

Abstract
The roles of the c-Jun N-terminal kinases (JNKs) in inflammatory arthritis have been investigated; however, the roles of each isotype (ie, JNK1 and JNK2) in rheumatoid arthritis and conclusions about whether inhibition of one or both is necessary for amelioration of disease are unclear. By using JNK1- or JNK2-deficient mice in the collagen-induced arthritis and the KRN T-cell receptor transgenic mouse on C57BL/6 nonobese diabetic (K/BxN) serum transfer arthritis models, we demonstrate that JNK1 deficiency results in protection from arthritis, as judged by clinical score and histological evaluation in both models of inflammatory arthritis. In contrast, abrogation of JNK2 exacerbates disease. In collagen-induced arthritis, the distinct roles of the JNK isotypes can, at least in part, be explained by altered regulation of CD86 expression in JNK1- or JNK2-deficient macrophages in response to microbial products, thereby affecting T-cell–mediated immunity. The protection from K/BxN serum–induced arthritis in Jnk1−/− mice can also be explained by inept macrophage function because adoptive transfer of wild-type macrophages to Jnk1−/− mice restored disease susceptibility. Thus, our results provide a possible explanation for the modest therapeutic effects of broad JNK inhibitors and suggest that future therapies should selectively target the JNK1 isoform.
Rheumatoid arthritis (RA) is a chronic autoimmune disease that affects multiple joints. RA is characterized by infiltration of immune cells, inflammation, pannus formation, and joint destruction. It is generally accepted that pro-inflammatory cytokines play an important role in the pathogenesis of RA.1 However, despite intense investigation, the cause of RA remains unknown.
The mitogen-activated protein kinases known as the c-Jun N-terminal kinases (JNKs) participate in RA,2 and their role has been investigated in several in vivo studies.3–5 The inhibition of JNK by using SP600125, which prevents both JNK1 and JNK2 activity, in a rat model of adjuvant arthritis resulted in a modest decrease in paw swelling and inflammation but showed a significant inhibition of radiographical damage.3 The influence of JNK2 was investigated in passive collagen-induced arthritis (CIA) using JNK2-deficient mice.4 In this study, the severity of arthritis was slightly increased in the JNK2-deficient animals, whereas the degree of synovial inflammation was comparable to that in wild-type (wt) mice. The influence of JNK1 was investigated in yet another model of autoimmune inflammation, tumor necrosis factor (TNF)-α–mediated inflammatory joint disease.5 Results from this study suggested that JNK1 is not required for TNF-α–mediated arthritis. However, none of these studies has compared JNK1- with JNK2-deficient animals in the same model of inflammatory arthritis. Thus, the conclusion from these studies about whether selective inhibition of one isoform or nonselective inhibition of JNK1 and JNK2 is required for major blockade of inflammatory arthritis calls for speculation. Recently, however, Guma and coworkers6,7 have suggested that JNK1, but not JNK2, is critical for the induction of antibody-induced arthritis (AIA) and serum-induced arthritis; they speculate that this is due to diminished macrophage migration in the AIA model and mast cell degranulation and cytokine production in the serum-induced arthritis model.
In the present study, we chose two well-defined mouse models of inflammatory arthritis, CIA and the KRN T-cell receptor transgenic mouse on C57BL/6 × nonobese diabetic (K/BxN) serum transfer model, to directly compare the importance of JNK1 with JNK2 during the autoimmune initiation phase and the T-cell–independent effector phase of experimental arthritis, respectively.
CIA is an autoimmune disease of the joints requiring both T- and B-cell immunity to type II collagen and macrophages for disease manifestation.8 Although T cells play a prominent role in classic CIA, the primary mechanism for the immunopathogenesis seems to be autoantibodies to type II collagen that subsequently bind to the joint cartilage and activate the complement system.9 This model was originally described for genetically susceptible strains bearing the major histocompatibility complex (MHC) haplotypes H-2q or H-2r, respectively (ie, DBA/1, B10.Q and B10.RIII). Recently, an altered protocol for the development of CIA in H-2b–bearing C57BL/6 mice was described.10 This disease resembles classic CIA both clinically and histologically and requires CD4+ T and B cells for development. Interestingly, CIA in B6 mice is milder but more chronic, with a more pronounced and persistent T-cell response, compared with classic CIA in DBA/1 mice.11 The K/BxN model of serum-induced arthritis is independent of both B and T cells, external antigen, and adjuvant for disease development.12 Thus, this serum transfer system allows us to focus on the inflammatory effector phase of disease, without the complicating influences of the autoimmune initiation phase. By using these models, we provide evidence showing that JNK1 is required for the initiation and effector phases of inflammatory arthritis, whereas deficiency in JNK2 exacerbates the clinical signs of the inflammatory response.
Materials and Methods
Mice
All mice were bred and maintained in the animal facility of the Panum Institute, University of Copenhagen, Copenhagen, Denmark. Jnk1−/− and Jnk2−/− mice were previously described13,14 and were backcrossed to C57BL/6 mice for at least eight generations. KRN T-cell receptor transgenic mice have also been described.15 These studies have been reviewed and approved by the local ethical committee.
CIA Model
The initiation of CIA in C57BL/6 mice has been previously described.10 Briefly, Complete Freund's Adjuvant (CFA) was prepared in our laboratory by dissolving 100 mg of heat-killed tuberculosis (H37Ra; Difco Laboratories, Detroit, MI) in 20 mL IFA (incomplete Freund's adjuvant) (Sigma, St Louis, MO). A 2 mg/mL chick type II collagen preparation (Sigma) was dissolved in 10 mmol/L acetic acid by gentle agitation overnight at 4°C and then emulsified with an equal volume of CFA. Mice were injected intradermally into the base of the tail with a total volume of 100-μL emulsion containing 100 μg collagen type II (CII) and 250 μg of Mycobacterium tuberculosis. Booster immunizations were given 21 days later. To quantify the intensity of CIA, each animal was assessed for redness and swelling of limbs and assigned a clinical score three times per week. The score was composed of the sum of involvement of each limb. The scale for each limb was as follows: 0, no clinical symptoms; 1, slight swelling; 2, extensive swelling; and 3, joint distortion and/or rigidity. The maximum score per mouse was 12.
Histological Assessment of CIA
When sacrificed, the limbs of the mice were removed, fixed in 10% formalin, and decalcified in EDTA. Paraffin-embedded sections were stained with Mayer's H&E.16 The severity of arthritis was blindly evaluated using three parameters: infiltration of monomorphonuclear and polymorphonuclear cells, hyperplasia of the synovium, and bone destruction. Each parameter was scored on a scale from 0 to 3, as follows: 0, absent; 1, weak; 2, moderate; and 3, severe.
Serum-Induced Arthritis
Serum samples were collected from K/BxN mice, aged 1 to 3 months, and pooled. wt, Jnk1−/−, and Jnk2−/− male mice were injected i.p. with 200 μL of serum on days 0 and 2.17 The development of arthritis was assessed by caliper (Mitotoyo, Tokyo, Japan), with ankle thickening defined as the difference in the ankle thickness from the day 0 measure. A clinical index was evaluated for each paw, according to the following criteria: 0, normal; 0.5, mild swelling of the paws or a few digits affected; and 1.0, clear inflammation in the ankle or joint. The score of the mice was evaluated blindly.
Generation of BMMs
The bone marrow macrophages (BMMs) obtained from wt Jnk1−/− and Jnk2−/− mice were cultured for 6 days in Dulbecco's modified Eagle's medium (Sigma), supplemented with 25% (v/v) fetal bovine serum and 25% (v/v) L929 cell-conditioned media. The cells were synchronized by culture with 10% fetal bovine serum for 24 hours.
Adoptive Transfer of BMMs
The BMMs were isolated from wt donor mice and differentiated as previously described. On days -1, 2, and 4, 1 × 106 macrophages were injected into the tail vein of Jnk1−/− recipient mice. wt, Jnk1−/−, and Jnk1−/− BMM recipient male mice were injected i.p. with 200 μL of serum on days 0 and 2.
Enzyme-Linked Immunosorbent Assay for CII-Specific Antibodies
Serum was isolated from individual animals and stored at −80°C. The concentration of total anti-CII-specific IgG was determined according to the manufacturer's instructions (MD Biosciences GmbH, Zürich, Switzerland). For CII-specific IgG1 and IgG2a determinations, 2 μg/mL of CII was diluted in Tris-NaCl (pH 7.4) and used for overnight coating of enzyme-linked immunosorbent assay plates (Nunc, Roskilde, Denmark). Serum samples were diluted 1:10,000 and incubated for 1 hour at room temperature. After washing, the plates were incubated for 1 hour at room temperature with horseradish peroxidase–conjugated IgG1 (Bethyl Laboratories, Inc., Montgomery, TX), diluted 1:50,000, or horseradish peroxidase–conjugated goat anti-mouse IgG2a (Bethyl Laboratories, Inc.), diluted 1:30,000. The reaction was developed with TMB for 1 hour and discontinued with H2SO4. The absorbance was measured at 450 nm in an enzyme-linked immunosorbent assay reader.
Quantitative PCR
Total RNA was isolated from macrophages as previously described.18 Quantitative RT-PCR analysis was performed using the SYBR Green PCR Master Mix kit, according to the manufacturer's recommendations (Stratagene, AH Diagnostics, Aarhus, Denmark) using the following primers: TNF-α, 5′-GCACAGAAAGCATGACCCG-3′ (forward) and 5′-GCCCCCCATCTTTTGGG-3′ (reverse) (annealing temperature, 58°C); IL-1β, 5′-CAACCAACAAGTGATATTCTCCATG-3′ (forward) and 5′-GATCCACACTCTCCAGCTGCA-3′ (reverse) (annealing temperature, 61°C); and IL-10, 5′-GGTTGCCAAGCCTTATCGGA-3′ (forward) and 5′-ACCTGCTCCACTGCCTTGCT-3′ (reverse) (annealing temperature, 60°C). All primers were from DNA Technology (Risskov, Denmark). The PCR was initiated with 95°C for 10 minutes, and the PCR cycling conditions were as follows: 95°C for 30 seconds, the previously mentioned specific annealing temperature for 1 minute, and 72°C for 30 seconds for 40 cycles. The quantitative PCR values were normalized using a specific cDNA standard curve obtained using known amounts of cDNA. TNF-α, IL-1β, and IL-10 were then normalized to glyceraldehyde-3-phophate dehydrogenase.
Flow Cytometry
Cells were stained with fluorochrome-conjugated primary antibodies (Abs) for 30 minutes at 4°C in PBS containing 0.5% bovine serum albumin, washed twice, acquired on an FACSCalibur flow cytometer (BD Biosciences, Broendby, Denmark) and analyzed with FlowJo Software (Tree Star, OR), or acquired on a BD LSR II flow cytometer (BD Biosciences) and analyzed with BD FACSDiva software. Abs against F4/80, CD86, and I-Ek were obtained from Pharmingen (Leiden, The Netherlands), whereas Abs against CD4 and 7-aminoactinomycin D (7-AAD) were obtained from BD Biosciences.
Western Blot Analysis
Whole cell lysate was resolved by SDS-PAGE and electroblotted onto nitrocellulose membranes. The membranes were incubated with primary antibodies in 5% nonfat milk overnight at 4°C and secondary antibodies for 1 hour at room temperature. The electrochemiluminescence detection method was used for all Western blot experiments.
DTH Model
wt, Jnk1−/−, and Jnk2−/− mice were sensitized on day 0, as described for CIA. On day 17, delayed-type hypersensitivity (DTH) responses were induced intradermally in the right ear by injection of 10 μg CII in 2.5 mmol/L acetic acid. Vehicle, 20 μL, was injected in the left ear as a control. Ear thickness was measured using a micrometer before and 18, 24, and 48 hours after challenge with antigen.
Cutaneous Contact Hypersensitivity Model
On days 0 and 1, shaved dorsal skins of mice were painted with 0.5% 2,4-dinitrofluorobenzene (DNFB) in 25-μL acetone. On day 5, the right ear was challenged with 0.25% DNFB and the left ear was painted with vehicle (acetone). Ear thickness to the first cartilage ridge was measured 24 hours later. To account for acute hapten-induced irritation, background swelling was measured in parallel with that of nonsensitized mice. Specific ear swelling was calculated as follows: (treated ear thickness − control ear thickness) − background swelling.
T-Cell Proliferation Assay
The RAW 264.7 murine macrophage cell line from American Type Culture Collection (Manassas, VA) was cultured in Dulbecco's modified Eagle's medium (Sigma) for 24 hours before treatment with 25 μmol/L SP600125 JNK inhibitor (Sigma) (concentration determined in preceding trials) or dimethylsulfoxide (control) for 2 hours before stimulation with lipopolysaccharide (LPS; 1 μg/mL) for 24 hours. After LPS stimulation, the cells were treated for 30 minutes at 37°C with mitomycin C (Sigma), washed 5 times in ice-cold PBS, and cultured in a 96-well plate with 30,000 cells/well in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1% GlutaMax (Invitrogen, Hellerup, Denmark), and 1% penicillin-streptomycin.
Primary T cells were isolated from the spleen of 6- to 8-week-old C57BL/6JBom mice using negative selection (EasySep Mouse T Cell Enrichment Kit; StemCell Technologies, Grenoble, France), according to the manufacturer's protocol. The T cells were then labeled with 1 μmol/L carboxyfluorescein diacetate succinimyl ester (Molecular Probes; Invitrogen) for 10 minutes at 37°C, resuspended in 10× ice-cold PBS/10% fetal bovine serum, and incubated on ice for 5 minutes. Next, 100,000 T cells/well were co-cultured with mitomycin C–treated RAW 264.7 cells, supplemented with 0.3 μg/mL anti-mouse CD3e Abs (eBioscience, San Diego, CA) and 10 ng/mL murine recombinant IL-2 (R&D Systems, Minneapolis, MN). Assessment of proliferation was judged after 96 hours using flow cytometry.
Statistics
Quantitative data are expressed as the mean ± SEM if nothing else is stated in the figure legend. Significance analysis was performed using the Student's t-test or the U-test. All results were compared with wt mice with the exception of the BMM reconstitution experiment in which Jnk1−/− mice reconstituted with wt BMMs were compared with Jnk1−/− mice.
Results
Jnk1−/− Mice Are Resistant to CIA
To examine the role of JNK1 and JNK2 in a model of chronic autoinflammatory arthritis, C57BL/6 mice deficient for either JNK1 or JNK2 were compared with wt mice for incidence and severity of CIA, induced by intradermal injection of CII in CFA, followed by a boost injection 21 days later. Of wt mice, 67% (n = 15) developed arthritis, with a clinical score of 3.3 ± 1.2 at termination of the experiment (day 70 after immunization). JNK1-deficient mice (n = 20) were almost completely protected from CIA, although 15% (3/20) of the JNK1-deficient mice developed weak arthritis, with a score of 0.3 ± 0.5. In contrast, JNK2-deficient mice were highly susceptible to CIA, and all of these mice (n = 10) developed severe arthritis, showing a clinical score of 5.4 ± 1.3 at day 70 (Figure 1).
Figure 1.
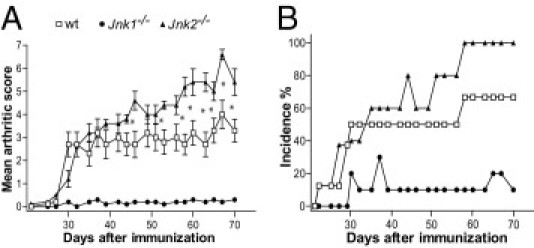
Jnk1−/− mice are resistant to CIA, whereas Jnk2−/− mice are highly susceptible. A: Clinical arthritis scores. B: Cumulative incidence of arthritis in wt, Jnk1−/−, and Jnk2−/− mice with CIA. The severity of arthritis was evaluated using a scoring system, as described in Materials and Methods. Results are presented as the mean ± SD. *P < 0.05 (n = 20 for Jnk1−/−, n = 10 for Jnk2−/−, and n = 15 for wt).
Histological Analysis Confirms the Lack of Inflammation in Joints from JNK1-Deficient Mice
To confirm the clinical analysis, H&E-stained sections of joints from wt, JNK1- deficient, and JNK2-deficient mice underwent histological examination. Joints from wt and Jnk2−/− mice showed severe inflammation and tissue destruction, with extensive hyperplasia of the synovium (Figure 2, A and B). However, although the arthritic score observed in Jnk2−/− mice was significantly higher compared with wt mice during the last 20 days of the disease course, no difference in histological analysis, including bone destruction, hyperplasia of the synovium, or infiltration of inflammatory cells, was detected between the groups. In contrast, joints from Jnk1−/− mice appeared normal and no (or minimal) tissue destruction and infiltration of inflammatory cells was observed (Figure 2, A and B).
Figure 2.
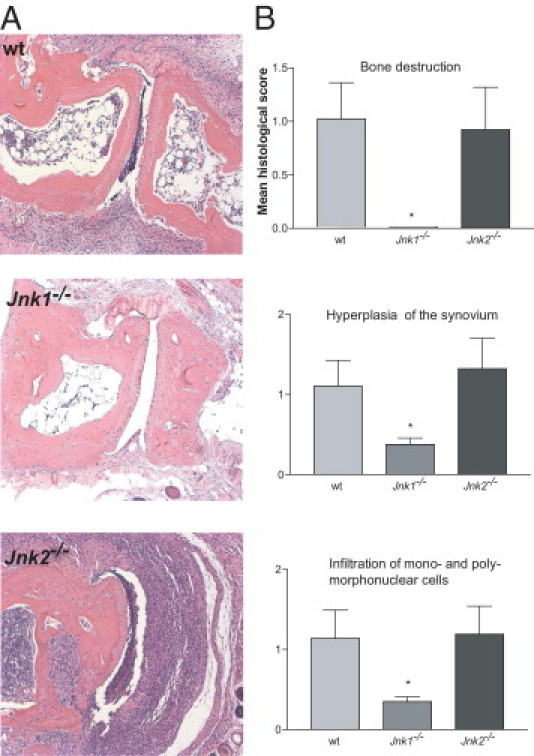
Histological analysis confirms the lack of inflammation in joints from Jnk1−/− mice with CIA. A: Representative H&E-stained sections from hind paws of wt, Jnk1−/−, and Jnk2−/− mice obtained at day 70 after immunization. There was absent or minimal inflammation in the Jnk1−/− section. B: The severity of arthritis was histologically evaluated using the following parameters: bone destruction, hyperplasia of the synovium, and infiltration of monomorphonuclear and polymorphonuclear cells. Each parameter was scored as described in Materials and Methods. *P < 0.05 (n = 10).
Altered Humoral Immunity in Response to CII in JNK1- and JNK2-Deficient Mice
CIA is dependent on both humoral and cellular immunity.19 To establish a possible explanation for the divergent roles of JNK1 and JNK2 observed during the progression of CIA, we compared the expression levels of CII-specific IgG between wt, Jnk1−/−, and Jnk2−/− mice during the disease course. As expected, Jnk1−/− mice showed dramatically reduced levels of total IgG compared with wt mice at days 30 and 70 after the first immunization (Figure 3A and data not shown, respectively). Surprisingly, the total IgG response in Jnk2−/− mice was also significantly reduced, although not to the same extent as observed in serum from Jnk1−/− mice, compared with that in wt mice at both time points studied.
Figure 3.
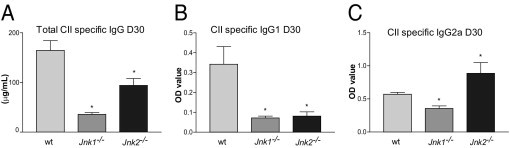
Altered humoral immunity in response to CII in Jnk1−/− and Jnk2−/− mice with CIA indicates a substantial Th1 response in the JNK2-deficient mice. A: Total CII-specific IgG serum levels in wt, Jnk1−/−, and Jnk2−/− mice on day 30 after immunization. IgG1 (B) and IgG2a (C) levels in wt, Jnk1−/−, and Jnk2−/− mice were measured on day 30 after immunization. *P < 0.05 (n = 10).
To assess the IgG subclass composition of the Ab response, we measured the levels of the CII-specific IgG1 and IgG2a isotypes in serum on day 30 after the first immunization. wt mice generated substantial CII-specific IgG1 and IgG2a titers, and the ratio of IgG2a/IgG1 was 1.7 (Figure 3B). In contrast, the titers of IgG1 and IgG2a in serum from Jnk1−/− mice were marginal compared with wt serum with an IgG2a/IgG1 ratio of 5.0. Interestingly, although Jnk2−/− mice showed significantly reduced IgG1 titers compared with wt mice, the levels of CII-specific IgG2a were significantly increased, revealing an IgG2a/IgG1 ratio of 11.1 (Figure 3B), strongly suggesting a predominant type 1 helper T cell (Th1) immune response.
Altered Cellular Immunity in JNK-Deficient Mice
To investigate the importance of JNK1 and JNK2 for the generation of T-cell–mediated immunity, we induced DTH responses by immunizing the mice intradermally with CII in CFA, followed by intradermal challenge in the ear using CII without the adjuvant 12 days later. At 24 hours after challenge, wt and Jnk2−/− mice showed a strong response, with 50% and 75% ear swelling, respectively. In contrast, Jnk1−/− mice were almost unresponsive (<10% ear swelling) (Figure 4A). Next, we wanted to investigate whether the lack of response in Jnk1−/− mice was directly related to a deficient T-cell response or if this might be due to an inability of myeloid cells (ie, macrophages and/or dendritic cells) to respond to microbial products present in CFA. We addressed this by challenging wt, Jnk1−/−, and Jnk2−/− mice in an alternative model of DTH that is independent of CFA. Thus, we assessed cutaneous contact hypersensitivity in animals using DNFB, which generates a specific cutaneous T-cell–mediated allergic response on repeated allergen contact.20 Similar to what we observed using CII, Jnk2−/− mice showed a strong response to DNFB 24 hours after challenge. However, although the response in JNK2-deficient mice was significantly increased compared with wt mice, there was no difference in ear swelling between wt and Jnk1−/− mice (Figure 4B). Thus, although there is an alteration in cellular immunity in Jnk1−/− mice when CFA is used for immunization, this is not the case in the absence of the adjuvant, suggesting that JNK1-deficient antigen-presenting cells might have an inadequate capability to respond to certain microbial agents.
Figure 4.
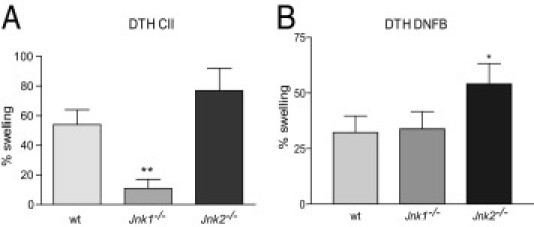
Altered cellular immunity in JNK-deficient mice. A: DTH response in wt, Jnk1−/−, and Jnk2−/− mice 24 hours after challenge with CII. B: The results at 24 hours after challenge with DNFB, suggesting a deficiency in the ability of Jnk1−/− antigen-presenting cells in responding to certain microbial products. Antigen-induced ear swelling is presented as the percentage increase in ear thickness of the stimulated ear compared with the control ear. *P < 0.05, **P < 0.01 (n = 7 for CII and n = 6 for DNFB).
In addition, because IL-17 produced by inflammatory Th17 may be involved in the pathogenesis of RA and in CIA,21 we wanted to explore the ability of JNK1- and JNK2-deficient T cells to differentiate into IL-17–producing effector cells. Thus, we purified naïve CD4+CD62L+ T cells from wt, Jnk1−/−, and Jnk2−/− mice and differentiated them in the presence of wt activated protein C, anti-CD3, transforming growth factor-β, and IL-6. Five days after stimulation, the T cells were restimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin and subjected to intracellular fluroscence-activated cell sorting (FACS) staining for the expression of IL-17. Although wt T cells produced significant amounts of IL-17 when restimulated, the amount of IL-17 produced from JNK1- and JNK2-deficient T cells was even higher (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Failure of Jnk1−/− Macrophages to Up-Regulate Costimulatory Molecules
To investigate whether antigen presenting cells (APC) from Jnk1−/− mice had a distorted capability to provide efficient stimulation for cellular immunity to occur, we generated BMMs from wt, Jnk1−/−, and Jnk2−/− mice and stimulated them with the bacterial cell wall protein LPS. Similar numbers of BMMs were generated from wt, Jnk1−/−, and Jnk2−/− bone marrow, and no difference in the expression level (mean fluorescence intensity) of F4/80, MHC II, or CD80 was detected between the groups (Figure 5A). Moreover, JNK has phosphorylated the PU.1 transcription factor, which is critical for differentiation of macrophages and is required for the expression of the macrophage colony-stimulating factor receptor (CSF-1R).22 However, we observed no difference in the expression levels of PU.1 or the CSF-1R in wt and Jnk1−/− BMMs (Figure 5D). We also did not detect differences in the expression levels of CSF-1R or the ability of JNK1-deficient BMMs to phosphorylate PU.1 in response to LPS (Figure 5D). LPS stimulation has previously induced a rapid down-regulation of the CSF-1R in macrophages.23 Accordingly, LPS treatment of BMMs for 45 minutes induced a marked down-regulation of the CSF-1R, and no difference was detected between wt and JNK1-deficient BMMs. Collectively, these data suggest that JNK1 or JNK2 is sufficient for macrophage differentiation and maturation.
Figure 5.
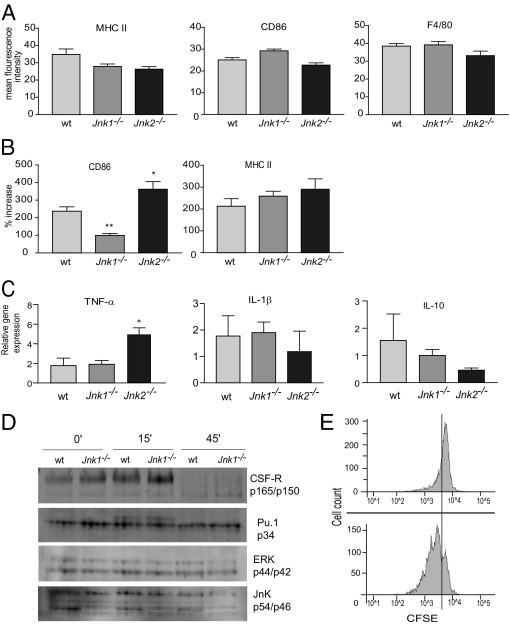
Failure of Jnk1−/− macrophages to up-regulate costimulatory molecule CD86. A: FACS analysis of MHC II (I-Ek), CD86, and F4/80 expression levels (mean fluorescence intensity) in unstimulated BMMs from wt, Jnk1−/−, and Jnk2−/− mice (n = 6). B: Percentage increase in mean fluorescence intensity in BMMs stimulated with LPS (10 ng/mL) for 24 hours (n = 6). C: The BMMs were stimulated for 6 hours with LPS (10 ng/mL), and cytokine mRNA levels were measured by real-time PCR analysis. *P < 0.05, **P < 0.001 (n = 6). D: Protein extracts were prepared from wt and Jnk1−/− BMMs stimulated with LPS (10 ng/mL) for the indicated time points and analyzed by immunoblotting. Data suggest that JNK1 or JNK2 is sufficient for macrophage differentiation and maturation. ERK, extracellular signal–regulated kinase. E: RAW 264.7 cells were treated with SP600125 JNK inhibitor (25 μmol/L) for 1 hour before 24-hour LPS stimulation (1 μg/mL) and co-cultured with carboxyfluorescein diacetate succinimyl ester–marked primary murine T cells for 96 hours. Although the dividing CD4+ cells in the control go through at least three cell divisions, most CD4+ cells stimulated with SP600125-treated RAW cells remain quiescent (76.8% versus 30.5% dividing cells). CFSE, carboxyfluorescein diacetate succinimyl ester.
When wt BMM was stimulated with LPS for 24 hours, the expression levels (mean fluorescence intensity) of MHC II and CD86 increased by 212% and 237%, respectively (Figure 5B). In Jnk2−/− BMMs, these numbers were even more pronounced (290% and 362%, respectively). In contrast, although Jnk1−/− BMMs were able to up-regulate MHC II to similar levels as wt mice (258%), a comparably modest increase in CD86 expression (99%) was observed (Figure 5B). We also analyzed the expression of CD86 after stimulation of wt and JNK1-deficient BMMs with mycobacterium. Similar to what we observed using LPS, although not as pronounced, mycobacterium induced a significant up-regulation of CD86 in wt BMMs (117%), whereas the increase in expression was modest in JNK1-deficient macrophages (43%) (data not shown). These results suggest that JNK1-deficient macrophages have an inferior costimulatory capacity compared with wt and Jnk2−/− macrophages and might be of particular importance because CD86 has been the prominent costimulatory molecule required for efficient activation of naïve CD4+ T cells in vivo.24 To validate these results, we cultured a murine macrophage cell line (ie, RAW 264.7 cells) and pretreated the cells with the specific JNK inhibitor SP600125 for 1 hour before stimulation for 24 hours with 1 μg/mL LPS. Hereafter, the cells were stained with CD86 Abs and run on a flow cytometer. A dose-dependent down-regulation of CD86 was shown (see Supplemental Figure S2 at http://ajp.amjpathol.org). Next, we made a co-culture of RAW cells and primary T cells to determine whether SP600125-treated RAW cells were able to activate primary naïve CD4+ T cells. The results are shown in Figure 5E. As judged by the percentage of dividing cells, the SP600125-pretreated RAW cells failed to induce significant activation of CD4+ T cells compared with control-treated RAW cells (30.5% versus 76.8% dividing cells). Furthermore, although the CD4+ cells activated by control-treated RAW cells went through at least three cell divisions, only a few of the CD4+ cells cultured with SP600125-treated RAW cells completed one cell cycle; most of them never divided.
Next, we evaluated the ability of BMMs from wt, Jnk1−/−, and Jnk2−/− mice to produce cytokines in response to LPS. Although the lack of JNK1 did not seem to affect the production of any of the cytokines investigated (ie, TNF-α, IL-1, IL-10, and IL-6; Figure 5C and data not shown), lack of JNK2 resulted in a significant increase in TNF-α production. Thus, although JNK1 deficiency appears to have a negative effect on the costimulatory capacity in macrophages, lack of JNK2 provides more efficient costimulation and increases the production of the pro-inflammatory cytokine TNF-α in response to LPS.
Divergent Roles of JNK1 and JNK2 in the Development of Serum-Induced Arthritis
To determine the role of the JNK signaling pathway in the effector phase of arthritis, we used the serum transfer model of inflammatory arthritis (K/BxN). This model is independent of both B and T cells and does not require adjuvants for the development of arthritis.12 After serum transfer, wt and Jnk2−/− mice displayed marked physical inflammation, as judged by clinical score and ankle swelling. The development of inflammatory arthritis in JNK2-deficient animals was significantly more rapid and severe compared with the wt control (P < 0.001 at days 5 through 9, as judged by δ ankle thickness, and P < 0.05 at days 3 through 8, as judged by clinical score; Figure 6A).
Figure 6.
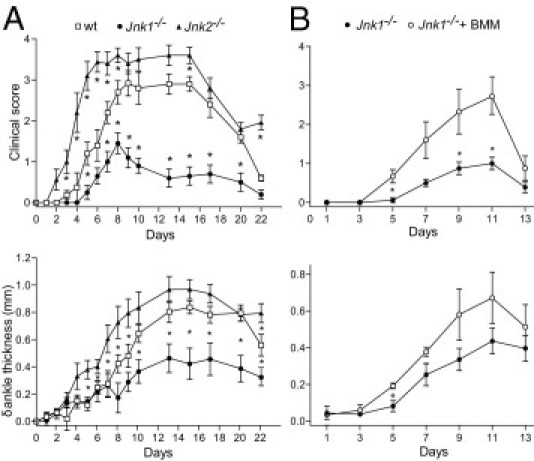
Divergent roles of JNK1 and JNK2 in the development of serum-induced arthritis. Clinical arthritis in wt, Jnk1−/−, and Jnk2−/− mice after injection of K/BxN serum i.p., as described in Materials and Methods. A: The mice were assessed for arthritis development by clinical index score and ankle thickness. B: wt BMMs were grown in culture, as described in Materials and Methods, and i.v. injected via tail vein into Jnk1−/− mice 1 day before and 1 and 3 days after serum transfer. The values represent the mean ± SEM. *P < 0.05 compared with A (wt mice) or B (mice not receiving BMMs under parallel conditions).
In contrast, Jnk1−/− mice developed weak arthritis compared with wt animals (P < 0.05 at days 10 through 22, as judged by δ ankle thickness, and P < 0.005 at days 10 through 20, as judged by clinical score). Although we could not detect a deficiency in the development and maturation of BMMs from Jnk1−/− mice, their inability to up-regulate CD86 in response to LPS suggests that JNK1-deficient macrophages are dysfunctional, at least in some aspects.
To further investigate whether the modest inflammatory response observed in JNK1-deficient mice could be due to inefficient macrophage function, we adoptively transferred wt BMMs to Jnk1−/− mice before the administration of K/BxN serum. This approach resulted in a significant increase in clinical score (P < 0.05 at days 5, 9, and 11; Figure 6B). An increase in ankle thickness was also observed, although this was only significant at day 5 after serum transfer (Figure 6B).
Discussion
In this study, we have shown distinct roles for JNK1 and JNK2 in two separate mouse models of RA. Although JNK1 is critically required and able to substitute for lack of JNK2 in the initiation and effector phase of inflammatory arthritis and even exacerbate clinical signs of arthritis, JNK2 does not possess these abilities. An important function for JNK during inflammatory arthritis appears to be control of innate immunity through macrophage activation and cytokine production.
The role of the JNK kinases, in inflammatory arthritis using JNK2-deficient mice and JNK-specific inhibitors (ie, SP600125, recognizing both JNK1 and JNK2), has been previously addressed.3,4 Blocking JNK1 and JNK2 using SP600125 in a rat model of adjuvant-mediated arthritis had a notable effect on inhibition of radiographical damage through inhibition of matrix-destroying metalloproteinases. This was confirmed in JNK1- and JNK2-deficient mouse synovial cell lines, although JNK2 seemed to be the relatively more important isotype for matrix-destroying metalloproteinase production. In contrast, chemical inhibition of both JNK1 and JNK2 in this model had a modest effect on the inflammatory response, as judged by paw swelling.3 In an additional study,4 using JNK2 knockout mice and a mouse model for passive CIA, the same group showed enhanced clinical arthritis in JNK2-deficient mice compared with the wt control, although synovial inflammation did not differ and cartilage and bone destruction levels were slightly better in JNK2-deficient mice compared with wt mice. In contrast, in a study5 of TNF-α–mediated joint disease, knockout of Jnk1 did not change the disease course, as judged by clinical and histological features. Recently, Guma and coworkers6,7 showed reduced disease severity in Jnk1−/− mice in two separate models of AIA when comparing mice deficient in JNK1 and JNK2 with wt animals.
We show that JNK1-deficient mice are almost completely protected from CIA, whereas JNK2-deficient mice developed more severe clinical signs of arthritis, with a higher incidence compared with wt mice. This is opposite of the results reported for the Jnk1−/− hTNFtg mouse; however, the TNF-α overexpression model does not involve the autoimmune disease phase and this may explain the differing results in the models.5 Thus, it seems likely that, although inhibition of both JNK1 and JNK2 is required for inhibition of matrix-destroying metalloproteinases from synovial cells during the effector phase of arthritis, the two isotypes have different roles in other aspects of the inflammatory response: JNK1 and JNK2 isoforms may compete for substrates and, although JNK1 is able to compensate for lack of JNK2 by up-regulating its activity, JNK2 is not able to do the same, producing apparent opposing phenotypes of the JNK1- and JNK2-deficient mice.25 The study5 of JNK1-deficient hTNFtg mice is in favor of this theory, in which the number of cells expressing phosphorylated JNK was significantly reduced in Jnk1−/− hTNFtg mice compared with Jnk1+/+ hTNFtg mice. However, a difference in phosphorylation of cJun within the synovial membrane could not be detected. Likewise, JNK2 was equally expressed in the synovial tissue, indicating that JNK2 was able to phosphorylate cJun in vivo. Indeed, in vitro studies of Jaeschke et al25 did show that JNK2 is able to phosphorylate cJun; however, its function is redundant compared with JNK1. Thus, lack of JNK1 may result in attenuation of inflammatory disease because of a reduced ability of JNK2 to efficiently phosphorylate its substrates compared with JNK1. Alternatively, JNK1 and JNK2 may have opposing effects under certain conditions.
JNK has previously been involved in T-cell activation and differentiation.13,14,26 Although JNK2 was required for sufficient interferon-γ production to induce Th1 differentiation,14,26 inhibition of JNK1 results in preferential differentiation to Th2 cells, through enhanced production of Th2 cytokines, leading to an inability to heal skin lesions on Leshmania infection.27
In CIA, mycobacterium adjuvant (ie, CFA) induces a strong immune response, leading to a cytokine environment favoring antigen-specific Th1 differentiation.28 This suggested that JNK1 deficiency, through enhanced Th2 cytokine production, might offer protection against CIA. In our studies, JNK1-deficient mice were protected from CIA. However, the minimal CII-specific IgG levels suggested that no functional T-cell response is generated in Jnk1−/− mice. Surprisingly, the total CII-specific IgG levels in Jnk2−/− mice were also lower compared with wt mice, although not to the same extent as in Jnk1−/− mice. More important, the IgG2a/IgG1 ratio in serum from Jnk2−/− mice was sevenfold higher compared with what we observed in wt mice and the level of IgG2a alone was significantly higher than in wt mice (Figure 3B). In the Jnk1−/− mice, the IgG2a/IgG1 ratio in serum was threefold higher than in wt mice, but the level of IgG2a alone was significantly reduced compared with Jnk2−/− and wt mice. This indicates a relative dominance of Th1 cytokines compared with wt mice in both Jnk1−/− and Jnk2−/− mice, which is opposite to what other studies13,14 have shown. However, because mycobacterium in CFA has induced a Th1 response,11 it seems possible that, under the existing experimental conditions, the T cells were able to differentiate into Th1 cells in the Jnk2−/− and Jnk1−/− mice, although this was less pronounced for Jnk1−/− mice, in agreement with the substantial Th2 response seen in other studies.13 Furthermore, because the severity of CIA is positively correlated with the IgG autoantibody response to CII in general29 and the predominance of autoreactive IgG2a Abs in particular,30 this could, at least partly, explain the resistance to CIA in Jnk1−/− mice and the enhanced severity of arthritis observed in Jnk2−/− mice compared with wt mice. Furthermore, we show that Jnk1−/− mice are able to generate a functional T-cell–mediated immune response against DNFB in a model of cutaneous contact hypersensitivity that is independent of an adjuvant (ie, CFA) and that JNK1-deficient T cells are able to differentiate into IL-17–producing effector cells. These data are in agreement with observations of comparable T-cell proliferation in Jnk1−/− and Jnk2−/− mice in the AIA model7 and collectively suggest that the protective effect of JNK1 inhibition is dependent on an inability of Jnk1−/− antigen-presenting cells to respond to certain microbial agents, rather than insufficiency of cell-mediated immunity in Jnk1−/− mice. A recent study31 has proposed that JNK1 activity is induced in myeloid cells in response to microbial pathogens and shows that Jnk1−/− mice are almost completely protected from the induction of experimental autoimmune encephalomyelitis. In addition, the migration of macrophages was impaired in the AIA model.7 Macrophages are required for CIA and have been the primary antigen-presenting cells in the immune response toward type II collagen.32–34
Both JNK1 and JNK2 are constitutively expressed in macrophages, and a recent study,35 using chemical inhibition of all JNK isoforms, has suggested that JNK signaling is required for differentiation of the monocyte/macrophage lineage, expression of the CSF-1R, and phosphorylation of the PU.1 transcription factor, which is important for development of the monocyte/macrophage lineage.22,36,37 We show that JNK1 (or JNK2) is sufficient for differentiation of the monocyte/macrophage lineage, expression of the CSF-1R, and phosphorylation of the PU.1 transcription factor. However, JNK1-deficient macrophages challenged with microbial pathogens (eg, LPS or mycobacterium) demonstrate dramatically reduced CD86 expression compared with wt macrophages. In line with this result, the murine macrophage cell line RAW 264.7 showed dose-dependent down-regulation of CD86 and could not stimulate primary murine naïve CD4+ T cells to divide when treated with a JNK inhibitor. In contrast, JNK2-deficient macrophages demonstrated a significant increase in CD86 expression compared with wt. Thus, JNK1 is important for adjuvant induced up-regulation of B7 costimulation on macrophages, whereas the role for JNK2 is inferior or possibly converse. One previous study38 has addressed the relative importance of the JNK isoforms for expression of B7 on macrophages in response to LPS. However, in this study, inhibition of JNK1 or JNK2 by the respective small-interfering RNA had a similar, but weak, inhibitory effect on expression of CD80, which possibly can be explained by lack of complete inhibition.
LPS acts primarily on toll-like receptor 4 on macrophages to activate JNK,39–41 and JNK has, in addition to regulation of costimulation, been involved in LPS-mediated expression of chemokines and cytokines (ie, TNF-α and IL-1) in macrophages.42,43 In our study, JNK1-deficient and wt BMMs produced similar levels of the pro-inflammatory cytokines, TNF-α and IL-1, in response to LPS, whereas JNK2-deficient BMMs produced markedly elevated levels of TNF-α and lower levels of IL-10 compared with wt BMMs. These results correlate with a previous study demonstrating similar levels of IL-1 and IL-10 in wt and JNK1-deficient BMMs after LPS stimulation31 and similar levels of IL-1β after TNF-α stimulation.7 We propose that, in an adjuvant and T-cell–dependent model of inflammation, JNK1 is a potent activator of innate immunity by triggering up-regulation of costimulation required for initiation of an adaptive T-cell response. In contrast, JNK2 activity alone most likely cannot induce satisfactory expression of costimulatory molecules and production of pro-inflammatory cytokines to induce arthritis in this model.
Han et al4 show enhanced clinical arthritis in JNK2-deficient mice compared with the wt control using a T-cell–independent mouse model for passive CIA. Interestingly, LPS was used in that study to exacerbate the inflammatory response.4 Our results, showing enhanced TNF-α production from JNK2-deficient macrophages in response to LPS, can partly explain the outcome in that study.
To compare the role of JNK1 with JNK2 in an arthritis model that is independent of the autoimmune induction phase, we chose the K/BxN model of serum-induced arthritis. This model is T-cell independent44,45 but dependent on immune complexes, complement,44 neutrophils, mast cells,46,47 macrophages,48 and the production of pro-inflammatory cytokines (ie, IL-1β and TNF-α) for progression.49 Surprisingly, although this model is independent of an adjuvant, such as mycobacterium or LPS, we observed a significant protection in JNK1-deficient mice, whereas an exacerbated response was detected in JNK2-deficient mice. Although the precise mechanistic explanation for the divergent roles of JNK1 and JNK2 during serum-induced arthritis remains to be investigated, the protective role of JNK1 deficiency is macrophage dependent, because adoptive transfer of wt BMMs to the JNK1-deficient mice restored their susceptibility to serum-induced arthritis. This supplements the results shown by Guma and coworkers,6 who also saw a protection in Jnk1−/− mice, in this case suspected to depend on diminished mast cell degranulation and cytokine production, collectively suggesting a dual effect on innate immunity during the inflammatory response in the model.
In conclusion, our results demonstrate divergent abilities for the JNK kinases in two distinct models of arthritis and suggest that the least common denominator determining the outcome of JNK1 and JNK2 deficiency during the autoimmune initiation phase, and during the effector phase of inflammatory arthritis, is related to macrophage function. Finally, the previously suggested approach of achieving major blockade of JNK signaling in inflammatory arthritis through blockade of both JNK1 and JNK24,5 should be revised, because isoform-specific inhibition of JNK1 likely will be a more efficient way of inhibiting the inflammatory response.
Acknowledgment
The KRN mice were provided by Diane Mathis and Christophe Benoist (IGMBC, Illkirk, France).
Footnotes
Supported by grants from The Danish Medical Research Councils (S.S.F.), The Leo Research Foundation, The Rheumatism Society (Gigtforeningen), Denmark, The Becket Foundation, The Lundbeck Foundation, The Novo Nordisk Research Foundation, and The University of Copenhagen.
K.D. and S.R. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2011.06.019.
Current address of J.M.L., Department of Systems Biology, Center for Biological Sequence Analysis, Technical University of Denmark, Kongens Lyngby, Denmark; of C.Ø., Novo Nordisk A/S, Maaloev, Denmark; of P.S., ALK ABELLÓ, Hoersholm, Denmark.
Supplementary data
FACS analysis of intracellular IL-17 staining level (mean fluorescence intensity) in activated T cells from wt, Jnk1−/−, and Jnk2−/− mice, showing substantial production of IL-17 from wt T cells. Despite this, the production from JNK1- and JNK2-deficient cells was even higher. Naive CD4+CD62L+ T cells were purified from wt, Jnk1−/−, and Jnk2−/− mice (n = 6) and were differentiated in the presence of wt activated protein C, anti-CD3, transforming growth factor-β, and IL-6. Five days later, the T cells were restimulated with PMA and ionomycin and subjected to intracellular FACS staining for IL-17.
Treatment of RAW 264.7 cells with JNK inhibitor SP600125 before LPS stimulation results in dose-dependent inhibition of CD86 up-regulation, as judged by percentage of CD86+ cells compared with untreated LPS-stimulated control. RAW cells were treated with different doses of SP600125 for 1 hour before the addition of 1 μg/mL LPS. After 24 hours, the cells were stained with CD86 antibodies and evaluated by FACS.
References
- 1.van den Berg W.B. Anti-cytokine therapy in chronic destructive arthritis. Arthritis Res. 2001;3:18–26. doi: 10.1186/ar136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schett G., Tohidast-Akrad M., Smolen J.S., Schmid B.J., Steiner C.W., Bitzan P., Zenz P., Redlich K., Xu Q., Steiner G. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000;43:2501–2512. doi: 10.1002/1529-0131(200011)43:11<2501::AID-ANR18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 3.Han Z., Boyle D.L., Chang L., Bennett B., Karin M., Yang L., Manning A.M., Firestein G.S. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han Z., Chang L., Yamanishi Y., Karin M., Firestein G.S. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. 2002;46:818–823. doi: 10.1002/art.10104. [DOI] [PubMed] [Google Scholar]
- 5.Koller M., Hayer S., Redlich K., Ricci R., David J.P., Steiner G., Smolen J.S., Wagner E.F., Schett G. JNK1 is not essential for TNF-mediated joint disease. Arthritis Res Ther. 2005;7:R166–R173. doi: 10.1186/ar1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guma M., Kashiwakura J., Crain B., Kawakami Y., Beutler B., Firestein G.S., Kawakami T., Karin M., Corr M. JNK1 controls mast cell degranulation and IL-1{beta} production in inflammatory arthritis. Proc Natl Acad Sci U S A. 2010;107:22122–22127. doi: 10.1073/pnas.1016401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guma M., Ronacher L.M., Firestein G.S., Karin M., Corr M. JNK-1 deficiency limits macrophage-mediated antigen-induced arthritis. Arthritis Rheum. 2011;63:1603–1612. doi: 10.1002/art.30271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams R.O. Collagen-induced arthritis in mice. Methods Mol Med. 2007;136:191–199. doi: 10.1007/978-1-59745-402-5_14. [DOI] [PubMed] [Google Scholar]
- 9.Joe B., Griffiths M.M., Remmers E.F., Wilder R.L. Animal models of rheumatoid arthritis and related inflammation. Curr Rheumatol Rep. 1999;1:139–148. doi: 10.1007/s11926-999-0011-7. [DOI] [PubMed] [Google Scholar]
- 10.Campbell I.K., Hamilton J.A., Wicks I.P. Collagen-induced arthritis in C57BL/6 (H-2b) mice: new insights into an important disease model of rheumatoid arthritis. Eur J Immunol. 2000;30:1568–1575. doi: 10.1002/1521-4141(200006)30:6<1568::AID-IMMU1568>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 11.Inglis J.J., Criado G., Medghalchi M., Andrews M., Sandison A., Feldmann M., Williams R.O. Collagen-induced arthritis in C57BL/6 mice is associated with a robust and sustained T-cell response to type II collagen. Arthritis Res Ther. 2007;9:R113. doi: 10.1186/ar2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monach P.A., Benoist C., Mathis D. The role of antibodies in mouse models of rheumatoid arthritis, and relevance to human disease. Adv Immunol. 2004;82:217–248. doi: 10.1016/S0065-2776(04)82005-4. [DOI] [PubMed] [Google Scholar]
- 13.Dong C., Yang D.D., Wysk M., Whitmarsh A.J., Davis R.J., Flavell R.A. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 14.Yang D.D., Conze D., Whitmarsh A.J., Barrett T., Davis R.J., Rincon M., Flavell R.A. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–585. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- 15.Kouskoff V., Korganow A.S., Duchatelle V., Degott C., Benoist C., Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 16.Schett G., Tuerk B. Bone histomorphometry in arthritis models 1. Methods Mol Med. 2007;135:269–283. doi: 10.1007/978-1-59745-401-8_17. [DOI] [PubMed] [Google Scholar]
- 17.Monach P.A., Mathis D., Benoist C. The K/BxN arthritis model. Curr Protoc Immunol. 2008 doi: 10.1002/0471142735.im1522s81. Chapter 15:Unit 15.22. [DOI] [PubMed] [Google Scholar]
- 18.Bonnesen B., Orskov C., Rasmussen S., Holst P.J., Christensen J.P., Eriksen K.W., Qvortrup K., Odum N., Labuda T. MEK kinase 1 activity is required for definitive erythropoiesis in the mouse fetal liver. Blood. 2005;106:3396–3404. doi: 10.1182/blood-2005-04-1739. [DOI] [PubMed] [Google Scholar]
- 19.Seki N., Sudo Y., Yoshioka T., Sugihara S., Fujitsu T., Sakuma S., Ogawa T., Hamaoka T., Senoh H., Fujiwara H. Type II collagen-induced murine arthritis, I: induction and perpetuation of arthritis require synergy between humoral and cell-mediated immunity. J Immunol. 1988;140:1477–1484. [PubMed] [Google Scholar]
- 20.Wang B., Feliciani C., Freed I., Cai Q., Sauder D.N. Insights into molecular mechanisms of contact hypersensitivity gained from gene knockout studies. J Leukoc Biol. 2001;70:185–191. [PubMed] [Google Scholar]
- 21.Nakae S., Nambu A., Sudo K., Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 22.Zhang D.E., Hetherington C.J., Chen H.M., Tenen D.G. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. 1994;14:373–381. doi: 10.1128/mcb.14.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schroder K., Lichtinger M., Irvine K.M., Brion K., Trieu A., Ross I.L., Ravasi T., Stacey K.J., Rehli M., Hume D.A., Sweet M.J. PU.1 and ICSBP control constitutive and IFN-gamma-regulated Tlr9 gene expression in mouse macrophages. J Leukoc Biol. 2007;81:1577–1590. doi: 10.1189/jlb.0107036. [DOI] [PubMed] [Google Scholar]
- 24.Lang T.J., Nguyen P., Peach R., Gause W.C., Via C.S. In vivo CD86 blockade inhibits CD4+ T cell activation, whereas CD80 blockade potentiates CD8+ T cell activation and CTL effector function. J Immunol. 2002;168:3786–3792. doi: 10.4049/jimmunol.168.8.3786. [DOI] [PubMed] [Google Scholar]
- 25.Jaeschke A., Karasarides M., Ventura J.J., Ehrhardt A., Zhang C., Flavell R.A., Shokat K.M., Davis R.J. JNK2 is a positive regulator of the cJun transcription factor. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 26.Sabapathy K., Hu Y., Kallunki T., Schreiber M., David J.P., Jochum W., Wagner E.F., Karin M. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr Biol. 1999;9:116–125. doi: 10.1016/s0960-9822(99)80065-7. [DOI] [PubMed] [Google Scholar]
- 27.Constant S.L., Dong C., Yang D.D., Wysk M., Davis R.J., Flavell R.A. JNK1 is required for T cell-mediated immunity against Leishmania major infection. J Immunol. 2000;165:2671–2676. doi: 10.4049/jimmunol.165.5.2671. [DOI] [PubMed] [Google Scholar]
- 28.Stasiuk L.M., Abehsira-Amar O., Fournier C. Collagen-induced arthritis in DBA/1 mice: cytokine gene activation following immunization with type II collagen. Cell Immunol. 1996;173:269–275. doi: 10.1006/cimm.1996.0277. [DOI] [PubMed] [Google Scholar]
- 29.Williams P.J., Jones R.H., Rademacher T.W. Correlation between IgG anti-type II collagen levels and arthritic severity in murine arthritis. Autoimmunity. 1998;27:201–207. doi: 10.3109/08916939808993831. [DOI] [PubMed] [Google Scholar]
- 30.Watson W.C., Townes A.S. Genetic susceptibility to murine collagen II autoimmune arthritis: proposed relationship to the IgG2 autoantibody subclass response, complement C5, major histocompatibility complex (MHC) and non-MHC loci. J Exp Med. 1985;162:1878–1891. doi: 10.1084/jem.162.6.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran E.H., Azuma Y.T., Chen M., Weston C., Davis R.J., Flavell R.A. Inactivation of JNK1 enhances innate IL-10 production and dampens autoimmune inflammation in the brain. Proc Natl Acad Sci U S A. 2006;103:13451–13456. doi: 10.1073/pnas.0601155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell I.K., Rich M.J., Bischof R.J., Hamilton J.A. The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J Leukoc Biol. 2000;68:144–150. [PubMed] [Google Scholar]
- 33.Guery L., Chiocchia G., Batteux F., Boissier M.C., Fournier C. Collagen II-pulsed antigen-presenting cells genetically modified to secrete IL-4 down-regulate collagen-induced arthritis. Gene Ther. 2001;8:1855–1862. doi: 10.1038/sj.gt.3301613. [DOI] [PubMed] [Google Scholar]
- 34.Michaelsson E., Holmdahl M., Engstrom A., Burkhardt H., Scheynius A., Holmdahl R. Macrophages, but not dendritic cells, present collagen to T cells. Eur J Immunol. 1995;25:2234–2241. doi: 10.1002/eji.1830250818. [DOI] [PubMed] [Google Scholar]
- 35.Himes S.R., Sester D.P., Ravasi T., Cronau S.L., Sasmono T., Hume D.A. The JNK are important for development and survival of macrophages. J Immunol. 2006;176:2219–2228. doi: 10.4049/jimmunol.176.4.2219. [DOI] [PubMed] [Google Scholar]
- 36.Mao C., Ray-Gallet D., Tavitian A., Moreau-Gachelin F. Differential phosphorylations of Spi-B and Spi-1 transcription factors. Oncogene. 1996;12:863–873. [PubMed] [Google Scholar]
- 37.Valledor A.F., Borras F.E., Cullell-Young M., Celada A. Transcription factors that regulate monocyte/macrophage differentiation. J Leukoc Biol. 1998;63:405–417. doi: 10.1002/jlb.63.4.405. [DOI] [PubMed] [Google Scholar]
- 38.Lim W., Gee K., Mishra S., Kumar A. Regulation of B7.1 costimulatory molecule is mediated by the IFN regulatory factor-7 through the activation of JNK in lipopolysaccharide-stimulated human monocytic cells. J Immunol. 2005;175:5690–5700. doi: 10.4049/jimmunol.175.9.5690. [DOI] [PubMed] [Google Scholar]
- 39.Poltorak A., He X., Smirnova I., Liu M.Y., Van H.C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 40.Muzio M., Natoli G., Saccani S., Levrero M., Mantovani A. The human toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6) J Exp Med. 1998;187:2097–2101. doi: 10.1084/jem.187.12.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qureshi S.T., Lariviere L., Leveque G., Clermont S., Moore K.J., Gros P., Malo D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beutler B., Cerami A. Tumor necrosis, cachexia, shock, and inflammation: a common mediator. Annu Rev Biochem. 1988;57:505–518. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 43.Dinarello C.A. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- 44.Ji H., Ohmura K., Mahmood U., Lee D.M., Hofhuis F.M., Boackle S.A., Takahashi K., Holers V.M., Walport M., Gerard C., Ezekowitz A., Carroll M.C., Brenner M., Weissleder R., Verbeek J.S., Duchatelle V., Degott C., Benoist C., Mathis D. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–168. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 45.Choe J.Y., Crain B., Wu S.R., Corr M. Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J Exp Med. 2003;197:537–542. doi: 10.1084/jem.20021850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee D.M., Friend D.S., Gurish M.F., Benoist C., Mathis D., Brenner M.B. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 47.Wipke B.T., Allen P.M. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 48.Solomon S., Rajasekaran N., Jeisy-Walder E., Snapper S.B., Illges H. A crucial role for macrophages in the pathology of K/B x N serum-induced arthritis. Eur J Immunol. 2005;35:3064–3073. doi: 10.1002/eji.200526167. [DOI] [PubMed] [Google Scholar]
- 49.Ditzel H.J. The K/BxN mouse: a model of human inflammatory arthritis. Trends Mol Med. 2004;10:40–45. doi: 10.1016/j.molmed.2003.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FACS analysis of intracellular IL-17 staining level (mean fluorescence intensity) in activated T cells from wt, Jnk1−/−, and Jnk2−/− mice, showing substantial production of IL-17 from wt T cells. Despite this, the production from JNK1- and JNK2-deficient cells was even higher. Naive CD4+CD62L+ T cells were purified from wt, Jnk1−/−, and Jnk2−/− mice (n = 6) and were differentiated in the presence of wt activated protein C, anti-CD3, transforming growth factor-β, and IL-6. Five days later, the T cells were restimulated with PMA and ionomycin and subjected to intracellular FACS staining for IL-17.
Treatment of RAW 264.7 cells with JNK inhibitor SP600125 before LPS stimulation results in dose-dependent inhibition of CD86 up-regulation, as judged by percentage of CD86+ cells compared with untreated LPS-stimulated control. RAW cells were treated with different doses of SP600125 for 1 hour before the addition of 1 μg/mL LPS. After 24 hours, the cells were stained with CD86 antibodies and evaluated by FACS.