

Abstract
Interferon (IFN)-γ is present in lesions of patients with Lyme disease and positively correlates with the severity of manifestations. To investigate the role of IFNγ in the development of Lyme carditis, wild-type and IFNγ-deficient C57BL/6 mice were infected with the causative bacterium, Borrelia burgdorferi. Histological analysis revealed no change in the severity of carditis between wild-type and IFNγ-deficient mice at 14, 21, 25, and 28 days after infection. However, a distinct shift in the types of leukocytes within the hearts of IFNγ-deficient mice was observed at 25 days. In the absence of IFNγ, the number of neutrophils in the heart was increased, whereas the number of T lymphocytes was decreased. Bacterial loads within hearts were the same as in wild-type mice. Macrophages secrete chemokines that recruit immune cells, which could contribute to the accumulation of leukocytes in murine Lyme carditis. The ability of IFNγ and B. burgdorferi to activate murine macrophages was examined, and the two stimuli synergistically induced chemoattractants for mononuclear cells (ie, CXCL9, CXCL10, CXCL11, CXCL16, and CCL12) and decreased those for neutrophils (ie, CXCL1, CXCL2, and CXCL3). IFNγ and B. burgdorferi also synergistically enhanced secretion of CXCL9 and CXCL10 by murine cardiac endothelial cells. These results indicate that IFNγ influences the composition of inflammatory infiltrates in Lyme carditis by promoting the accumulation of leukocytes associated with chronic inflammation and suppressing that of cells that typify acute inflammation.
Lyme disease is characterized by the infiltration of leukocytes into tissues infected by the spirochetal bacterium Borrelia burgdorferi. B. burgdorferi is transmitted to the skin of a human host through the bite of an Ixodes scapularis tick. This infection initiates a local inflammatory event, resulting in a rash known as erythema migrans.1–3 In infected lesions, the innate immune response includes the recruitment of neutrophils, macrophages, and lymphocytes.4,5 If patients are left untreated, the bacteria can enter the bloodstream and disseminate throughout the body. Over time, the infection can develop into a multisystemic disorder, in which the immune response causes arthritis, myocarditis, and complications within the nervous system.2
Lyme disease in humans may develop into a chronic inflammatory disorder, with macrophages and T lymphocytes found in the synovium of infected joints.2 Neutrophils are present in synovial fluid but are rarely noted in the synovium itself.2,3 Type 1 helper T cells (Th1s) compose most lymphocytes found in synovial fluid.6 These cells stimulate the cellular immune response through production of interferon (IFN)-γ and other pro-inflammatory cytokines. The Th1/Th2 cell ratio in the synovial fluid directly correlates with the severity of Lyme arthritis.6 Serum levels of IFNγ also correlate positively with the severity of disease in individuals infected with B. burgdorferi.7 These data suggest that Th1 cells, which secrete IFNγ, have a crucial role in the pathological features of Lyme disease in humans and potentially contribute to the development of a chronic inflammatory state.
The vascular endothelium plays an integral role in the recruitment of Th1 cells and other circulating leukocytes to areas of infection through the up-regulation of adhesion molecules on its surface and the secretion of chemoattractant cytokines (chemokines).8 B. burgdorferi induces the expression of adhesion molecules (eg, E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1) on the surface of primary human umbilical vein endothelial cells (HUVECs)9 and stimulates endothelium to release the chemokines CXCL810 and CCL2.11 An in vitro model of the blood vessel wall, consisting of monolayers of HUVECs grown on acellular amniotic connective tissue,12 has demonstrated that B. burgdorferi acts on endothelium to promote the transmigration of neutrophils,9,10 T lymphocytes,13 and monocytes.11 In addition, the populations of CD4+ and CD8+ T cells that traverse HUVECs stimulated with B. burgdorferi are enriched for those that produce IFNγ.14
Thus, there is both in vivo and in vitro evidence that secretion of IFNγ by T lymphocytes may contribute to the progression of human Lyme disease. Therefore, our laboratory previously investigated whether IFNγ alters the phenotype of endothelium exposed to B. burgdorferi. We found that incubation of HUVECs with a combination of IFNγ and B. burgdorferi promotes synergistic secretion of seven chemokines. All but one of these attract T cells and macrophages, which are typically found in lesions of those with chronic inflammation. Concurrent stimulation of HUVEC-amnion cultures by IFNγ and B. burgdorferi also increases transendothelial migration of T cells compared with stimulation with either agent alone. In contrast, the migration of neutrophils across B. burgdorferi–activated HUVECs is decreased when IFNγ is added.15 Given that neutrophils typify acute inflammatory lesions, these in vitro observations support the hypothesis that IFNγ mediates a switch from acute to chronic inflammation in human Lyme disease.
In this study, we tested the hypothesis that IFNγ promotes chronic inflammation in Lyme disease using the murine model of the illness. The mouse model of Lyme disease has provided a means of investigating the progression of pathological features in the joints and heart.16 In infected mice, arthritic lesions are populated by many neutrophils, both in the lumina and connective tissues of the joint.17,18 At peak severity, ranging from 2 to 4 weeks after infection,19,20 lesions in mice with Lyme carditis contain mostly mononuclear leukocytes, predominately macrophages. Lymphocytes are also observed, but in most instances relatively few neutrophils are noted.19–21 Compared with murine Lyme arthritis, the cellular infiltrates seen in Lyme carditis in the mouse more closely resemble those found in human Lyme arthritis and carditis.2,5 Therefore, we chose to use murine Lyme carditis as a model to study the potential role of IFNγ in the switch from acute to chronic inflammation in human Lyme disease.
Herein, we demonstrate that mice deficient in IFNγ show an increase in neutrophils and a decrease in T lymphocytes within lesions of Lyme carditis compared with wild-type animals. Thus, IFNγ modulates the immune response to B. burgdorferi by promoting the recruitment of leukocytes that are associated with chronic inflammation.
Materials and Methods
Infection of Mice
Wild-type C57BL/6 and B6.129S7-Ifngtm1Ts/J (IFNγ-deficient) mice were received from the Jackson Laboratory (Bar Harbor, ME) at the age of 8 weeks. Although C3H/He mice develop more severe Lyme carditis than do C57BL/6 animals,19 we were unable to identify a source for C3H/He strains that lack IFNγ or its receptor. All procedures involving mice were approved by Stony Brook University's Institutional Animal Care and Use Committee (Stony Brook, NY). Low-passage B. burgdorferi N40 was grown in Barbour-Stoenner-Kelly-H (BSK) medium (Sigma-Aldrich Co, St Louis, MO), supplemented with 6% rabbit serum (Sigma-Aldrich Co) for 5 to 7 days at 33°C. Spirochetes were counted using dark-field microscopy, and mice were inoculated i.d. on the back with 2.5 × 105 bacteria in 100 μL of BSK medium or with 100 μL of BSK medium alone as a control. To confirm infection, bladders were collected at sacrifice, and each was cultured separately in a tube containing 7 mL of BSK medium.18 After 7 days, cultures were examined for spirochetes using dark-field microscopy. Results verified the infection of all mice that had been inoculated with spirochetes.
Evaluation of Severity of Carditis
Five infected and two uninfected wild-type or IFNγ-deficient mice were sacrificed using a CO2 chamber preset at 5 psi at 14, 21, and 28 days after inoculation. Hearts were collected, perfused with PBS (Gibco/Invitrogen Corporation, Grand Island, NY), and cut sagittally such that all four chambers were bisected. Hearts were fixed in 10% neutral-buffered formalin for 24 hours at room temperature, embedded in paraffin, divided into sections (5-μm thick), and stained with H&E. The severity of carditis was graded by two independent observers (G.J.S. and S.J.H.) who were unaware of the origin of each section. The severity of inflammation within these sections was measured using the following criteria: grade 0, no inflammation; 1, minimal inflammation with fewer than two small foci of inflammation; 2, moderate inflammation with two or more foci; and 3, severe inflammation with focal and diffuse infiltration covering a large area.22
Identification of Leukocytic Subtypes in Cardiac Lesions
Five infected and two uninfected wild-type or IFNγ-deficient mice were sacrificed using CO2 at 14 and 25 days after inoculation. Hearts were collected, perfused with PBS, and bisected sagittally. Hearts were flash frozen in optimal cutting temperature embedding medium (Sakura Finetek USA, Inc., Torrance, CA) using isopentane cooled by liquid N2. Frozen tissues were serially divided into sections (5-μm thick) using a cryostat and stored at -80°C until stained. This experiment was repeated using five or six infected and four uninfected wild-type or IFNγ-deficient mice at the 25-day point only.
For immunofluorescent staining, sections were fixed in acetone for 5 minutes at -20°C, rehydrated in PBS lacking Ca2+ and Mg2+ (PBS-; Gibco/Invitrogen Corporation), blocked with 5% bovine serum albumin (Sigma-Aldrich Co) in PBS- for 1 hour, and washed. Sections were incubated for 30 minutes at room temperature with conjugated primary antibodies diluted in PBS-. Unconjugated primary antibodies were placed on sections overnight at 4°C. After washing, sections were incubated with conjugated secondary antibodies for 45 minutes at room temperature. All sections were mounted with VectaShield mounting medium containing DAPI (Vector Laboratories, Inc., Burlingame, CA). In addition, some tissue sections were stained with H&E to grade the severity of carditis, as previously described.
To identify T lymphocytes, fluorescein isothiocyanate (FITC)–conjugated rat anti-mouse CD3 (1:50) was used. Natural killer cells were identified by FITC mouse anti-mouse NK1.1 (1:50). Alexa Fluor 647 rat anti-mouse B220 (1:50) and phycoerythrin rat anti-mouse IgM (1:50) were used simultaneously to identify B cells. To identify dendritic cells, FITC hamster anti-mouse CD11c (1:50) and Alexa Fluor 647 rat anti-mouse CD11b (1:50) were used simultaneously. All of these antibodies were obtained from BD Biosciences, San Jose, CA. Unconjugated rat anti-mouse Gr-1 (1:100; AbD Serotec, Raleigh, NC) and goat anti-mouse F4/80 (1:50; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were used simultaneously to identify neutrophils and macrophages. Secondary antibodies for detection were Alexa Fluor 555 donkey anti-goat IgG (1:1000; Invitrogen Corporation, Carlsbad, CA), followed by Alexa Fluor 488 goat anti-rat IgG (1:1000; AbD Serotec). Fc Block (BD Biosciences) was used to prevent non-specific binding of antibodies to cell surface Fc receptors. Matched antibodies to irrelevant antigens from the same manufacturers were used as negative controls. For comparison, negative controls were prepared in parallel with the positively stained samples. The number of positively stained leukocytes was counted by an observer (G.J.S.) using coded samples. DAPI staining of leukocytes was first used to identify the areas where leukocytes had infiltrated into the atrial walls and the base of the heart, the typical locations of Lyme carditis in mice.19,21,22 Then, the number of leukocytes stained for particular markers was tallied in 10 fields (×630) selected at random from within these areas. Whenever possible, fields were selected from two sections separated by an interval of at least 30 μm.
Assessment of B. burgdorferi Burden in Hearts of Mice
Five infected and two uninfected wild-type or IFNγ-deficient mice were sacrificed using CO2 at 25 days after inoculation. Hearts were removed, perfused with PBS, blotted dry, and minced. DNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen Inc., Valencia, CA), according to the manufacturer's protocol, and stored at -20°C. Primers were used to amplify the B. burgdorferi recA gene (forward, 5′-GTGGATCTATTGTATTAGATGAGGCTCTCG-3′; and reverse, 5′-GCCAAAGTTCTGCAACATTAACACCTAAAG-3′) and mouse Nid1 (nidogen-1) gene (forward, 5′-CCAGCCACAGAATACCATCC-3′; and reverse, 5′-GGACATACTCTGCTGCCATC-3′). The amounts of B. burgdorferi recA DNA were analyzed relative to Nid1 on an ABI 7500 Real Time PCR System (Applied Biosystems, Carlsbad, CA) in 96-well Thermo-Fast detection plates (Thermo Fisher Scientific, Waltham, MA) using the ABsolute Blue SYBR Green Low ROX Master Mix (Thermo Fisher Scientific), according to the manufacturer's protocol, in a total volume of 25 μL. Results are presented as the expression of recA in infected mice relative to uninfected mice, as calculated by ABI System SDS software (Applied Biosystems). Reactions were performed in quadruplicate.
Microarray Analysis of Murine Macrophages
B. burgdorferi strain HBD11 at passages 44 to 49 was cultured at 33°C in low-endotoxin BSK medium9 that contained 5% EX-CYTE Growth Enhancement Media Supplement (Millipore, Billerica, MA). The infectious N40 strain, which was used to inoculate mice, did not grow in this medium; therefore, it was unsuitable for in vitro experiments with cells that respond vigorously to endotoxin. Three to five days after passage, spirochetes were centrifuged at 5000 × g for 20 minutes and resuspended in the appropriate experimental medium. Spirochetes were counted using dark-field microscopy and added to macrophages at a ratio of 10 live spirochetes per macrophage.
To isolate murine macrophages, C57BL/6 mice at the age of 8 weeks were euthanized using CO2. Femurs and tibiae were removed, and marrow was collected by flushing the inside of each bone with Dulbecco's modified Eagle's medium (DMEM; Gibco/Invitrogen Corporation) containing 5% fetal bovine serum (Thermo Fisher Scientific), 100 U/mL of penicillin, and 100 μg/mL of streptomycin. Cells were plated in bone marrow medium-high (BMMhigh), which consisted of DMEM with 2 mmol/L l-glutamine, 1 mmol/L sodium pyruvate (Gibco/Invitrogen Corporation), 20% heat-inactivated fetal bovine serum, and 30% medium conditioned by L929 cells.23 On the fifth day, macrophages were detached by rinsing with cold PBS- and plated in BMMlow, which was composed of DMEM with 1 mmol/L sodium pyruvate, 2 mmol/L l-glutamine, 10% heat-inactivated fetal bovine serum, and 15% L929 cell-conditioned medium.24,25 For each experimental group, 8 × 106 macrophages were plated in BMMlow for 24 hours at 37°C. Macrophages were then incubated for 8 hours at 37°C with BMMlow only, B. burgdorferi (10 spirochetes/cell), recombinant murine IFNγ (10 ng/mL; R&D Systems, Inc., Minneapolis, MN), or the same amounts of B. burgdorferi and IFNγ combined. The stimuli were removed, and RNAlater (Applied Biosystems) was added at 4°C overnight. RNA was isolated using an RNeasy Mini Kit (Qiagen Inc.), according to the manufacturer's protocol, and stored at -80°C. The quality and concentration of RNA samples were measured using an Agilent 2100 Bioanalyzer Automated Analysis System (Agilent Technologies, Inc., Santa Clara, CA). Microarray analysis was performed by the Stony Brook University DNA Microarray Core Facility using the Mouse Expression Array 430 2.0 (Affymetrix, Santa Clara, CA). Data were analyzed by the Affy and Limma packages from Bioconductor open source software for bioinformatics (http://bioconductor.org, last accessed March 1, 2008) run in the R statistical environment, and synergy scores were calculated as previously described.15
Measurement of Expression of Chemokines by Murine Macrophages
Murine macrophages were isolated as previously described and incubated with BMMlow only, B. burgdorferi (10 live spirochetes/cell), IFNγ (10 ng/mL), or B. burgdorferi and IFNγ together for 8 hours at 37°C. The stimuli were removed, and RNAlater was added at 4°C overnight. RNA was isolated using an RNeasy Mini Kit, according to the manufacturer's protocol, and stored at −80°C. RNA was converted into cDNA using a Verso cDNA Kit (Thermo Fisher Scientific), as specified by the manufacturer. Solaris quantitative PCR primer and probe pairs for chemokines of interest were used in conjunction with Solaris qPCR Gene Expression Low ROX Master Mix (Thermo Fisher Scientific) on an ABI 7500 Real Time PCR System in 96-well Thermo-Fast detection plates, according to the manufacturer's protocol, in a total volume of 25 μL. The Gapdh (glyceraldehyde-3-phosphate dehydrogenase) transcript was used as an endogenous control for all samples. The efficiency of each primer and probe set was analyzed and was within acceptable limits for fold-change calculation using the ΔΔCT method, as calculated by ABI System SDS software. To measure the expression of chemokines at the protein level, murine macrophages were stimulated as previously described for 24 hours at 37°C. Conditioned media were removed and centrifuged at 8000 × g for 10 minutes. Supernatants were analyzed for content of selected chemokines using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Inc.).
Measurement of Expression of Chemokines by MCECs
The base medium for culture of murine cardiac endothelial cells (MCECs) consisted of DMEM containing 20% heat-inactivated fetal bovine serum, 100 U/mL of penicillin, 100 μg/mL of streptomycin, 2 μg/mL of amphotericin B, and 25 mmol/L HEPES (Gibco/Invitrogen Corporation). To make complete MCEC medium, base medium was supplemented with 1× nonessential amino acids (Gibco/Invitrogen Corporation), 1× sodium pyruvate (Gibco/Invitrogen Corporation), 100 μg/mL of heparin sodium (Sigma-Aldrich Co), and 100 μg/mL of endothelial cell growth supplement (Biomedical Technologies, Stoughton, MA).
To obtain primary cultures of MCECs, hearts were harvested from 7- to 10-day-old C57BL/6 pups. To reduce contamination by fibroblasts, the great vessels and most of the atria were dissected away, as previously described.26 The ventricles were then bisected, and the pooled heart tissue was rinsed with HBSS (Gibco/Invitrogen Corporation). Heart tissue from up to eight pups was placed in a gentleMACS C tube (Miltenyi Biotec, Auburn, CA) in 5 mL of HBSS containing 600 U/mL of collagenase type 2 (Worthington Biochemical Corp, Lakewood, NJ) and 60 U/mL of DNase I (Roche, Indianapolis, IN). A gentleMACS Dissociator (Miltenyi Biotec) was used to process the tissue first with the preset program m_heart_01, followed by a 30-minute incubation at 37°C with inversion of the C tube every 5 minutes and a second dissociation with preset program m_heart_02. The resulting suspension was filtered with a 70-μm cell strainer (BD Biosciences). The filtrate was then centrifuged at 300 × g for 10 minutes at room temperature. The resulting pellet was resuspended in 2 mL of cold base medium, mixed with 8 mL of cold 30% Histodenz solution (Sigma-Aldrich Co) in a 15-mL conical tube, overlaid with 2 mL of cold base medium, and centrifuged at 1500 × g for 20 minutes at 4°C with the brake off. The cells at the low-density interface were transferred to a fresh tube and washed once with 10 mL of cold MACS buffer (PBS-containing 0.5% bovine serum albumin and 2 mmol/L EDTA). The pellet was resuspended in 100 μL of MACS buffer, 1 μg of FITC anti-mouse CD31 (eBioscience, San Diego, CA) per 2 × 106 cells was added, and the sample was incubated in the dark for 10 minutes at 4°C. The cells were then washed with 2 mL of MACS buffer, resuspended in 100 μL of MACS buffer, and incubated with 20 μL of anti-FITC MicroBeads (Miltenyi Biotec) in the dark at 4°C for 15 minutes. Cells were washed with 2 mL of MACS buffer, resuspended in 500 μL of MACS buffer, and positively selected using an LS column (Miltenyi Biotec) equilibrated with 3 mL of MACS buffer and affixed to a midiMACS Separator (Miltenyi Biotec). No more than 6 × 106 cells were applied per column. The columns were rinsed three times with 3 mL of MACS buffer, and labeled cells were collected by detaching the column from the magnetic separator and flushing the column with 5 mL of cold MACS buffer. The collected MCEC fraction was centrifuged and resuspended in 3 mL of warm complete medium per four to five hearts, and cells were plated on gelatin-coated 6-cm dishes at 3 mL of suspension per dish. MCECs were incubated overnight in a humidified CO2 incubator at 37°C. The day after isolation, the cultures were washed three times, fed with complete medium, and refed every 2 to 3 days.
A second enrichment step targeting trypsin-resistant CD102 was performed when cultures were approximately 90% confluent (usually 4 to 5 days after isolation). MCECs were detached with 0.125% trypsin in PBS- with 2 mmol/L EDTA for 3 minutes at 37°C. Trypsin was neutralized with 2 mL of base medium, and the cells were washed with 2 mL of MACS buffer and resuspended in 100 μL of MACS buffer. FITC anti-mouse CD102 (BD Biosciences) at 2 μg per 2 × 106 cells was added, and the preparation was incubated in the dark for 10 minutes at 4°C. Magnetic labeling with anti-FITC MicroBeads and collection over LS columns were performed as previously described. MCECs at passage 2 were used for experiments, with a purity of 89% to 94%, as determined by flow cytometry to assess expression of CD102. Staining for CD11b demonstrated that contamination by myelomonocytic cells ranged from 1.0% to 2.2%. Secretion of chemokines by MCECs exposed to live B. burgdorferi, IFNγ, or the two agents combined was analyzed by ELISA, exactly as for murine macrophages.
Statistics
Data assessing the degree of severity of Lyme carditis were analyzed by the U-test (two tailed). All other data were analyzed using an unpaired analysis of variance, followed by the Tukey-Kramer multiple-comparisons test, using InStat software (GraphPad, La Jolla, CA). P < 0.05 was considered statistically significant.
Results
To determine the role of IFNγ in the development of Lyme carditis in mice, wild-type and IFNγ-deficient C57BL/6 animals were injected i.d. with B. burgdorferi N40. Three individual experiments were conducted to compare the severity of carditis in the two types of mice at times ranging from 14 to 28 days after infection. Lesions consisting of infiltrated leukocytes were seen in all infected mice (Figure 1, A–D) but not in sham-inoculated animals (Figure 1E). The results of a histological examination of sections stained with H&E revealed that most infiltrating leukocytes were located at the base of the heart and root of the aorta, consistent with previous reports.19,21,22 The leukocytes were observed along the endocardium and in the myocardium (Figure 1, A–D). The severity of carditis in wild-type and IFNγ-deficient mice was assessed semiquantitatively using a previously established scale of 0 to 3.22 An initial study examined hearts harvested from mice 14, 21, and 28 days after infection; severity scores did not differ between wild-type and IFNγ-deficient mice at any of these times (Figure 2A). A second study measured the severity of carditis at 14 and 25 days after infection, and a third study was performed at 25 days only. The combined results of the second and third studies are shown in Figure 2B. Consistent with the initial experiment, there was no difference in the severity of carditis between wild-type and IFNγ-deficient mice at 14 or 25 days after infection. Overall, the size and density of lesions within hearts of infected wild-type and IFNγ-deficient mice were comparable at all times examined.
Figure 1.
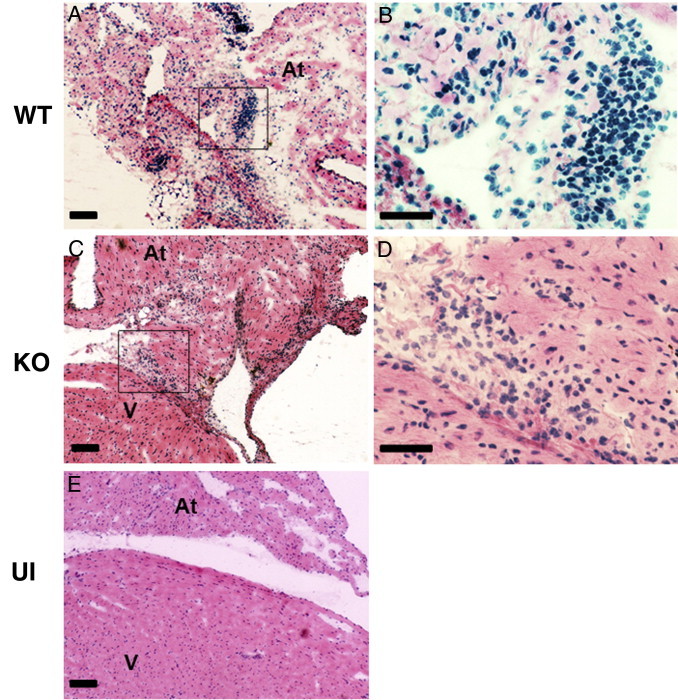
The histopathological features of Lyme carditis are similar in wild-type (WT) and IFNγ-deficient mice. The WT and IFNγ-deficient (KO) mice were infected with B. burgdorferi N40 or inoculated with BSK medium as uninfected (UI) controls. Representative sections stained with H&E from hearts of infected WT (A and B), infected KO (C and D), and UI WT (E) mice at 25 days after infection are shown. Boxed regions in A and C are magnified in B and D, respectively. Scale bars: 100 μm (A, C, and E); 50 μm (B and D). At, atrium; V, base of ventricle.
Figure 2.
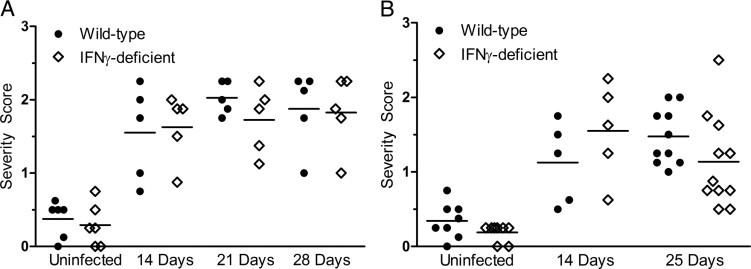
IFNγ-deficient mice do not exhibit a change in severity of carditis compared with wild-type mice when infected with B. burgdorferi. Wild-type and IFNγ-deficient mice were infected with B. burgdorferi N40. Five to eleven mice of each type were sacrificed at 14, 21, and 28 days (A) or 14 and 25 days (B) after infection. Two to four mice of each strain, inoculated with BSK medium only, were sacrificed at the same time points. The severity of carditis was assessed by two independent observers, as described in Materials and Methods. Horizontal bars indicate the mean score of carditis severity. Each data point represents the averaged severity score of an individual mouse. Data from uninfected mice at the various time points were combined. In B, data from mice infected for 25 days were combined from two separate experiments. There was no significant difference between wild-type and IFNγ-deficient mice in any of the experimental groups.
Although the absence of IFNγ did not alter the severity of murine Lyme carditis, we investigated the possibility that IFNγ affects the composition of leukocytic infiltrates within these cardiac lesions. This possibility is supported by our in vitro studies, wherein IFNγ increases the migration of T cells but decreases that of neutrophils across endothelium activated by B. burgdorferi.15 Therefore, we used immunofluorescent microscopy to analyze the leukocytic subtypes that constitute the lesions of Lyme carditis in wild-type and IFNγ-deficient mice. The numbers of B cells, natural killer cells, dendritic cells, T cells, neutrophils, and macrophages were counted in regions of inflammation using frozen sections prepared from the same hearts that were graded for severity of carditis in Figure 2B. B lymphocytes were not detected in lesions of Lyme carditis of wild-type or IFNγ-deficient mice at either 14 or 25 days after infection (data not shown). In addition, natural killer cells (Figure 3, A and B) and dendritic cells (Figure 3, C and D) were rarely seen at these points in either type of mouse. T cells were more prevalent within lesions than dendritic cells and natural killer cells, but they were still in the minority in both wild-type (Figure 3E) and IFNγ-deficient (Figure 3F) mice. Simultaneous staining for neutrophils and macrophages revealed that both were present within lesions of carditis in the two strains of mice, but macrophages predominated (Figure 3, G and H).
Figure 3.
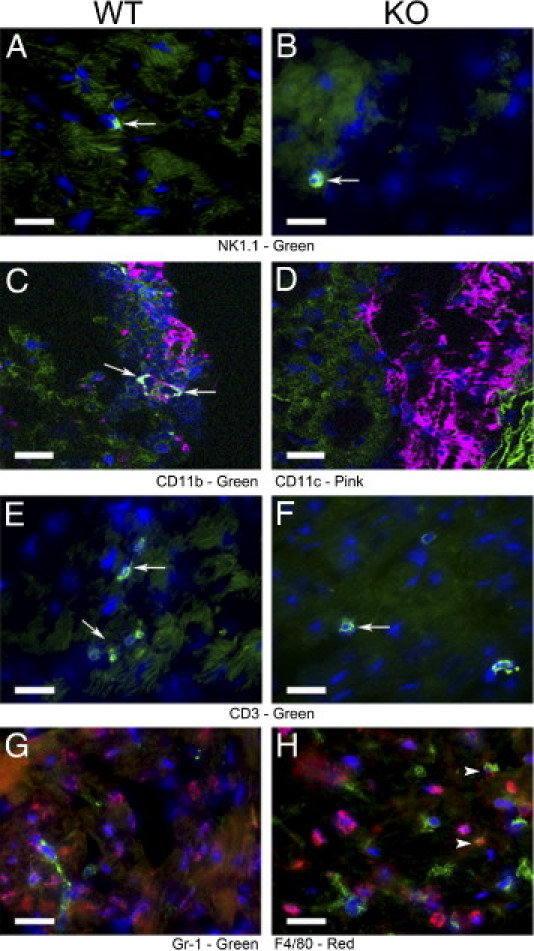
The composition of leukocytic infiltrates differs in the hearts of wild-type (WT) and IFNγ-deficient mice infected with B. burgdorferi. The WT and IFNγ-deficient (KO) mice were infected with B. burgdorferi N40. Representative sections from hearts of WT (A, C, E, and G) and IFNγ-deficient (B, D, F, and H) mice at 25 days after infection are shown. Sections were stained with anti-NK1.1 (green) to detect natural killer cells (A and B). To distinguish dendritic cells, anti-CD11c (green) and anti-CD11b (pink) were used concurrently (C and D). Anti-CD3 (green) was used to detect T cells (E and F). Samples were incubated with anti-Gr-1 (green) and anti-F4/80 (red) concurrently to detect neutrophils and macrophages, respectively (G and H). Arrows indicate examples of positively stained cells. Dendritic cells expressing both CD11c and CD11b are indicated by white staining (C). Arrowheads indicate examples of cells expressing both Gr-1 and F4/80 (H). Scale bars: 20 μm (A–H).
Quantitative analysis of the immunostained sections determined that there was an increase in the number of CD3+ T cells in hearts of infected wild-type mice at 25 days after inoculation, relative to uninfected mice (P < 0.001, Figure 4A). Gr-1+ neutrophils were increased relative to sham-inoculated controls in both strains of mice at 14 days after infection (P < 0.01) but only in IFNγ-deficient mice at 25 days (P < 0.001, Figure 4B). F4/80+ macrophages were elevated in both strains of mice after infection for either 14 or 25 days (P < 0.001, Figure 4C).
Figure 4.
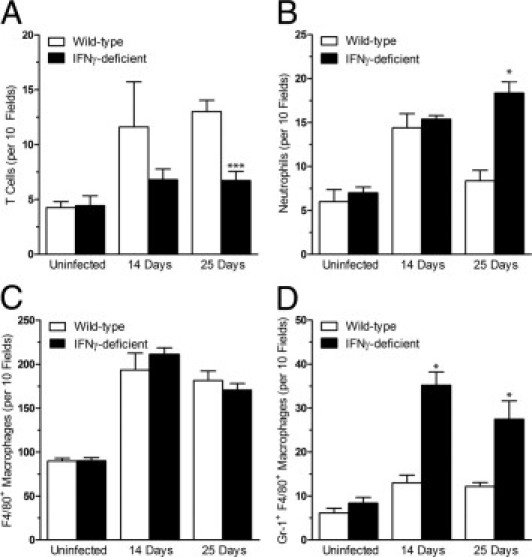
IFNγ increases T cells and decreases neutrophils within lesions of murine Lyme carditis. Wild-type and IFNγ-deficient mice were infected with B. burgdorferi N40 or inoculated with BSK medium as uninfected controls. Hearts were collected at 14 and 25 days after infection, divided into sections, and stained with fluorescent antibodies against CD3 or both Gr-1 and F4/80 concurrently. Results show the mean ± SD total number of cells in 10 fields (×630) expressing CD3 (A), Gr-1 (B), F4/80 (C), or both Gr-1 and F4/80 (D). There was no difference between controls at 14 and 25 days after infection, so data from the two time points were combined. Uninfected groups, n = 8 from two independent experiments; 14-day groups, n = 5 from one experiment; 25-day groups, n = 10 or 11 from two experiments. *P < 0.001 versus wild-type at the same time point.
When amounts of T lymphocytes, neutrophils, and macrophages were compared between wild-type and IFNγ-deficient mice, there were no significant differences at 14 days after infection. However, at 25 days after infection, a shift in subtypes of leukocytes within lesions of Lyme carditis was observed. In the hearts of infected IFNγ-deficient mice, there was a decrease in the number of T cells compared with infected wild-type mice (Figure 4A). In contrast, there was an increase in the number of neutrophils in IFNγ-deficient mice relative to wild-type mice (Figure 4B). The population of F4/80+ macrophages was increased in infected mice, but differences in genotype had no influence on amounts (Figure 4C). During the counting of neutrophils and macrophages, cells that were positively stained for both Gr-1 and F4/80 were observed. This species of myeloid cell27 was present to some degree in all mice. Notably, though, the number of Gr-1+ F4/80+ cells was greatly increased in the cardiac lesions of infected IFNγ-deficient mice compared with infected wild-type mice at both 14 and 25 days (Figure 4D).
The shift in the populations of neutrophils and T cells in IFNγ-deficient mice at 25 days could be the result of an altered number of spirochetes within the heart. Therefore, it was important to compare the spirochetal load in the hearts of infected wild-type and IFNγ-deficient mice. Through the use of quantitative real-time PCR, it was determined that hearts of wild-type and IFNγ-deficient mice contained comparable numbers of B. burgdorferi (Figure 5). From these data, it is evident that alterations in the composition of leukocytic infiltrates in the absence of IFNγ are not simply because of a change in spirochetal burden.
Figure 5.
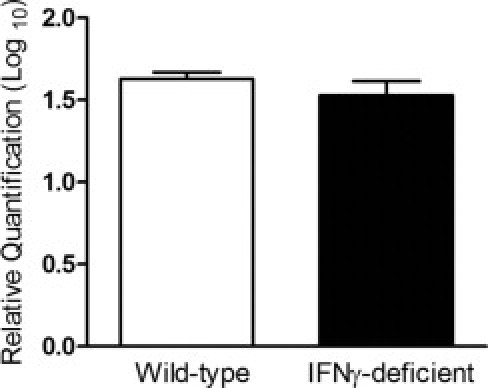
Deficiency of IFNγ does not alter spirochetal burden in hearts of infected mice. Wild-type and IFNγ-deficient mice were infected with B. burgdorferi N40 or inoculated with BSK medium as uninfected controls. Hearts were collected at 25 days after infection. DNA was extracted from the tissue and assessed using real-time PCR. The gene recA was used to measure the presence of B. burgdorferi. Samples were normalized to mouse Nid1, and data were plotted as mean ± SD relative to uninfected controls of the same genotype. Wild-type group, n = 4; IFNγ-deficient group, n = 5.
Like endothelial cells, macrophages, when activated, secrete various pro-inflammatory cytokines, which may recruit additional leukocytes to areas of infection. Furthermore, macrophages are present in lesions of Lyme disease in both humans28 and mice18–20 (Figure 4C) and are activated by B. burgdorferi.29,30 Therefore, we hypothesized that, as for HUVECs,15 IFNγ could be acting as a switch to promote the production by B. burgdorferi–stimulated macrophages of chemokines that attract leukocytes associated with chronic inflammation. To determine whether IFNγ alters the expression of chemokine genes in murine macrophages exposed to spirochetes, microarray analysis was performed. Primary murine bone marrow–derived macrophages were stimulated with B. burgdorferi, IFNγ, or B. burgdorferi and IFNγ concurrently for 8 hours. Among the genes encoding chemokines, those for CXCL9, CXCL11, CXCL16, and CCL12 were synergistically up-regulated by B. burgdorferi and IFNγ, whereas those for CXCL1, CXCL3, and CXCL5 were synergistically down-regulated. The up-regulated genes all encode attractants for mononuclear cells, whereas the down-regulated genes encode neutrophil chemoattractants.31
Six of these chemokines were chosen for further analysis using real-time RT-PCR. Transcripts of chemokines that typically attract neutrophils (ie, CXCL1 and CXCL3) were highly up-regulated by B. burgdorferi alone compared with cells activated by IFNγ alone (Figure 6, A and D). Conversely, genes encoding chemokines that attract mononuclear cells (ie, CXCL9, CCL12, CXCL11, and CXCL16) were up-regulated by stimulation with IFNγ, whereas B. burgdorferi alone had little effect (Figure 6, B, C, E, and F). The response of macrophages to IFNγ and B. burgdorferi combined also differed between the two categories of chemokines. The addition of both stimuli resulted in reduced expression of chemokines that attract neutrophils, compared with B. burgdorferi alone (Figure 6, A and D). On the contrary, B. burgdorferi and IFNγ together promoted increased expression of chemokines that attract mononuclear leukocytes, compared with IFNγ only (Figure 6, B, C, E, and F).
Figure 6.
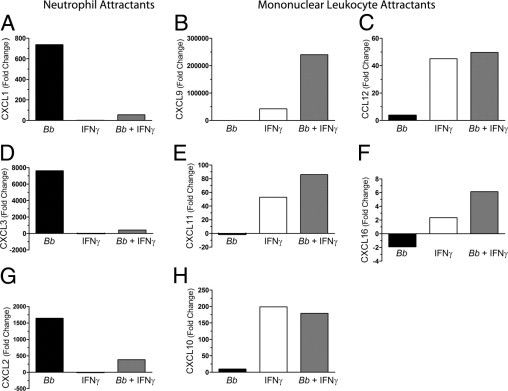
Incubation of murine macrophages with B. burgdorferi (Bb) and IFNγ alters the expression of chemokine genes. Murine macrophages were incubated with medium alone, B. burgdorferi, IFNγ, or B. burgdorferi and IFNγ concurrently for 8 hours. RNA was extracted from cells and analyzed using real-time RT-PCR for the following transcripts: CXCL1 (A), CXCL9 (B), CCL12 (C), CXCL3 (D), CXCL11 (E), CXCL16 (F), CXCL2 (G), and CXCL10 (H). CXCL1, CXCL2, and CXCL3 attract neutrophils; the remainder attract mononuclear leukocytes. Fold changes are relative to the level of transcripts in macrophages incubated with medium alone.
As measured by microarray analysis, transcripts for CXCL2 and CXCL10 were synergistically up-regulated by B. burgdorferi and IFNγ in HUVECs15 but not in murine macrophages. Nevertheless, we examined the transcription of these chemokine genes in the macrophages using real-time RT-PCR. When macrophages were activated by B. burgdorferi alone, expression of CXCL2, a chemokine that recruits neutrophils, was increased. As was the case for other neutrophil chemoattractants, a decrease in expression was observed with the further addition of IFNγ (compare Figure 6G with Figure 6, A and D). The expression of CXCL10, an attractant for mononuclear cells, was enhanced by stimulation of macrophages with IFNγ alone, but incubation with B. burgdorferi and IFNγ concurrently did not further increase the level of transcripts (Figure 6H).
The secretion of CXCL1, CXCL2, CXCL9, and CXCL10 from stimulated macrophages was measured by ELISA. In agreement with the RT-PCR results, the production of CXCL1 and CXCL2 was decreased when macrophages were incubated concurrently with IFNγ and B. burgdorferi compared with B. burgdorferi alone (Figure 7, A and C). In contrast, amounts of CXCL9 were synergistically increased by the two stimuli combined (Figure 7B). Although transcripts for CXCL10 in macrophages incubated with IFNγ were not further increased when B. burgdorferi was added (Figure 6H), the secretion of this chemokine was synergistically elevated by the two stimuli together (Figure 7D). Within macrophages activated by B. burgdorferi, IFNγ thus promotes the production of chemokines that attract mononuclear leukocytes but suppresses that of chemokines that recruit neutrophils.
Figure 7.
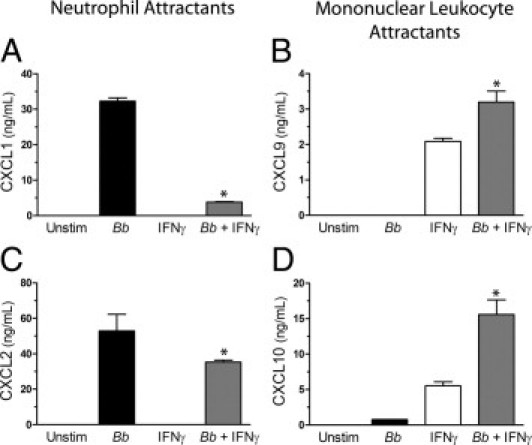
IFNγ alters the pattern of chemokines secreted by B. burgdorferi (Bb)–stimulated macrophages. Murine macrophages were incubated with medium alone, B. burgdorferi, IFNγ, or B. burgdorferi and IFNγ concurrently for 24 hours. Conditioned media were assessed by ELISA for the concentrations of CXCL1 (A), CXCL9 (B), CXCL2 (C), and CXCL10 (D). CXCL1 and CXCL2 attract neutrophils, whereas CXCL9 and CXCL10 attract mononuclear leukocytes. Data represent the mean ± SD of triplicate samples. This experiment was repeated two more times for CXCL1, CXCL2, and CXCL10 with similar results. The experiment for CXCL9 was repeated once more, yielding similar results. *P < 0.001 versus B. burgdorferi or IFNγ alone.
To further investigate the mechanisms driving the selective recruitment of leukocytes in murine Lyme carditis, the secretion of chemokines by endothelial cells isolated from hearts of C57BL/6 mice was quantified by ELISA. B. burgdorferi alone stimulated MCECs to produce CXCL1 and CXCL2, which attract neutrophils. However, IFNγ had no effect on secretion of these chemokines, either alone or combined with the spirochetes (Figure 8, A and C). Conversely, the production of two chemokines that attract T cells, CXCL9 and CXCL10, was induced by IFN-γ but not B. burgdorferi. Furthermore, secretion of CXCL9 and CXCL10 was synergistically enhanced when MCECs were costimulated with B. burgdorferi and IFNγ (Figure 8, B and D). In summary, murine macrophages (Figure 7), MCECs (Figure 8), and HUVECs15 all displayed synergistic up-regulation of CXCL9 and CXCL10 when challenged simultaneously with IFNγ and B. burgdorferi. Although spirochetes stimulated all three types of cell to secrete chemokines that attract neutrophils, IFNγ suppressed this secretion in only the macrophages.
Figure 8.
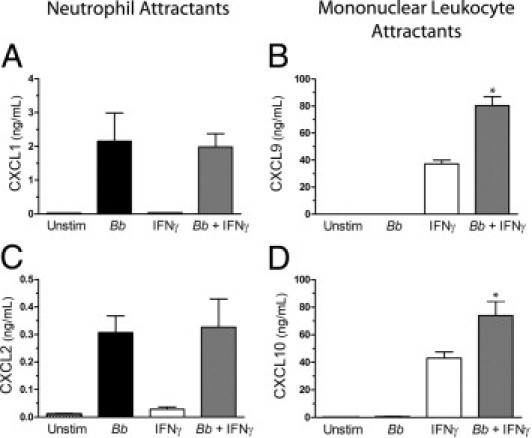
IFNγ alters the pattern of chemokines secreted by B. burgdorferi (Bb)–stimulated MCECs. The MCECs were incubated with medium alone, B. burgdorferi, IFNγ, or B. burgdorferi and IFNγ concurrently for 24 hours. Conditioned media were assessed by ELISA for the concentrations of CXCL1 (A), CXCL9 (B), CXCL2 (C), and CXCL10 (D). CXCL1 and CXCL2 attract neutrophils, whereas CXCL9 and CXCL10 attract mononuclear leukocytes. Data represent the mean ± SD of triplicate samples. This experiment was repeated two more times for CXCL1, CXCL2, and CXCL10. Results were similar, with the exception of one assessment for CXCL1, in which the secretion was slightly, but significantly, increased by the addition of IFNγ to B. burgdorferi–stimulated MCECs. Assessment of CXCL9 was repeated once more, with similar results. *P < 0.001 versus B. burgdorferi or IFNγ alone.
Discussion
A previous study showed that IFNγ synergizes with B. burgdorferi to promote secretion of seven chemokines from human endothelial cells. Six of the seven chemokines attract leukocytes typical of chronic inflammation. Moreover, IFNγ increases the migration of T cells, while decreasing that of neutrophils, across B. burgdorferi–activated HUVECs.15 These in vitro observations suggest that IFNγ may influence recruitment of leukocytes to tissues infected with B. burgdorferi. The current study used the murine model of Lyme disease to address this question in vivo. Our results indicate that IFNγ influences the composition of leukocytic infiltrates in hearts of infected mice because lesions in IFNγ-deficient animals have more neutrophils and fewer T cells than those in wild-type mice. Whether this altered composition affects the time needed for clearance of the spirochetes or resolution of carditis is an important question for future investigation.
The presence of IFNγ positively correlates with the severity of Lyme disease in humans.7 However, studies of the role of IFNγ in murine Lyme arthritis have reached conflicting conclusions. Treatment of infected mice with anti-IFNγ antibodies reduces joint swelling.32,33 In addition, neutralization of IL-4, which opposes the function of IFNγ, exacerbates arthritis.32 These data support the notion that IFNγ contributes to the evolution of inflammation. On the other hand, IFNγ- and IFNγ receptor–deficient mice develop the same severity of Lyme arthritis as wild-type animals, suggesting that IFNγ is not required for the progression of disease.34,35
The expression of IFNγ is increased in the hearts of mice infected with B. burgdorferi,36,37 but examination of its role in murine Lyme carditis has been largely through indirect means. Stat1-deficient mice, which have defects in signaling pathways induced by IFNα, IFNβ, and IFNγ, show an increase in the extent of carditis, but not arthritis, at 21 days after infection.22 This observation suggests that IFNγ may play a protective role in murine Lyme carditis. In apparent contradiction, mice deficient in IL-4, which produce abnormally high levels of IFNγ, develop more severe carditis after infection with B. burgdorferi, implying that IFNγ may exacerbate carditis.38 In both instances, spirochetal loads are similar in hearts of wild-type and knockout animals, even though the severity of carditis differs. To directly examine the effects of IFNγ on the progression of murine Lyme carditis, we infected mice that were incapable of producing IFNγ with B. burgdorferi. Hearts were scored for overall severity of carditis, and no difference between wild-type and IFNγ-deficient strains was measured at any time. Similar to the previous reports,22,38 spirochetal burdens in hearts of the two strains of mice were the same. One previous publication also directly examined the role of IFNγ in Lyme carditis using IFNγ receptor–deficient mice. In disagreement with our results, these researchers37 observed that loss of the receptor produced a slight increase in severity and a greater spirochetal load in the heart at 14 days after infection. The differences in strain of mouse, strain of B. burgdorferi, and route of inoculation might account for the disparity.
Although IFNγ did not alter the severity of murine carditis in our study, we speculated that it might still be influencing the types of leukocytes that infiltrate cardiac lesions. Indeed, at 25 days after infection, cardiac lesions of IFNγ-deficient mice contained more neutrophils and fewer T cells than those of their wild-type counterparts. It is likely that the larger population of neutrophils observed in IFNγ-deficient mice is a consequence of enhanced recruitment of the leukocytes because they are short-lived cells in tissues.39 Consistent with other publications,20–22 macrophages were the predominant leukocyte observed in both strains of infected mice. Unlike T cells and neutrophils, F4/80+ macrophages were found in similar numbers in infected IFNγ-deficient and wild-type mice at both time points examined. F4/80 is not highly expressed on all macrophages,40 so it is possible that some of these leukocytes were not counted in our analysis. Strikingly, there was an increase in the amount of F4/80+ cells that also expressed Gr-1 in the lesions of IFNγ-deficient mice at both 14 and 25 days. IFNγ is required for macrophages to mature into inflammatory M1 macrophages. This subset of mature macrophages is a prominent secretor of pro-inflammatory cytokines.27 The Gr-1+ F4/80+ leukocytes that were observed are potentially myeloid-derived suppressor cells, an immature population that can dampen inflammation and promote healing of damaged cardiac tissue.27 Gr-1+ F4/80+ cells were not prevalent in infected wild-type mice in our study. Therefore, they are unlikely to play a major role in the progression of Lyme disease.
We found that IFNγ and B. burgdorferi synergistically induced macrophages, like endothelium,15 to produce chemokines that attract cells typical of chronic inflammation, including CXCL9, CXCL10, and CXCL11. Notably, both CXCL9 and CXCL10 are observed in the synovial fluid of patients with antibiotic-refractory Lyme disease41 and septic arthritis,42,43 whereas CXCL9 is prevalent in autoimmune arthritis.44 In contrast, the addition of IFNγ to murine macrophages stimulated by B. burgdorferi synergistically decreased expression of several attractants for neutrophils, including CXCL1 and CXCL2. CXC chemokine receptor 2, which is a receptor for both CXCL1 and CXCL2, is required for the migration of neutrophils into joints during murine Lyme arthritis.45 In addition, CXCL1 regulates the infiltration of neutrophils into lesions of murine Lyme carditis.46 CCL2 is a major attractant for mononuclear leukocytes in many disease models.47 However, its expression, as measured by microarray analysis, was not synergistically altered by spirochetes and IFN-γ in HUVECs15 or murine macrophages. In accordance, secretion of CCL2 was not synergistically enhanced by the two stimuli in either murine macrophages or MCECs (data not shown).
Our current and published15 in vitro observations support the idea that chemokines derived from macrophages and endothelial cells shape the composition of leukocytic infiltrates in Lyme carditis. In hearts of infected mice, IFNγ promoted the accumulation of T lymphocytes. Likewise, IFNγ enhanced the production of attractants for T cells by endothelial cells15 and macrophages exposed to B. burgdorferi. IFNγ also suppressed recruitment of neutrophils into lesions of Lyme carditis, and it reduced production of chemoattractants for neutrophils by spirochete-stimulated macrophages. Nevertheless, IFN-γ had no effect on secretion of the neutrophil attractants CXCL1 and CXCL2 by MCECs activated with B. burgdorferi. Similarly, IFNγ does not reduce secretion of CXCL8, a major neutrophil attractant in humans, by HUVECs activated with B. burgdorferi. Moreover, levels of transcripts for CXCL2 in HUVECs are synergistically induced by the two stimuli.15 Collectively, these results raise the possibility that chemokines secreted by macrophages play a more important role in recruitment of neutrophils during Lyme carditis than those produced by endothelial cells. Furthermore, IFNγ dampens migration of neutrophils across spirochete-stimulated HUVECs in vitro, despite the fact that expression of two chemokines important for their recruitment is not decreased.15 Perhaps, then, alterations in other factors necessary for extravasation, such as endothelial cell adhesion molecules, also are involved in the mechanism by which IFNγ down-modulates accumulation of neutrophils in murine Lyme carditis.
B. burgdorferi elicits inflammatory responses that mirror other forms of chronic inflammation. Arthritic lesions in Lyme disease and rheumatoid arthritis have similar pathological features, and both contain T cells, which are primarily helper T cells, distributed throughout the affected tissue.48 As with B. burgdorferi, IFNγ synergizes with ligands of Toll-like receptors, such as peptidoglycan, double-stranded RNA, flagellin, and lipopolysaccharide, to increase the secretion of CXCL9 and CXCL11 from fibroblasts42 and CXCL9, CXCL10, and CXCL11 from human microvascular endothelial cells.44 In our study, IFNγ was required for an enrichment of T cells and reduction of neutrophils as Lyme carditis progressed, and it controls a shift in the profile of infiltrating leukocytes in other inflammatory conditions as well. In cardiac allografts in IFNγ-deficient mice, T cells and other mononuclear leukocytes that normally accumulate are replaced by many neutrophils.49 In addition, the absence of IFNγ results in more neutrophils infiltrating into the central nervous system of mice during experimental autoimmune encephalomyelitis.50 As we observed in murine Lyme carditis, IFNγ is altering the composition of leukocytic populations in these instances by promoting the infiltration of mononuclear cells and suppressing that of neutrophils. These results support the premise that IFNγ is acting as a molecular switch in the progression of acute to chronic inflammation in Lyme disease and other inflammatory conditions.
Acknowledgments
We thank John Schwedes (Stony Brook University DNA Microarray Core Facility) and Lance Palmer for performing and analyzing murine microarrays, Stephanie Burke (Pathology Translational Research Laboratory, Stony Brook University Medical Center) for her indispensable knowledge of histological techniques, Silvia Leonor (Stony Brook University Hospital Non-Transplant Tissue Bank) for processing sample blocks and slides, Galina Romanov for supplying macrophages, and Indralatha Jayatilaka for excellent laboratory support.
Footnotes
Supported by grants from the Long Island Chapter of the Arthritis Foundation and the NIH (PO1 AI055621).
S.J.H. and S.C.M. contributed equally to this work.
References
- 1.Benach J.L., Bosler E.M., Hanrahan J.P., Coleman J.L., Habicht G.S., Bast T.F., Cameron D.J., Ziegler J.L., Barbour A.G., Burgdorfer W., Edelman R., Kaslow R.A. Spirochetes isolated from the blood of two patients with Lyme disease. N Engl J Med. 1983;308:740–742. doi: 10.1056/NEJM198303313081302. [DOI] [PubMed] [Google Scholar]
- 2.Duray P.H., Steere A.C. Clinical pathologic correlations of Lyme disease by stage. Ann N Y Acad Sci. 1988;539:65–79. doi: 10.1111/j.1749-6632.1988.tb31839.x. [DOI] [PubMed] [Google Scholar]
- 3.Duray P.H. Histopathology of clinical phases of human Lyme disease. Rheum Dis Clin North Am. 1989;15:691–710. [PubMed] [Google Scholar]
- 4.Steere A.C., Glickstein L. Elucidation of Lyme arthritis. Nat Rev Immunol. 2004;4:143–152. doi: 10.1038/nri1267. [DOI] [PubMed] [Google Scholar]
- 5.Duray P.H. The surgical pathology of human Lyme disease: an enlarging picture. Am J Surg Pathol. 1987;11(Suppl 1):47–60. doi: 10.1097/00000478-198700111-00005. [DOI] [PubMed] [Google Scholar]
- 6.Gross D.M., Steere A.C., Huber B.T. T helper 1 response is dominant and localized to the synovial fluid in patients with Lyme arthritis. J Immunol. 1998;160:1022–1028. [PubMed] [Google Scholar]
- 7.Salazar J.C., Pope C.D., Sellati T.J., Feder H.M., Jr, Kiely T.G., Dardick K.R., Buckman R.L., Moore M.W., Caimano M.J., Pope J.G., Krause P.J., Radolf J.D. Coevolution of markers of innate and adaptive immunity in skin and peripheral blood of patients with erythema migrans. J Immunol. 2003;171:2660–2670. doi: 10.4049/jimmunol.171.5.2660. [DOI] [PubMed] [Google Scholar]
- 8.Krishnaswamy G., Kelley J., Yerra L., Smith J.K., Chi D.S. Human endothelium as a source of multifunctional cytokines: molecular regulation and possible role in human disease. J Interferon Cytokine Res. 1999;19:91–104. doi: 10.1089/107999099314234. [DOI] [PubMed] [Google Scholar]
- 9.Sellati T.J., Burns M.J., Ficazzola M.A., Furie M.B. Borrelia burgdorferi upregulates expression of adhesion molecules on endothelial cells and promotes transendothelial migration of neutrophils in vitro. Infect Immun. 1995;63:4439–4447. doi: 10.1128/iai.63.11.4439-4447.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burns M.J., Sellati T.J., Teng E.I., Furie M.B. Production of interleukin-8 (IL-8) by cultured endothelial cells in response to Borrelia burgdorferi occurs independently of secreted IL-1 and tumor necrosis factor alpha and is required for subsequent transendothelial migration of neutrophils. Infect Immun. 1997;65:1217–1222. doi: 10.1128/iai.65.4.1217-1222.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burns M.J., Furie M.B. Borrelia burgdorferi and interleukin-1 promote the transendothelial migration of monocytes in vitro by different mechanisms. Infect Immun. 1998;66:4875–4883. doi: 10.1128/iai.66.10.4875-4883.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang A.J., Furie M.B., Nicholson S.C., Fischbarg J., Liebovitch L.S., Silverstein S.C. Effects of human neutrophil chemotaxis across human endothelial cell monolayers on the permeability of these monolayers to ions and macromolecules. J Cell Physiol. 1988;135:355–366. doi: 10.1002/jcp.1041350302. [DOI] [PubMed] [Google Scholar]
- 13.Gergel E.I., Furie M.B. Activation of endothelium by Borrelia burgdorferi in vitro enhances transmigration of specific subsets of T lymphocytes. Infect Immun. 2001;69:2190–2197. doi: 10.1128/IAI.69.4.2190-2197.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gergel E.I., Furie M.B. Populations of human T lymphocytes that traverse the vascular endothelium stimulated by Borrelia burgdorferi are enriched with cells that secrete gamma interferon. Infect Immun. 2004;72:1530–1536. doi: 10.1128/IAI.72.3.1530-1536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dame T.M., Orenzoff B.L., Palmer L.E., Furie M.B. IFN-gamma alters the response of Borrelia burgdorferi-activated endothelium to favor chronic inflammation. J Immunol. 2007;178:1172–1179. doi: 10.4049/jimmunol.178.2.1172. [DOI] [PubMed] [Google Scholar]
- 16.Barthold S.W., Beck D.S., Hansen G.M., Terwilliger G.A., Moody K.D. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis. 1990;162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 17.Barthold S.W., Persing D.H., Armstrong A.L., Peeples R.A. Kinetics of Borrelia burgdorferi dissemination and evolution of disease after intradermal inoculation of mice. Am J Pathol. 1991;139:263–273. [PMC free article] [PubMed] [Google Scholar]
- 18.Barthold S.W., de Souza M.S., Janotka J.L., Smith A.L., Persing D.H. Chronic Lyme borreliosis in the laboratory mouse. Am J Pathol. 1993;143:959–971. [PMC free article] [PubMed] [Google Scholar]
- 19.Armstrong A.L., Barthold S.W., Persing D.H., Beck D.S. Carditis in Lyme disease susceptible and resistant strains of laboratory mice infected with Borrelia burgdorferi. Am J Trop Med Hyg. 1992;47:249–258. doi: 10.4269/ajtmh.1992.47.249. [DOI] [PubMed] [Google Scholar]
- 20.Ruderman E.M., Kerr J.S., Telford S.R., III, Spielman A., Glimcher L.H., Gravallese E.M. Early murine Lyme carditis has a macrophage predominance and is independent of major histocompatibility complex class II-CD4+ T cell interactions. J Infect Dis. 1995;171:362–370. doi: 10.1093/infdis/171.2.362. [DOI] [PubMed] [Google Scholar]
- 21.Montgomery R.R., Booth C.J., Wang X., Blaho V.A., Malawista S.E., Brown C.R. Recruitment of macrophages and polymorphonuclear leukocytes in Lyme carditis. Infect Immun. 2007;75:613–620. doi: 10.1128/IAI.00685-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown C.R., Blaho V.A., Fritsche K.L., Loiacono C.M. Stat1 deficiency exacerbates carditis but not arthritis during experimental Lyme borreliosis. J Interferon Cytokine Res. 2006;26:390–399. doi: 10.1089/jir.2006.26.390. [DOI] [PubMed] [Google Scholar]
- 23.Stanley E.R., Heard P.M. Factors regulating macrophage production and growth: purification and some properties of the colony stimulating factor from medium conditioned by mouse L cells. J Biol Chem. 1977;252:4305–4312. [PubMed] [Google Scholar]
- 24.Celada A., Gray P.W., Rinderknecht E., Schreiber R.D. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J Exp Med. 1984;160:55–74. doi: 10.1084/jem.160.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forestal C.A., Gil H., Monfett M., Noah C.E., Platz G.J., Thanassi D.G., Benach J.L., Furie M.B. A conserved and immunodominant lipoprotein of Francisella tularensis is proinflammatory but not essential for virulence. Microb Pathog. 2008;44:512–523. doi: 10.1016/j.micpath.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim Y.C., Luscinskas F.W. Isolation and culture of murine heart and lung endothelial cells for in vitro model systems. Methods Mol Biol. 2006;341:141–154. doi: 10.1385/1-59745-113-4:141. [DOI] [PubMed] [Google Scholar]
- 27.Fairweather D., Cihakova D. Alternatively activated macrophages in infection and autoimmunity. J Autoimmun. 2009;33:222–230. doi: 10.1016/j.jaut.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steere A.C., Coburn J., Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radolf J.D., Arndt L.L., Akins D.R., Curetty L.L., Levi M.E., Shen Y., Davis L.S., Norgard M.V. Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides activate monocytes/macrophages. J Immunol. 1995;154:2866–2877. [PubMed] [Google Scholar]
- 30.Ma Y., Weis J.J. Borrelia burgdorferi outer surface lipoproteins OspA and OspB possess B-cell mitogenic and cytokine-stimulatory properties. Infect Immun. 1993;61:3843–3853. doi: 10.1128/iai.61.9.3843-3853.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Y., Zhou Y., Iribarren P., Wang J. Chemokines and chemokine receptors: their manifold roles in homeostasis and disease. Cell Mol Immunol. 2004;1:95–104. [PubMed] [Google Scholar]
- 32.Keane-Myers A., Nickell S.P. Role of IL-4 and IFN-gamma in modulation of immunity to Borrelia burgdorferi in mice. J Immunol. 1995;155:2020–2028. [PubMed] [Google Scholar]
- 33.Matyniak J.E., Reiner S.L. T helper phenotype and genetic susceptibility in experimental Lyme disease. J Exp Med. 1995;181:1251–1254. doi: 10.1084/jem.181.3.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown C.R., Reiner S.L. Experimental Lyme arthritis in the absence of interleukin-4 or gamma interferon. Infect Immun. 1999;67:3329–3333. doi: 10.1128/iai.67.7.3329-3333.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glickstein L., Edelstein M., Dong J.Z. Gamma interferon is not required for arthritis resistance in the murine Lyme disease model. Infect Immun. 2001;69:3737–3743. doi: 10.1128/IAI.69.6.3737-3743.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyle M.K., Telford S.R., III, Criscione L., Lin S.R., Spielman A., Gravallese E.M. Cytokines in murine Lyme carditis: th1 cytokine expression follows expression of proinflammatory cytokines in a susceptible mouse strain. J Infect Dis. 1998;177:242–246. doi: 10.1086/517364. [DOI] [PubMed] [Google Scholar]
- 37.Olson C.M., Jr, Bates T.C., Izadi H., Radolf J.D., Huber S.A., Boyson J.E., Anguita J. Local production of IFN-gamma by invariant NKT cells modulates acute Lyme carditis. J Immunol. 2009;182:3728–3734. doi: 10.4049/jimmunol.0804111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satoskar A.R., Elizondo J., Monteforte G.M., Stamm L.M., Bluethmann H., Katavolos P., Telford S.R., III Interleukin-4-deficient BALB/c mice develop an enhanced Th1-like response but control cardiac inflammation following Borrelia burgdorferi infection. FEMS Microbiol Lett. 2000;183:319–325. doi: 10.1111/j.1574-6968.2000.tb08978.x. [DOI] [PubMed] [Google Scholar]
- 39.Summers C., Rankin S.M., Condliffe A.M., Singh N., Peters A.M., Chilvers E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010;31:318–324. doi: 10.1016/j.it.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirsch S., Gordon S. The use and limitation of monoclonal antibodies against mononuclear phagocytes. Immunobiology. 1982;161:298–307. doi: 10.1016/S0171-2985(82)80086-7. [DOI] [PubMed] [Google Scholar]
- 41.Shin J.J., Glickstein L.J., Steere A.C. High levels of inflammatory chemokines and cytokines in joint fluid and synovial tissue throughout the course of antibiotic-refractory Lyme arthritis. Arthritis Rheum. 2007;56:1325–1335. doi: 10.1002/art.22441. [DOI] [PubMed] [Google Scholar]
- 42.Proost P., Verpoest S., Van de Borne K., Schutyser E., Struyf S., Put W., Ronsse I., Grillet B., Opdenakker G., Van D.J. Synergistic induction of CXCL9 and CXCL11 by Toll-like receptor ligands and interferon-gamma in fibroblasts correlates with elevated levels of CXCR3 ligands in septic arthritis synovial fluids. J Leukoc Biol. 2004;75:777–784. doi: 10.1189/jlb.1003524. [DOI] [PubMed] [Google Scholar]
- 43.Proost P., Vynckier A.K., Mahieu F., Put W., Grillet B., Struyf S., Wuyts A., Opdenakker G., Van D.J. Microbial Toll-like receptor ligands differentially regulate CXCL10/IP-10 expression in fibroblasts and mononuclear leukocytes in synergy with IFN-gamma and provide a mechanism for enhanced synovial chemokine levels in septic arthritis. Eur J Immunol. 2003;33:3146–3153. doi: 10.1002/eji.200324136. [DOI] [PubMed] [Google Scholar]
- 44.Loos T., Dekeyzer L., Struyf S., Schutyser E., Gijsbers K., Gouwy M., Fraeyman A., Put W., Ronsse I., Grillet B., Opdenakker G., Van D.J., Proost P. TLR ligands and cytokines induce CXCR3 ligands in endothelial cells: enhanced CXCL9 in autoimmune arthritis. Lab Invest. 2006;86:902–916. doi: 10.1038/labinvest.3700453. [DOI] [PubMed] [Google Scholar]
- 45.Brown C.R., Blaho V.A., Loiacono C.M. Susceptibility to experimental Lyme arthritis correlates with KC and monocyte chemoattractant protein-1 production in joints and requires neutrophil recruitment via CXCR2. J Immunol. 2003;171:893–901. doi: 10.4049/jimmunol.171.2.893. [DOI] [PubMed] [Google Scholar]
- 46.Ritzman A.M., Hughes-Hanks J.M., Blaho V.A., Wax L.E., Mitchell W.J., Brown C.R. The chemokine receptor CXCR2 ligand KC (CXCL1) mediates neutrophil recruitment and is critical for development of experimental Lyme arthritis and carditis. Infect Immun. 2010;78:4593–4600. doi: 10.1128/IAI.00798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yadav A., Saini V., Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clin Chim Acta. 2010;411:1570–1579. doi: 10.1016/j.cca.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 48.Steere A.C., Duray P.H., Butcher E.C. Spirochetal antigens and lymphoid cell surface markers in Lyme synovitis: comparison with rheumatoid synovium and tonsillar lymphoid tissue. Arthritis Rheum. 1988;31:487–495. doi: 10.1002/art.1780310405. [DOI] [PubMed] [Google Scholar]
- 49.Miura M., El-Sawy T., Fairchild R.L. Neutrophils mediate parenchymal tissue necrosis and accelerate the rejection of complete major histocompatibility complex-disparate cardiac allografts in the absence of interferon-gamma. Am J Pathol. 2003;162:509–519. doi: 10.1016/s0002-9440(10)63845-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zehntner S.P., Brickman C., Bourbonniere L., Remington L., Caruso M., Owens T. Neutrophils that infiltrate the central nervous system regulate T cell responses. J Immunol. 2005;174:5124–5131. doi: 10.4049/jimmunol.174.8.5124. [DOI] [PubMed] [Google Scholar]