites reveals increased nitrotyrosine levels in glial cells. B: Upper panel: IHC for PECAM-1 (CD31) on sections of brain from AAV-tauP301L–injected mice at 21 days after infection. Lower panel: Quantification of PECAM-1–positive blood-vessels in brain of AAV-tauP301L–injected mice (n = 6) and AAV-EGFP–injected mice (n = 4) at 21 days after infection, expressed as mean ± SEM ratio of the ipsilateral versus contralateral sides.
After confirming that inflammation and oxidative stress were associated with tau-induced neurodegeneration, we analyzed other vascular markers. Injured neurons and activated glial cells release various proinflammatory and anti-inflammatory factors and angiogenic substances, whereas inflammation and oxidative stress regulate angiogenesis.33 We analyzed expression of the platelet endothelial cell adhesion molecule 1 (PECAM-1), also denoted as CD31, which is an important marker of neuroinflammation and BBB damage.34,35 Quantitative IHC for CD31 revealed significantly increased expression of PECAM-1 in the AAV-tauP301L–injected hemisphere in WT mice than in AAV-EGFP–injected mice (Figure 11B). This was interpreted to mean that the neurodegeneration and associated selective BBB damage actively involved the endothelial cells in the neurovascular unit, supposedly by emitting trophic factors that stimulate an angiogenesis-related reaction.
The combined phenomena are proposed to reflect both cell autonomous and nonautonomous mechanisms of dendritic disease. The observed accumulations of autophagic vacuoles within dendrites implicate an important role for autophagy in the process of degeneration, if not causal, at least by trying to cope with the evident accumulating neuronal debris. It was concluded that the actual mode of pyramidal cell death in this model involves several mechanisms including autophagy, as has been proposed previously.1,5
Discussion
Primary Tauopathies versus AD
In primary tauopathies, it is evident that protein tau is responsible for the deleterious effects that lead neurons eventually to cell death. The unanswered question remains why it takes many decades, or even almost a human lifetime, before tauopathy results in clinical dementia. Epigenetic-, age-, and lifestyle-related factors and peripheral disease progressively become co-morbid with protein tau in the primary tauopathies.
Conversely, amyloidopathy is accepted as the primary defect in the secondary tauopathy of AD; however, the role of protein tau and the contribution of tauopathy are issues that remain debated in AD research. Nevertheless, clinical studies and animal models support the supposition that the tauopathy in AD is an essential pathologic factor in the overall process of synaptic and cognitive demise and of neurodegeneration and dementia.4,5,36–43
Major differences among tauopathies are the actual brain regions concerned, eg, frontotemporal versus frontomedial cortex or limbic regions versus hindbrain. Whether this reflects essential differences in molecular mechanisms or in cellular triggers is not clear. We accept that increased phosphorylation of protein tau is essential in all tauopathies but that it is subject to modulation by mutation, deviating isoform ratio, amyloid burden, α-synuclein, inflammation, and other factors that remain to be defined. Eventually, they all end with the typical tauopathy recognized postmortem as argyrophilic protein tau aggregates in soma and neuropil.24
We believe that protein tau is central to neurodegeneration and dementia, which, however, does not equate to the long-held thesis that the large protein tau aggregates are the neurotoxic species per se. The AAV-tau model examined in the present study and previously1,5 demonstrates that clearly, and is supported by other tau models including rTg4510 mice,42 biGT mice,3 rTGWT mice,43 and tau255 mice,39 among others.5,9,10,41
The AAV-based model for tau-mediated hippocampal neurodegeneration that has been developed recently1 was further explored in the present study. The objective was to define the earliest factors that contribute, genetically and epigenetically, molecularly and cellularly, and centrally and peripherally, to neuronal death and brain damage triggered by protein tau.
Tau-Mediated Neurodegeneration Is Preceded by Dendritic Defects and Autophagy
In the AAV model, hippocampal expression of human WT tau4R or mutant tauP301L effected extensive CA1 pyramidal neurodegeneration in WT mice.1 In the present study, we similarly injected AAV-tauP301L intracerebrally in YFP-expressing transgenic mice, which not only confirmed the neurodegenerative phenotype in an independent genetic background but also highlighted a major aspect of the mechanism, ie, early dendritic damage and degeneration. The YFP-positive CA1 pyramidal cell bodies were still intact when their dendritic compartments were demonstrating disease, which implicates protein tau as initiating its actions in distal dendritic compartments.
Ultrastructurally, neuronal processes in the hippocampal subfields of the stratum radiatum, stratum pyramidale, stratum oriens, and alveus accumulated multivesicular bodies, indicative of intense autophagy in these regions that express the extra human protein tau. Damaged processes were surrounded by electron-lucent astrocytes, which suggests that additional non-cell autonomous mechanisms contribute to the neuronal injury and demise. Similar cytoplasmic vacuolization in a human tau–expressing transgenic mouse model was interpreted to suggest a necrotic mechanism of decay, although other morphologic criteria implied more divergent pathways of cell death.44 In our AAV-based model, dendrites and axoplasm of degenerating neurons appeared similarly dark, indicating that the condition eventually affected all neuronal processes that contained human protein tau. One consequence of the extreme vacuolization was the nearly complete loss of axoplasm in neurons in specified hippocampal regions, an aspect reminiscent of spongiform degeneration.25 Overexpression of protein tau to high levels induced structural abnormalities in the cortex of rTg4510 mice, including progressive neuropathy with axons having split and ballooned myelin sheaths and vacuoles reminiscent of spongiform degeneration.42,45
Tau-Mediated Neurodegeneration Is Closely Linked to Neuroinflammation
Neuroinflammation, most often combined with defective BBB functions, is evident in most neurologic disorders, although it is not generally accepted as essentially contributive.13,14 Microvascular endothelium forms with astrocytes, pericytes, and extracellular matrix, the neurovascular unit, which ensures effective shielding from the periphery, which is needed for normal functioning of the central nervous system. In primary tauopathies, microvascular conditions have not been well studied in clinical or experimental detail. We explored the effects of tau-mediated neuronal damage on vascular dysfunctions in the recently developed AAV-tauP301L model.1 We addressed the controversial issues of dendritic degeneration and autophagy raised in the previous paragraph, not directly by premeditation or intention but because analysis of the mouse model presents clear-cut experimental evidence that is impossible to overlook. Moreover, the ultrastructural analysis of the neurovascular unit presented additional evidence for extensive and early involvement of dendritic disease. These were observed closely relative to the vascular aspects, and, therefore, are discussed partially interwoven with the neurovascular defects (see Tau-Mediated Neurodegeneration is Closely Linked to Neurovascular Defects).
Inflammation and signs of vascular dysfunction were observed early after infection and were closely associated with and, therefore, a likely direct consequence of the beginning degeneration of axons and dendrites, when the neuronal cell bodies were still intact. Most degenerating neuronal processes were enclosed by glial cell bodies and processes, and astroglia are also well known intricate partners in the BBB. The combinations enabled us to propose the most likely sequence of events, as follows. AAV-mediated neuronal expression of protein tau causes axonal and dendritic interference with microtubule-mediated transport, resulting in presynaptic and postsynaptic dysfunction of neurons that release factors that activate microglia and astroglia. The resulting inflammatory and vascular problems contribute to, and even fuel, the vicious cycle that is generally believed to operate in neurodegeneration and dementia. Early inflammatory response has been suggested to precede neuronal loss in a similar model of AAV9-mediated expression of protein tau in the substantia nigra.46,47
We extensively probed the mechanisms that activate microglia because they are most closely concerned in the AAV-tau–induced neurodegeneration, corroborating the observations in the cdk5/p25 model for hippocampal sclerosis.1,27 CX3CR1EGFP/EGFP mice were analyzed because fractalkine CX3CL1 is produced by neurons and its receptor CX3CR1 is exclusively expressed by microglia. Disruption of signaling between neurons and microglia by CX3CL1-CX3CR1 induced neurotoxicity in various mouse models.48 Conversely, this fractalkine system was neuroprotective in a mouse model of focal cerebral ischemia49 and in the triple-transgenic mouse model of AD.50 Apparently, the outcome depends on the nature of the damaging trigger and then on the fine balancing of the factors involved. In our model, CX3CR1 deficiency did not prevent microglial recruitment and activation, which suggest that other neuron-microglia signaling pathways are involved.51 Although microglia are considered the innate immune system in the brain, the role of astroglia in general and in AAV-tau–mediated neurodegeneration should not to be overlooked. It was observed that astrogliosis preceded the surge of MHCII-positive activated microglia, which suggests that astrocytes were more rapidly responding to the initial insult. Microglial activation occurred closely with the onset and peak of neurodegeneration, when the neuronal cell bodies were becoming damaged. Moreover, microgliosis was transient, and subsided when most pyramidal neurons were annihilated in the CA1 subregions, whereas astroglia remained activated over the entire course of AAV-tauP301L expression.1 We propose that astrogliosis seems to be more reactive to the dendritic damage or to the viral aspects of the model, whereas microgliosis is closely related in time and space to the neurodegeneration of the CA pyramidal cell bodies.
Tau-Mediated Neurodegeneration Is Closely Linked to Neurovascular Defects
The astrocytic processes surrounding the brain microvasculature were swollen even before neurons began to degenerate, constricting small capillaries and hampering microcirculation. Constriction of capillaries will decrease edema that follows the increased permeability of the BBB; however, the reduced oxygen supply to the brain will result in further damage to neurons. The mechanism of protein tau–induced BBB damage may involve the contribution of proinflammatory cytokine tumor necrosis factor-α and chemokine MCP-1 released from activated microglial cells.52
Extravasation of plasma proteins α2-macroglobulin and immunoglobulins was identified in brain of AAV-tauP301L mice, which suggests that the BBB is compromised. However, with no evidence for other small plasma proteins, albumin and transferrin, and the tight junction being intact, major breakdown of the BBB was ruled out. These specific effects do not yet entice us to implicate immunoglobulin and α2-macroglobulin in the mechanism of neurodegeneration. More experimental work is needed to understand these findings. In a rat SOD1 model of amyotrophic lateral sclerosis, IgG and hemosiderin were detected in presymptomatic stages, and Evans blue dye extravasation was evident in symptomatic stages.53 Recent findings in pericyte-deficient mice demonstrated that BBB breakdown causes secondary dendritic damage and neuronal loss.54 This suggests that accumulation of serum proteins in the AAV-tauP301L model may further contribute to dendritic degeneration.
Vascular dysfunction in AD is generally related to abluminal amyloid depositions around cerebral blood vessels that are thought to negatively affect microvascular permeability, likely in both directions. In AD brain parenchyma, microglia are considered to attempt to eliminate the extracellular amyloid deposits via phagocytosis, which is a late event that contributes to the chronic negative aspect of neuroinflammation. The purely phagocytic action of microglia evidently is not at stake in the early phases of the model analyzed in the present study. Nevertheless, activated microglia release proinflammatory factors, cytokines, chemokines, reactive oxygen species, prostaglandins, and other mediators that can help, but also harm, neurons. Activated astrocytes often co-localize with neurons and microglia at damaged sites; however, a precise or specific contribution is not evident in AD.
The continued analysis of our AAV-tauP301L–based model of neurodegeneration reveals early damage to axonal and dendritic neuronal compartments and to spines and synapses. The damage seems to trigger intense autophagocytic activity that eventually contributes to degeneration of the processes, which subsequently is reflected in degenerating soma of pyramidal neurons in CA1/2 of the hippocampus.1 This proposed sequence of neuronal events is closely linked to inflammatory reactions and vascular alterations, likely triggered by the initial neuronal injury after expression of protein tau. The AAV-based model, therefore, is most complementary to the existing transgenic mouse models, which largely lack the neurodegeneration and inflammation aspects for as yet unknown reasons.4,5 The data further validate the AAV-based model for mechanisms that underlie tau-induced neurodegeneration downstream of the amyloid connection in AD and are relevant in primary tauopathies.
Acknowledgments
We thank many collaborators and scientists for technical assistance, advice, materials, and scientific support, in particular, Monika Zebski for assistance with virus production, Eugeen Vanmechelen for generous gifts of monoclonal antibodies, and Annick Vogels, Evelyne Sauvage, and Gabriela Casteels for assistance with administration.
Footnotes
Supported by Fonds Wetenschappelijk Onderzoek-Vlaanderen, Instituut voor Wetenschap en Techniek, EEC-Framework Programs, Katholieke Universiteit Leuven (KULeuven) Research Fund, and KULeuven Research and Development. T.J. is the recipient of EU Marie Curie doctoral fellowship MEST-CT-2005-020589 from the European Graduate School of Neuroscience (EURON).
References
- 1.Jaworski T., Dewachter I., Lechat B., Croes S., Termont A., Demedts D., Borghgraef P., Devijver H., Filipkowski R.K., Kaczmarek L., Kügler S., Van Leuven F. AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in WT mice. PLoS One. 2009;4:e7280. doi: 10.1371/journal.pone.0007280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak E., Braak H. Alzheimer's disease: transiently developing dendritic changes in pyramidal cells of sector CA1 of the Ammon's horn. Acta Neuropathol. 1997;93:323–325. doi: 10.1007/s004010050622. [DOI] [PubMed] [Google Scholar]
- 3.Terwel D., Muyllaert D., Dewachter I., Borghgraef P., Croes S., Devijver H., Van Leuven F. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. 2008;172:786–798. doi: 10.2353/ajpath.2008.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaworski T., Kügler S., Van Leuven F. Modeling of tau-mediated synaptic and neuronal degeneration in Alzheimer's disease. Int J Alzheimer Dis. 2010 doi: 10.4061/2010/573138. pii:573138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaworski T., Dewachter I., Seymour C.M., Borghgraef P., Devijver H., Kügler S., Van Leuven F. Alzheimer's disease: old problem, new views from transgenic and viral models. Biochim Biophys Acta. 2010;1802:808–818. doi: 10.1016/j.bbadis.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Braak E., Braak H., Mandelkow E.M. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- 7.Kajiwara R., Wouterlood F.G., Sah A., Boekel A.J., Baks-te Bulte L.T., Witter M.P. Convergence of entorhinal and CA3 inputs onto pyramidal neurons and interneurons in hippocampal area CA1: an anatomical study in the rat. Hippocampus. 2008;18:266–280. doi: 10.1002/hipo.20385. [DOI] [PubMed] [Google Scholar]
- 8.Braak H., Braak E. Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
- 9.Götz J., Ittner L.M. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- 10.Duyckaerts C., Potier M.C., Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnaud L., Robakis N.K., Figueiredo-Pereira M.E. It may take inflammation, phosphorylation and ubiquitination to “tangle” in Alzheimer's disease. Neurodegener Dis. 2006;3:313–319. doi: 10.1159/000095638. [DOI] [PubMed] [Google Scholar]
- 12.Lucin K.M., Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwab C., Klegeris A., McGeer P.L. Inflammation in transgenic mouse models of neurodegenerative disorders. Biochim Biophys Acta. 2010;1802:889–902. doi: 10.1016/j.bbadis.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Banks W.A. Mouse models of neurological disorders: a view from the blood-brain barrier. Biochim Biophys Acta. 2010;1802:881–888. doi: 10.1016/j.bbadis.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng G., Mellor R.H., Bernstein M., Keller-Peck C., Nguyen Q.T., Wallace M., Nerbonne J.M., Lichtman J.W., Sanes J.R. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 16.Jung S., Aliberti J., Graemmel P., Sunshine M.J., Kreutzberg G.W., Sher A., Littman D.R. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shevtsova Z., Malik J.M., Michel U., Bähr M., Kügler S. Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol. 2005;90:53–59. doi: 10.1113/expphysiol.2004.028159. [DOI] [PubMed] [Google Scholar]
- 18.Franklin K.B.J., Paxinos G. Academic Press; San Diego, CA: 1997. The Mouse Brain in Stereotaxic Coordinates. [Google Scholar]
- 19.Chen X., Lan X., Roche I., Liu R., Geiger J.D. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J Neurochem. 2008;107:1147–1157. doi: 10.1111/j.1471-4159.2008.05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ujiie M., Dickstein D.L., Carlow D.A., Jefferies W.A. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003;10:463–470. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- 21.Van Dooren T., Muyllaert D., Borghgraef P., Cresens A., Devijver H., Van der Auwera I., Wera S., Dewachter I., Van Leuven F. Neuronal or glial expression of human apolipoprotein e4 affects parenchymal and vascular amyloid pathology differentially in different brain regions of double- and triple-transgenic mice. Am J Pathol. 2006;168:245–260. doi: 10.2353/ajpath.2006.050752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewachter I., Ris L., Croes S., Borghgraef P., Devijver H., Voets T., Nilius B., Godaux E., Van Leuven F. Modulation of synaptic plasticity and Tau phosphorylation by wild-type and mutant presenilin1. Neurobiol Aging. 2008;29:639–652. doi: 10.1016/j.neurobiolaging.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell T.W., Nissanov J., Han L.Y., Mufson E.J., Schneider J.A., Cochran E.J., Bennett D.A., Lee V.M., Trojanowski J.Q., Arnold S.E. Novel method to quantify neuropil threads in brains from elders with or without cognitive impairment. J Histochem Cytochem. 2000;48:1627–1638. doi: 10.1177/002215540004801206. [DOI] [PubMed] [Google Scholar]
- 24.Duyckaerts C., Delatour B., Potier M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 25.Liberski P.P., Brown D.R., Sikorska B., Caughey B., Brown P. Cell death and autophagy in prion diseases. Folia Neuropathol. 2008;46:1–25. [PubMed] [Google Scholar]
- 26.Nakashiba T., Young J.Z., McHugh T.J., Buhl D.L., Tonegawa S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science. 2008;319:1260–1264. doi: 10.1126/science.1151120. [DOI] [PubMed] [Google Scholar]
- 27.Muyllaert D., Terwel D., Kremer A., Sennvik K., Borghgraef P., Devijver H., Dewachter I., Van Leuven F. Neurodegeneration and neuroinflammation in cdk5/p25-inducible mice: a model for hippocampal sclerosis and neocortical degeneration. Am J Pathol. 2008;172:470–485. doi: 10.2353/ajpath.2008.070693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiss N., Miller F., Cazaubon S., Couraud P.O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta. 2009;1788:842–857. doi: 10.1016/j.bbamem.2008.10.022. [DOI] [PubMed] [Google Scholar]
- 29.Zlokovic B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Blacker D., Wilcox M.A., Laird N.M., Rodes L., Horvath S.M., Go R.C., Perry R., Watson B., Jr, Bassett S.S., McInnis M.G., Albert M.S., Hyman B.T., Tanzi R.E. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet. 1998;19:357–630. doi: 10.1038/1243. [DOI] [PubMed] [Google Scholar]
- 31.Deane R., Sagare A., Zlokovic B.V. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer's disease. Curr Pharm Des. 2008;14:1601–1605. doi: 10.2174/138161208784705487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zecca L., Youdim M.B., Riederer P., Connor J.R., Crichton R.R. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 33.West X.Z., Malinin N.L., Merkulova A.A., Tischenko M., Kerr B.A., Borden E.C., Podrez E.A., Salomon R.G., Byzova T.V. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–976. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalinowska A., Losy J. PECAM-1, a key player in neuroinflammation. Eur J Neurol. 2006;13:1284–1290. doi: 10.1111/j.1468-1331.2006.01640.x. [DOI] [PubMed] [Google Scholar]
- 35.Woodfin A., Voisin M.B., Nourshargh S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. 2007;27:2514–2523. doi: 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- 36.van Leuven F. Single and multiple transgenic mice as models for Alzheimer's disease. Prog Neurobiol. 2000;61:305–312. doi: 10.1016/s0301-0082(99)00055-6. [DOI] [PubMed] [Google Scholar]
- 37.Roberson E.D., Scearce-Levie K., Palop J.J., Yan F., Cheng I.H., Wu T., Gerstein H., Yu G.Q., Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 38.Zempel H., Thies E., Mandelkow E., Mandelkow E.M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B.C., Christie M.J., Napier I.A., Eckert A., Staufenbiel M., Hardeman E., Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 40.Vossel K.A., Zhang K., Brodbeck J., Daub A.C., Sharma P., Finkbeiner S., Cui B., Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashe K.H., Zahs K.R. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K.H. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoover B.R., Reed M.N., Su J., Penrod R.D., Kotilinek L.A., Grant M.K., Pitstick R., Carlson G.A., Lanier L.M., Yuan L.L., Ashe K.H., Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andorfer C., Acker C.M., Kress Y., Hof P.R., Duff K., Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ludvigson A.E., Luebke J.I., Lewis J., Peters A. Structural abnormalities in the cortex of the rTg4510 mouse model of tauopathy: a light and electron microscopy study. Brain Struct Funct. 2011;216:31–42. doi: 10.1007/s00429-010-0295-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klein R.L., Dayton R.D., Tatom J.B., Diaczynsky C.G., Salvatore M.F. Tau expression levels from various adeno-associated virus vector serotypes produce graded neurodegenerative disease states. Eur J Neurosci. 2008;27:1615–1625. doi: 10.1111/j.1460-9568.2008.06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang D.B., Dayton R.D., Zweig R.M., Klein R.L. Transcriptome analysis of a tau overexpression model in rats implicates an early pro-inflammatory response. Exp Neurol. 2010;224:197–206. doi: 10.1016/j.expneurol.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardona A.E., Pioro E.P., Sasse M.E., Kostenko V., Cardona S.M., Dijkstra I.M., Huang D., Kidd G., Dombrowski S., Dutta R., Lee J.C., Cook D.N., Jung S., Lira S.A., Littman D.R., Ransohoff R.M. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 49.Dénes A., Ferenczi S., Halász J., Környei Z., Kovács K.J. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- 50.Fuhrmann M., Bittner T., Jung C.K., Burgold S., Page R.M., Mitteregger G., Haass C., LaFerla F.M., Kretzschmar H., Herms J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zitvogel L., Kepp O., Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798–804. doi: 10.1016/j.cell.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 52.Kovac A., Zilkova M., Deli M.A., Zilka N., Novak M. Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J Alzheimer Dis. 2009;18:897–906. doi: 10.3233/JAD-2009-1197. [DOI] [PubMed] [Google Scholar]
- 53.Nicaise C., Mitrecic D., Demetter P., De Decker R., Authelet M., Boom A., Pochet R. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. 2009;1301:152–162. doi: 10.1016/j.brainres.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 54.Bell R.D., Winkler E.A., Sagare A.P., Singh I., Larue B., Deane R., Zlokovic B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]