

Abstract
Aged mice in a murine model of myocardial infarction exhibit less effective myocardial repair. We hypothesized that the deficiency arises from altered lineage choice of endogenous mesenchymal stem cells (MSCs) and faulty maturation of myofibroblasts. Examination of cardiac MSCs revealed a substantial reduction in the pluripotency marker Nanog in cells from aged mice. In addition, the aged MSCs demonstrated a redirected lineage choice that favored adipocytic commitment over fibroblast or myofibroblast differentiation. Fibroblasts derived from aged MSCs demonstrated reduced expression of transforming growth factor-β (TGF-β) receptors I and II and diminished SMAD3 phosphorylation, associated with attenuated contractility and migration. Overexpression of constitutively active TGF-β receptor I in aged cardiac fibroblasts ameliorated their defective motility but did not improve their contractility. Culturing of MSCs and fibroblasts with AICAR (5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside) to activate adenosine monophosphate–activated kinase resulted in TGF-β–dependent development of myofibroblasts with markedly enhanced contractility despite no reduction in adipocytic commitment or increased expression of TGF-β receptors and SMAD3 phosphorylation. The data suggest an adenosine monophosphate–activated kinase–dependent gain of function as mediated by phosphorylation of TGF-β–activated kinase 1 and p38 mitogen–activated protein kinase, which amplifies the response to TGF-β1 via a non-canonical pathway, thus compensating for the reduced expression of TGF-β receptors.
Cardiac fibroblasts are the most prevalent cell type in the heart. These cells exert a critical role in regulating normal myocardial function and in the adverse myocardial remodeling that occurs after myocardial infarction (MI).1 Irreversible cardiomyocyte damage owing to cessation of oxygen supply during MI leads to necrosis, which stimulates inflammatory reactions that trigger reparative pathways and activate cells to form a scar. Cytokines released by inflammatory infiltrating leukocytes promote endogenous mesenchymal stem cell (MSC) proliferation and migration toward the infarct site, followed by differentiation into fibroblasts that deposit scar-forming collagen. The fibroblasts mature into myofibroblasts, expressing scar-contracting α–smooth muscle actin (α-SMA).2 Resident fibroblasts also become activated and participate in this process. After several weeks, a mature scar is formed, and most of the myofibroblasts undergo apoptosis.3–5
We have previously established in a model of mouse MI that, compared with young animals, aged mice demonstrate greater infarct expansion and less effective myocardial repair.6 Defective scar formation arises from a decreased number of myofibroblasts and diminished collagen deposition in the infarct, which results in a structurally unstable scar formed by loose connective tissue.7
Evidence indicates that multipotent cells can be generated in vitro from several adult organs including the heart.8 Tissue-resident progenitor cells of mesenchymal origin can differentiate into myogenic, adipocytic, chondrocytic, osteoblastic, and fibroblastic lineages.9–11 The potential of those stem cells to differentiate decreases with age.11,12 Multipotentiality of stem cells has been strongly associated with expression of certain transcription factors such as Nanog, Oct3/4, Klf4, and Sox2.13,14 It has been proposed that Nanog has a key role in maintaining embryonic stem cell pluripotency15; however, stem cell pluripotency is also expressed in adult stem cells.8
The ability of the progenitor cells to participate in scar formation, in particular insofar as maturation of fibroblasts into myofibroblasts, decreases with age.6,7 We isolated cardiac resident MSCs from young and aged mice and compared their multipotentiality. MSCs derived from aged animals exhibited diminished expression of Nanog and increased adipocytic potential. Those cells converted into dysfunctional fibroblasts with reduced expression of transforming growth factor-β (TGF-β) receptor types I and II (TβRI and TβRII, respectively). Choy et al16 described the mechanism by which TGF-β1 inhibits adipocyte formation and suggested that decreased responsiveness to TGF-β1 might account for enhanced adipogenesis and impaired myofibroblast maturation.
AICAR (5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside)17 increases the expression of pluripotent markers such as Nanog in murine embryonic stem cells18 and inhibits adipocytic differentiation by reducing expression of fatty acid synthase and acetyl-CoA carboxylase.19 AICAR is an adenosine monophosphate mimetic and activator of adenosine monophosphate–activated kinase (AMPK). The present study, to our surprise, demonstrated that culture of aged MSCs using AICAR-stimulated AMPK did not alter their adipocytic lineage choice. However, AMPK phosphorylation markedly increased myofibroblast contractile function in response to TGF-β1. The results demonstrated an AMPK-generated non-canonical pathway involving TGF-β–activated kinase (Tak1) phosphorylation and p38 mitogen–activated protein kinase (p38MAPK) activation that restores myofibroblast function.
Materials and Methods
Reagents
Reagents used included AICAR (Toronto Research Chemicals, Inc., Toronto, ON, Canada), Compound C [6-(4)-(2-piperidin-1-ylethoxy)phenyl(−3-pyridin-4-ylpyrazolo)1,5-a)pyrimidine] and metformin (1,1-dimethylbiguanidine hydrochloride) (Sigma-Aldrich Corp., St. Louis, MO), SB203580 and (5Z)−7-oxozeaenol (EMD Chemicals, Inc., Gibbstown, NJ), and SP600125 and PP2 (Enzo Life Sciences, Inc., Farmingdale, NY).
Animals
Male C57BL/6 mice aged 2 to 4 months were obtained from the Center for Comparative Medicine, Baylor College of Medicine. Male C57BL/6 mice aged 14, 24, and 30 months were purchased from the National Institute on Aging (National Institutes of Health, Bethesda, MD). All animals were used in agreement with guidelines of the Baylor College of Medicine Animal Care and Research Advisory Committee.
Cell Isolation
Mouse hearts were cut into 1-mm3 pieces, digested using Liberase TH (Roche Diagnostics Corp., Indianapolis, IN), and incubated in a 37°C shaking water bath with regular trituration by pipet to obtain a single cell suspension (all non-myocytes). Cells were centrifuged at 250 × g for 5 minutes. The cell pellet was washed and suspended in cell growth medium.2
Tissue Culture
Fibroblast Culture
Cells were cultured in Dulbecco's modified Eagle's medium and F-12 nutrient (DMEM/F12) in a ratio of 1:1 (Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific Pierce Protein Research Products, Rockford, IL) and antibiotic-antimycotic (Invitrogen Corp.).
Stem Cell Culture
To maintain the undifferentiated state of any stem cells in the preparation, immediately after isolation, cells were placed in stem cell medium comprising DMEM/F12 in a ratio of 3:1 (Invitrogen Corp.) supplemented with 20 ng/mL epidermal growth factor (R&D Systems, Inc., Minneapolis, MN), 10 ng/mL basic fibroblast growth factor (Invitrogen Corp.), 10 ng/mL leukemia inhibitory factor (Sigma-Aldrich Corp.), and 1X B27 (Invitrogen Corp.).20
Differentiation
To induce MSC differentiation into the osteogenic lineage, cells were cultured in DMEM/F12 medium supplemented with 10% FBS, 10 nmol/L dexamethasone, 10 mmol/L β-glycerophosphate, 50 μg/mL ascorbate phosphate, and 10 nmol/L 1α,25-dihydroxyvitamin D3. To induce adipogenesis, cells were placed in DMEM/F12 medium supplemented with 10% FBS, 10 μmol/L dexamethasone, 1 μg/mL insulin, and 0.5 mmol/L isobutylmethylxanthine. For chondrogenesis, cells were differentiated in DMEM/F12 medium supplemented with 0.1 μmol/L dexamethasone, 50 μg/mL ascorbate phosphate, 40 μg/ml l-proline, 100 μg/mL sodium pyruvate, 1X ITS-3, and 10 ng/mL TGF-β3 (R&D Systems, Inc.). To induce the differentiation process, cells were cultured in the appropriate medium for 21 days.21
Osteogenesis was detected via histochemical staining of calcium deposits using Alizarin Red S. Adipogenesis was evaluated via the presence of neutral lipids in the cytoplasm stained with Oil Red O and by staining with an anti-perilipin A antibody. To analyze chondrogenic differentiation, sulfated matrix proteoglycans were stained using Alcian Blue. Unless otherwise stated, all reagents were acquired from Sigma-Aldrich Corp.
Whole-Cell Lysate Preparation
Cells were washed with cold PBS and harvested using PBS buffer containing a phosphatase and protease inhibitor cocktail (Halt Protease and Phosphatase Single-Use Inhibitor Cocktail, Thermo Scientific Pierce Protein Research Products). Next, cells were centrifuged at 250 × g for 5 minutes. The cell pellet was resuspended in 50 μL radioimmunoprecipitation buffer (Thermo Scientific Pierce Protein Research Products) supplemented with inhibitor cocktail, and subsequently were frozen, thawed, and centrifuged at 10,000 × g for 5 minutes. The supernatant was collected, and protein concentration was determined using a bicinchoninic acid assay (BCA Protein Assay Kit; Thermo Scientific Pierce Protein Research Products).
Western Blot Analysis
Total cell protein aliquots (35 μg) were mixed with reducing Laemmli sample buffer, boiled for 5 minutes, and separated on 4% to 15% Tris-HCl gel (Bio-Rad Laboratories, Inc., Hercules, CA). Proteins were transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc.) using a mini trans-blot electrophoretic transfer cell apparatus (Bio-Rad Laboratories, Inc.). For immunoblotting, the membranes were incubated with the blocking buffer, and the antibodies were diluted according to the manufacturer's protocol. To detect signal, membranes were incubated with peroxide and luminal enhancer solutions (SuperSignal West Pico Chemiluminescent Substrate; Thermo Scientific Pierce Protein Research Products).
Antibodies used included anti-TβRI and anti-TβRII (R&D Systems, Inc.), anti-actin (Abcam, Inc., Cambridge, MA), anti–α-SMA (Sigma-Aldrich Corp.), anti-SMAD3, anti-pSMAD3 (Ser423/Ser245), anti-AMPK, anti-pAMPK (Thr172), anti-p38MAPK, anti-p-p38MAPK (Thr180/Thr182), anti-Tak1, anti-p-Tak1 (Thr184/Thr187), anti-FAK, anti-pFAK (Tyr576/Tyr577), anti-JNK, and anti-pJNK (Thr183/Tyr185) (all from Cell Signaling Technology, Inc., Beverly, MA).
Collagen Pad Assay
Fibroblasts were passaged 3 days before the experiment and cultured in the presence of 10% FBS to enable expression of contractile proteins. The collagen solution was produced by mixing acid-soluble collagen type I (BD Biosciences, San Jose, CA), a twofold concentration of minimum essential medium (Invitrogen Corp.), and water in a ratio of 1:1.5:1, and the mixture was buffered to pH 7.5. The final concentration of collagen was 1.7 mg/mL. Fibroblasts were mixed into the collagen solution at a cell density of 2.0 × 105 cells/mL. The mixture was poured to form small pads (18 mm in diameter) and allowed to gel.
For free-floating gel experiments, fibroblasts were seeded in collagen pads, which were transferred to 35-mm dishes immediately (after they gelled) and incubated with DMEM supplemented with 5 mg/mL BSA (Sigma-Aldrich Corp.). Photographs were obtained at time 0 (when the collagen gelled) and at 24 hours later. The contraction of the gel was expressed as a percentage of the initial lattice area, with the surface area of the non-contracted state serving as 100%.
For attached gel experiments, polymerized restrained matrices were incubated for 48 hours in DMEM supplemented with 50 μg/mL ascorbic acid and 10% FBS.22 Contraction was initiated by releasing the attached gels using a spatula. Gels were incubated for 24 hours in DMEM with or without 10 ng/mL TGF-β1.
Migration
Migration of cardiac fibroblasts was assessed using a modified Boyden's chamber method.23 Cells were serum-starved for 24 hours before the experiment. The cells (1.5 × 105 cells per chamber) were plated into culture inserts (Millipore Corp., Billerica, MA) in serum-free medium. TGF-β1 (10 ng/mL) (BioLegend, Inc., San Diego, CA) or FBS (1%) was added to the medium in the lower chamber, and cells were allowed to migrate through 8-μm pores. The inserts were removed after 4 hours, washed, and stained with 0.1% crystal violet. Non-migratory cells from the interior of the insert were removed using a cotton swab. Stained cells attached to the insert underside were transferred to a well containing 70% methanol, and were incubated for 15 minutes. The concentration of dye released from cells was assessed at λ = 560 nm at spectrophotometry.
Wound Scratch Assay
Cells were grown in 24-well culture dishes until confluent, and were serum-starved for 24 hours before assay. Subsequently, the cell monolayer was wounded using a sterile plastic micropipet tip and photographed using phase contrast microscopy (at 0 hour), and complete medium was reintroduced. Cells were photographed every 24 hours until the gap was closed.
Immunohistochemistry
Immunohistochemistry was performed on cells fixed in 4% paraformaldehyde using a Vectastain ABC kit (Vector Laboratories, Inc., Burlingame, CA). Endogenous peroxidase activity was blocked via incubation in 0.3% hydrogen peroxide (Sigma-Aldrich Corp.). Nonspecific antibody binding was prevented via blocking in 1.5% rabbit or goat serum (Vectastain ABC kit). Cells were incubated with primary antibody for 1 hour at room temperature; then, secondary goat anti-rabbit antibody was applied for 30 minutes. The color was developed by using 3′3-diaminobenzine (Vector Laboratories, Inc.) as substrate. Antibodies included anti-Nanog and anti-Oct3/4 (R&D Systems, Inc.), and anti-perilipin A (Abcam, Inc.). IgG controls were purchased from Rockland Immunochemicals, Inc. (Gilbertsville, PA).
Immunofluorescence
Cells cultured on coverslips were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and rinsed in PBS. Then cells were incubated with primary antibodies for 1 hour. After 2 washes using PBS, cells were incubated for 45 minutes using an appropriate secondary antibody (Alexa Fluor 488– or Alexa Fluor 594–conjugated antibody; Invitrogen Corp.). After incubation with secondary antibody, cells were washed in PBS, and nuclei were counterstained using DAPI containing mounting medium (Slow Fade Gold Antifade Reagent with DAPI; Invitrogen Corp.).
Antibodies and reagents included anti-CD44 and rat IgG1κ (Beckman Coulter, Inc., Brea, CA), anti-collagen type I and goat IgG (Rockland Immunochemicals, Inc.), anti–discoidin domain receptor 2 (Genex Bioscience, Inc., Hayward, CA), anti–α-Sma (Sigma-Aldrich Corp.), and anti-flag (BioLegend, Inc.). DNase I (deoxyribonuclease I–Alexa Fluor 488) and phalloidin (phalloidin-rhodamine conjugate) were purchased from Invitrogen Corp., rabbit IgG from Cell Signaling Technology, Inc., and mouse IgG-FITC and rat IgG2aκ from BD Biosciences.
qPCR
Total RNA was isolated from whole hearts using TRizol reagent (Invitrogen Corp.), purified using an RNeasy kit (Qiagen, Inc., Valencia, CA), and transcribed to cDNA using an iScript cDNA Synthesis kit (Bio-Rad Laboratories, Inc.). Quantitative PCR (qPCR) was performed using an iQ5 Multicolor Real Time PCR Detection System using SYBR Green Super mix (both from Bio-Rad Laboratories, Inc.) and specific primers. Gene expression was measured using the comparative CT method to calculate the amount of target mRNA normalized to an endogenous reference (18S). Data from aging animals were expressed as the fold increase or decrease of mRNA concentration relative to mRNA expression detected in 4-month-old hearts (except in Figure 2A). Each sample was tested in triplicate to ensure reproducibility. Primers were designed using commercially available software (Beacon Designer software version 7.0; Premier Biosoft International, Palo Alto, CA).
Figure 2.
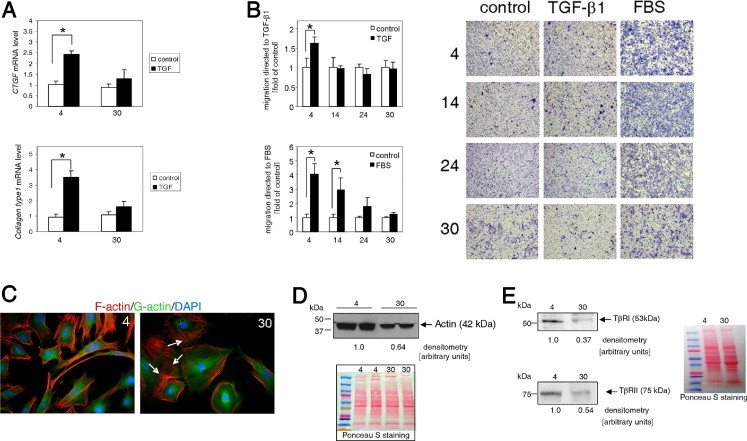
Progenitor cells isolated from aged animals give rise to dysfunctional fibroblasts. A: Cardiac fibroblasts isolated from 4- and 30-month-old mice were serum-starved for 48 hours, then stimulated with 10 ng/mL TGF-β1 for 24 hours, and mRNA levels were measured using qPCR. Expression of the gene of interest was normalized to 18S mRNA level, and untreated samples were used as a calibrator. B: Aged fibroblasts demonstrated reduced chemotaxis toward TGF-β1 and FBS. Cardiac fibroblasts were serum-starved for 24 hours before assay. A sample of 105 cells were placed in Boyden chambers and allowed to migrate through 8-μm pores in response to 10 ng/mL TGF-β1 or 1% FBS. Cells that migrated through the pores were stained, and photographs were obtained (right panel). The absorbed dye was released via extraction buffer. Optical density was measured at 560 nm, and the data were plotted into graphs (left and middle panels). Each experiment was performed in triplicate. Data are given as the mean ± SD of at least three independent experiments. *P < 0.05. C: Polymerized actin fibers were disorganized in fibroblasts derived from 30-month-old animals. F-actin distribution (white arrows) and expression was altered in aged fibroblasts (30 months old) compared with young fibroblasts (4 months old). Polymerized actin (F-actin) was stained using phalloidin–Alexa Fluor 595, and depolymerized actin (G-actin) was visualized using DNase I conjugated with Alexa Fluor 488. D: Western blot analysis of total actin in whole-cell lysates obtained from cardiac fibroblasts. E: Reduced expression of TβRI and TβRII in cardiac fibroblasts. Left panel, Western blot analysis of TβRI and TβRII expression (upper and lower panels, respectively). Right panel, Ponceau S staining of transferred proteins. Cells were derived from animals aged 4, 14, 24, or 30 months.
Primer Sequences
Primer sequences were as follows: Nanog, sense 5′-ATTTGGAGGTGAATTTGG-3′ and antisense 5′-TGTTGCGTAAGTCTCATA-3′; Bmp2, sense 5′-AGAGTGGTTGTCCAATCC-3′ and antisense 5′-GATCAGTAATGTCTGGTTCTTAT-3′; Bmp4, sense 5′-GTCAAGACACCATGATTC-3′ and antisense 5′-GGTCTCAGGTATCAAACT-3′; Dlk1, sense 5′-AAGGTGTCCATGAAAGAGC-3′ and antisense 5′-CAGGTTTCGCACTTGTTG-3′; connective tissue growth factor, sense 5′-ATCCCACCAAAGTGAGAACG-3′ and antisense 5′-TAATTTCCCTCCCCGGTTAC-3′; collagen type 1, sense 5-GTATGCTTGATCTGTATCT-3′ and antisense 5′-CGACTCCTACATCTTCTG-3′; and 18S RNA, sense 5′-ACCGCAGCTAGGAATAATGGA-3′ and antisense 5′-GCCTCAGTTCCGAAAACCA-3′.
Transfection
The constitutively active mutant of TβRI (T202D) in the pRK5-flag vector was created by Feng and Derynck24 and obtained from Addgene, Inc. (Cambridge, MA). pRK5-flag was used as a control vector. Fibroblast cultures were transfected using Fibroblast Transfection Reagent (Altogen Biosystems, Las Vegas, NV) according to the manufacturer's protocol. In brief, 1.5 × 105 cells were cultured in the presence of transfectant-DNA complexes (ratio, 3.34:1) for 72 hours; then functional assays were performed.
Statistical Analysis
Results are given as mean ± SD. A comparison between two groups was made using an unpaired Student's t-test. For multiple-group comparison, one-way analysis of variance was used. Differences were considered statistically significant at P < 0.05.
Densitometry
Densitometry was performed using commercially available software (SCION Image, Alpha version 4.0.3.2; Scion Corp., Frederick, MD).
Results
Aging Affects Multipotency and Lineage Choice of Cardiac Resident MSCs
Nonmyocyte cardiac cells were isolated and cultured in stem cell medium to support the growth of primitive cells in an undifferentiated state (mature or differentiated cells were unable to grow in it) (see Supplemental Figure S1A at http://ajp.amjpathol.org). Expression of markers on these cells was evaluated using immunohistochemistry and immunofluorescence staining. Cardiac MSCs were uniformly positive for CD44 (a hyaluronic acid receptor and marker of MSCs) and markers of undifferentiated embryonic stem cells such as Nanog and Oct3/4 (see Supplemental Figure S1, B and D, respectively. Available at http://ajp.amjpathol.org.).
To evaluate the degree of multipotentiality, cardiac MSCs from young (4 months old) and aged (30 months old) mice were subjected for 21 days to differentiation media supporting the osteogenic, chondrogenic, and adipogenic lineages (see Materials and Methods, Differentiation). Cells isolated from both age groups underwent osteogenic and chondrocytic differentiation to the same degree (Figure 1A, upper and middle panels). However, when stem cells from aged animals were challenged using adipocytic differentiation medium (dexamethasone, 3-isobutyl-1-methylxanthine, and a low concentration of insulin), they dramatically changed their phenotype, became round, and accumulated neutral fat droplets (stained using Oil Red O) (Figure 1A, lower panel). Concomitantly, they expressed perilipin A, the marker of fully mature adipocytes. Progenitor cells isolated from young animals subjected to the same conditions differentiated only marginally, with few cells accumulating small amounts of lipids (Figure 1A). To further corroborate these findings, we determined the expression of the early preadipocytic marker delta-like 1 homolog (Dlk1) in young and aged MSCs cultured in stem cell medium. The Dlk1 level was increased by 40-fold in middle-aged (14 months old) progenitor cells when compared with cells isolated from the young animals (Figure 1B), and by 70-fold in 24- and 30-month-old cells, which suggests that with age progression, cardiac progenitor cells drift toward a preadipocytic phenotype. Also observed was increased bone morphogenic protein 2 (Bmp2) mRNA levels (3.5-fold) in aged MSCs (Figure 1C), consistent with the role of Bmp2 in adipogenesis. Bmp4 and Bmp2 potentially may have a role in initiation of an adipocytic differentiation program25 via paracrine pathways. However, expression of Bmp4 mRNA was reduced by 90% in progenitor cells isolated from aged animals (Figure 1D). This unexpected result suggested that Bmp4 in cardiac MSCs has a different role than Bmp2 does, and rather than favoring adipogenic differentiation, it helps to maintain stem cells in a primitive state. This would be consistent with the reported role of Bmp4 in suppressing lineage commitment and, therefore, keeping murine embryonic stem cells in a pluripotent state.26 To explore further a possible change in the primitive status of these cells with aging, we analyzed the expression of Nanog using qPCR and Western blot analysis. Those studies demonstrated that both mRNA (Figure 1E) and protein concentrations (Figure 1F) were decreased by 80% in cells isolated from 30-month-old animals in comparison with cells derived from 4-month-old mice. The same degree of down-regulation was observed using both techniques, which suggested that Nanog expression was controlled on a transcriptional level in those cells, as has been reported by others.27,28 Because some protein expression [ie, lamin A, GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and actin, all used as loading indicators] is affected by aging (lamin A and GAPDH not shown), we included photographs of membranes stained using Ponceau S with our results of Western blot analysis to ensure equal protein loading and to enable comparison of protein concentrations.
Figure 1.

Alteration of lineage choice and expression of pluripotent markers in aged cardiac resident fibroblast progenitors. A: Cultured progenitor cells isolated from hearts of 4- and 30-month old mice were investigated for multiple lineage differentiation capacity. Calcium deposits in osteocytes were evaluated via alizarin red staining (upper panel). Chondrocytic differentiation was assessed via Alcian Blue staining (middle panel). Adipocytic conversion was visualized using perilipin A (black rim around lipid droplets) and Oil Red O, which stains neutral fat droplets (red) (lower panel). Note the higher degree of adipocytic differentiation in aged progenitor cells. Scale bars: 50 μm (upper and middle panels; 100 μm (lower panel). Right column shows control staining of cells cultured in DMEM/F12 supplemented with 10% FBS. B: Expression of Dlk1 preadipocytic marker in cardiac progenitors isolated from young (4 months old), middle-aged (14 months old), and aged (24 or 30 months old) mice evaluated using qPCR. C: Bmp2 mRNA level is increased in cardiac MSCs isolated from 30-month-old animals. D: Bmp4 mRNA level is reduced in cardiac MSCs isolated from aged animals. Animals were 4, 14, 24, or 30 months of age. Expression of Nanog is reduced in aged cardiac progenitor cells at both the transcriptional (E) and translational levels (F). Right panel in F shows Ponceau S staining of transferred proteins. All reported data were repeated at least three times in independent experiments. *P < 0.05. qPCR data were normalized to 18S.
In addition, we analyzed expression levels of these markers in fibroblast cultures (see Supplemental Table S1 at http://ajp.amjpathol.org) to ensure that these markers are associated with the undifferentiated state. Although Nanog, Dlk1, and Bmp4 were disparately expressed in cells from young and aged MSCs, there was almost no difference in quantity of target mRNA isolated from fibroblast cultures derived from young and aged animals.
Progenitor Cells Isolated from Aged Animals Differentiate into Dysfunctional Fibroblasts
Despite their up-regulation of a preadipocytic marker expression (Figure 1B) and increased adipocytic potential (Figure 1A), aged cardiac MSCs were able to differentiate into fibroblasts (see Supplemental Figure S2A at http://ajp.amjpathol.org) expressing canonical fibroblast markers such as collagen type l (see Supplemental Figure S2B at http://ajp.amjpathol.org) and discoidin domain receptor 2 (see Supplemental Figure S2C at http://ajp.amjpathol.org). Therefore, it was decided to challenge the function of fibroblasts derived from aged animals in a series of tests.
First tested was the ability of cardiac fibroblasts to express connective tissue growth factor (a potent enhancer of extracellular matrix deposition) and collagen type 1 (a member of the extracellular matrix component) in response to TGF-β1. Quiescent cultured cardiac fibroblasts derived from young animals when treated with TGF-β1 for 24 hours demonstrated increased expression of connective tissue growth factor and collagen type 1 by twofold to threefold, in agreement with the findings of others.29,30 In contrast, fibroblasts derived from 30-month-old mice cultured under the same conditions as the young fibroblasts demonstrated no substantial increase in connective tissue growth factor and collagen type 1 mRNA expression in response to TGF-β1 (Figure 2A). It has been previously demonstrated that 24-month-old mice exhibit reduced collagen deposition after MI.7
Next, we compared the directional motility of fibroblasts derived from young and aged mice. Quiescent cells were seeded on a plate insert and allowed to migrate through 8-μm pores in response to 10 ng/mL TGF-β1 or 1% FBS chemoattraction. The migratory capacity of cells derived from young and aged animals was noticeably different. Defective migration toward TGF-β1 was observed as early as age 14 months, although cells from these animals could still migrate toward the more potent chemoattractant FBS (Figure 2B). Fibroblasts derived from 24- and 30-month-old animals demonstrated compromised ability to migrate toward TGF-β1 and FBS. During migration, a cell is required to coordinate polarization, adhesion, and actin polymerization to move the membrane. Therefore, we examined actin structure in fibroblasts derived from young and aged animals. Polymerized actin (F-actin) was labeled using phalloidin, and depolymerized actin (G-actin) was visualized using DNase I. The appearance of cytoplasmic actin filaments correlated with the capacity of those cells to migrate; the young fibroblasts (Figure 2C) exhibited a stronger F-actin signal and generally exhibited long unidirectional actin filaments. In contrast, the cytoskeletal actin of the aged fibroblasts formed shorter filaments, some of them with a nonlinear orientation indicating disorganization. In addition, total actin expression was reduced by approximately 30% in aged fibroblasts (Figure 2D).
Defective directional migration of fibroblasts derived from aged animals may originate from impaired signaling. Compared with fibroblasts from young animals, in aged fibroblasts, expression of TβRI was reduced by approximately 60%, and of TBRII by approximately 50% (Figure 2E).
To further confirm the possibility of a movement defect, we tested the ability of the aged fibroblasts to undergo chemokinesis. They exhibited delayed migration or proliferation as assessed using a scratch-induced wound healing assay (see Supplemental Figure S3 at http://ajp.amjpathol.org). Cells derived from young and aged animals were serum-starved for 24 hours, and a scratch denuded an area of the confluent cell layer. Serum-containing medium was reintroduced, and fibroblast migration was photographed until the wound closed. The young cells closed the gap within 24 hours, middle-aged cells within 48 hours, and 24-month-old cells by 72 hours. Fibroblasts isolated from 30-month-old mice took more than 72 hours to fully close the wound.
Final Step of Fibroblast Maturation Is Impaired by Aging
Fibroblasts derived from young and aged hearts were challenged using myofibroblast functional assays. Contraction of free-floating matrices provided a model for mechanically relaxed tissue comparable to early-stage wound healing, whereas attached matrices simulated processes undergoing in granulation tissue.31
It was discovered that fibroblasts isolated from aged animals were less capable of contracting collagen lattices (Figure 3, A and B). Fibroblasts were cultured in the presence of 10% FBS to enable expression of contractile proteins, and subsequently were seeded in free-floating collagen pads, and the medium was changed to serum-free DMEM. Gel contraction was gradually diminished as donor age increased (Figure 3A), from a mean contraction of 87% (13% area of original size) in young cells to 18% (82% area of original size) in 30-month-old fibroblasts, which translates to a sixfold reduction in contractility. Also examined was contraction of attached collagen matrices populated with fibroblasts isolated from 4- and 30-month-old animals (Figure 3B) (see Materials and Methods, Collagen Pad Assay for detailed protocol). Released pads were incubated in DMEM without any supplement or were supplemented with 10 ng/mL TGF-β1, as indicated. Contraction of released matrices began within 1 hour, and full contraction was observed after 24 hours (as documented). Cells isolated from young animals were much more contractile than cells isolated from 30-month-old mice (Figure 3A and B).
Figure 3.
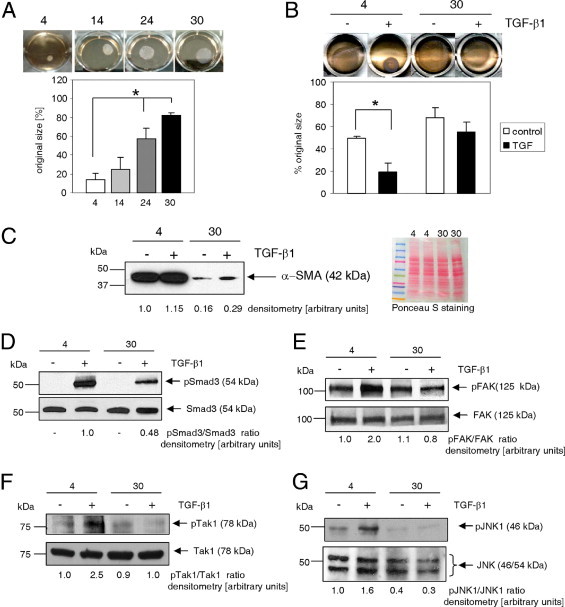
Myofibroblast maturation is impaired by aging. A: Fibroblasts derived from aged animals demonstrated reduced basal contraction of free-floating collagen lattices (see Materials and Methods). B: Cell contractility in response to TGF-β1 was assessed by reduction in the surface area of collagen gel pads. Stressed collagen matrices populated with cardiac fibroblasts isolated from young (4 months old) and aged (30 months old) mice were incubated for 48 hours and then released to initiate contraction into medium containing 10 ng/mL TGF-β1, as indicated. Representative photograph was obtained 24 hours later. C: Left panel, Western blot analysis of α-SMA expression. Compared with cells derived from young (4 months old) mice, fibroblasts derived from aged animals (30 months old) demonstrated reduced basal and TGF-β1–dependent α-SMA expression. Cells were treated with 10 ng/mL TGF-β1 for 72 hours. Right panel, Ponceau S staining of transferred proteins. D: Reduced TGF-β1–dependent Smad3 activation in fibroblasts derived from aged animals. Fibroblasts from young (4 months old) and aged (30 months old) mice were incubated with 10 ng/mL TGF-β1 for 30 minutes. Whole-cell lysates were subjected to Western blot analysis. pSmad3 antibody detects Smad3 phosphorylated at Ser423/Ser425. Fibroblasts isolated from young hearts and aged hearts were serum-starved for 48 hours and then stimulated with 10 ng/mL TGF-β1 for 30 minutes. Activation of FAK (phosphorylation of Tyr576/Tyr577) (E), Tak1 (phosphorylation of Thr184/Thr187) (F), and JNK (phosphorylation of Thr183/Tyr185) (G) was assessed using Western blot analysis. Cardiac fibroblasts were isolated from animals aged 4, 14, 24, or 30 months. Data are given as the mean ± SD of at least three independent experiments. *P < 0.05.
Next evaluated was expression of α-SMA, a myofibroblast marker. Cells isolated from young heart tissue and placed in fibroblast-supporting medium differentiated into fibroblasts and spontaneously matured into myofibroblasts expressing α-SMA. α-SMA was robustly expressed in fibroblasts derived from young animals even without addition of TGF-β1 (Figure 3C), whereas cells isolated from aged animals expressed markedly less (sixfold) α-SMA. TGF-β1 stimulation increased α-SMA expression only marginally in the young cells because of an already high basal level, whereas α-SMA expression in the aged fibroblasts was up-regulated less than twofold, which corresponds to approximately 25% of the protein concentration observed in the young myofibroblasts.
α-SMA expression is transcriptionally regulated by binding of Smad3 to the Smad binding element on the α-SMA promoter32 as a result of activation of TβRs. For Smad3 to be translocated to the nucleus and bind to Smad binding element, it must be phosphorylated, usually on Ser423/Ser425 residues. Thus, we investigated whether aging modulates phosphorylation of Smad3 protein in response to TGF-β1 stimulation. Smad3 proteins were phosphorylated in cells isolated from 4- and 30-month-old animals in response to TGF-β1 exposure. Smad3 phosphorylation in aged fibroblasts was reduced by approximately 50% (Figure 3D).
The importance of TGF-β1 activation of the non–Smad-dependent pathway has been demonstrated by others.33 Moreover, focal adhesion kinase (FAK), Tak1, and c-Jun N-terminal kinase (JNK) are essential for TGF-β–dependent α-SMA expression in fibroblasts and contraction of collagen matrices.34,35 Therefore, we analyzed the activation of these pathways in fibroblasts isolated from 4- and 30-month-old mice (Figure 3, E–G). Phosphorylation of FAK (Tyr576/577), Tak1 (Thr184/Thr187), and JNK (Thr183/Tyr185) in response to TGF-β1 stimulation was impaired in fibroblasts isolated from aged mice. Fibroblasts isolated from young mice responded to TGF-β1 by increasing phosphorylation of these kinases by approximately twofold.
When cultured on dishes treated with standard tissue culture, fibroblasts isolated from young mice demonstrated strong constitutive activation of FAK, Tak1, and JNK (data not shown), which explains their high basal α-SMA expression (Figure 3C). Hence, to document TGF-β1–dependent changes in kinase activation, cells were cultured on bacteriologic (non–tissue culture treated) Petri dishes to ensure lower adhesion (Figure 3, E–G). TGF-β1–dependent α-SMA expression in young fibroblasts cultured on Petri dishes demonstrated an approximately threefold difference in α-SMA expression between control and treated samples (see Supplemental Figure S4 at http://ajp.amjpathol.org).
It was hypothesized that in cells from aged hearts, defective function and poor maturation toward the myofibroblast phenotype result from reduced expression of TβRs. Therefore, we overexpressed a constitutively active TβRI (T202D) mutant in the cells and determined whether that improved their function. Transfection efficiency was assessed using anti-flag antibody staining (Figure 4A). Most cells were transfected; however, the degree to which they expressed flag varied. Overexpression of constitutively active TβRs in fibroblasts failed to restore their ability to contract collagen pads (Figure 4B), although motility of the transfected aged fibroblasts was remedied (Figure 4C) to the level observed in cells isolated from middle-aged mice (compare Figure 2A, lower panel, with Figure 4C).
Figure 4.

Overexpression of constitutively active TβRI (T202D) mutant and its effect on aged cardiac fibroblast function. A: Expression of flag (red immunofluorescence) confirms positive transfection efficiency. Nuclei counterstained with DAPI (blue). B: Contractility of fibroblasts transfected with TβRI (T202D) mutant was not improved in a free-floating collagen pad contraction assay despite positive transfection efficiency as visualized via anti-flag staining (red) of cells populating the collagen pad. C: Migration assay of aged fibroblasts transfected with TβRI (T202D) mutant or control vector (pRK5) toward 1% FBS. Cells were serum-starved for 24 hours before the assay. Cells that migrated through the pores of Boyden's chambers were stained, and photographs were obtained (left panel, representative image). The absorbed dye was released, and o.d. was measured. Data are given as the mean ± SD of three independent experiments. *P < 0.05. Rat IgG2aκ served as a negative control for flag staining. Scale bar = 20 μm.
Rescue of Defective Function Requires AMPK Activation
Defective function of fibroblasts derived from aged mice, as clearly demonstrated in Figures 2 and 3, might have been caused by impaired progenitor cell differentiation. Reduced expression of multipotency markers in aged mesenchymal progenitor cells (Figure 1), elevated expression of a preadipocytic marker (Figure 1B), and increased adipocytic potential (Figure 1A) could result in faulty fibroblast maturation. The aged heart changes its metabolism to glucose consumption, with repressed fatty acid use.36 Perhaps an increase in the availability of fatty acids for storage promotes commitment of stem cells toward the adipocyte lineage. It is not clear whether metabolic changes per se can trigger progenitor cell differentiation; however, AICAR blocks adipogenesis19,37 and augments expression of Nanog.18
To assess whether the AICAR effect on adipogenesis is relevant to fibroblast differentiation, aged progenitor cells (from 30-month-old mice) were cultured in stem cell medium with and without 0.5 mmol/L AICAR for 30 days. Substantial changes were observed in cell shape (Figure 5A) in cultures treated with AICAR, which were accompanied by positive staining for α-SMA (Figure 5B). Cells grown in stem cell medium without AICAR were negative for α-SMA. All cells, regardless of treatment, expressed collagen type I, in agreement with the findings of others38. These results demonstrate that activation of the AMPK pathway forced some progenitor cells to mature into myofibroblasts despite their maintenance in stem cell medium. AICAR did not substantially alter the adipocytic potential of aged MSCs (Figure 5C).
Figure 5.
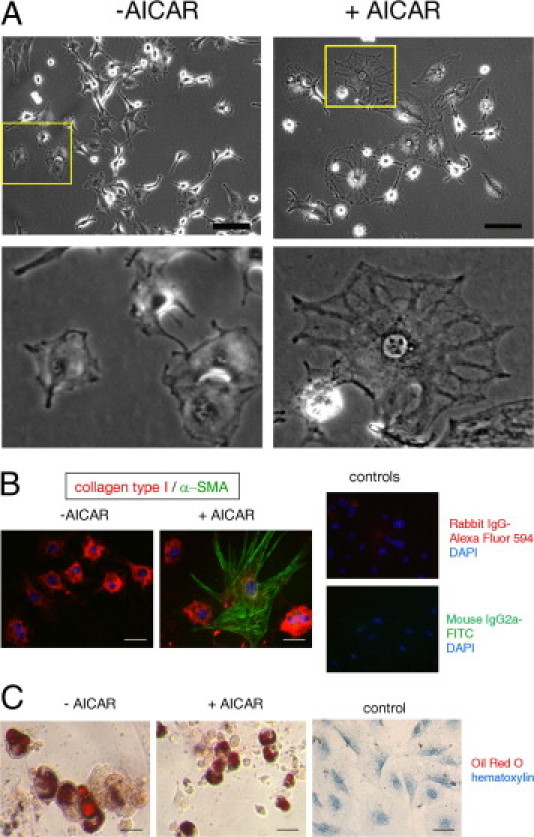
AICAR induces myofibroblast differentiation but does not affect adipogenesis. A: Microscopic images of progenitor cells cultured in stem cell medium in the absence (left panel) and presence (right panel) of 0.5 mmol/L AICAR. Lower panel, Magnified image of the cells marked by yellow squares. Note the change in cell structure. B: lmmunofluorescence detection of progenitor cells untreated (left panel) and treated (right panel) with 0.5 mmol/L AICAR, red stained for collagen type I and green stained for α-SMA. Nuclei were visualized using DAPI stain (blue). Far right panel, IgG controls. C: Adipocytic potential of cardiac stem cells isolated from 30-month-old mice. Cells were incubated for 21 days in differentiation medium in the presence (+AICAR) or absence (−AICAR) of 0.5 mmol/L AICAR. All cells used for those experiments were isolated from 30-month-old mice. Right panel, Cells incubated in control medium. Scale bar = 50 μm.
To assess the functional consequences of the AICAR effects on cell phenotype, fibroblast cultures isolated from 30-month-old animals were treated with AICAR for 30 days to enable progenitor cells to differentiate, and then subsequently were treated with TGF-β1 for 3 days to amplify myofibroblast maturation. Cells were then seeded into collagen matrices. AICAR enhanced TGF-β1–mediated collagen lattice contraction by approximately 40% (Figure 6A). Compound C, an AMPK inhibitor, completely blocked TGF-β1/AICAR-dependent contractility, which suggested that this phenomenon is mediated via AMPK. Moreover, TGF-β1 was necessary for AICAR-dependent contraction; contractility of aged fibroblasts was not improved using AICAR alone. Concomitantly, α-SMA expression was up-regulated by twofold in cells treated with AICAR compared with age-matched untreated cells, and was further enhanced by fivefold after TGF-β1 treatment (Figure 6B). AICAR treatments, both a short 1-hour exposure (Figure 6C) and a long-term application (Figure 6D), resulted in phosphorylation of the AMPK subunit on Thr172, which corresponds to its activation. AICAR application did not affect total AMPK expression. TGF-β1 increased AICAR-mediated AMPK phosphorylation by approximately 60% (Figure 6D). Phosphorylation of AMPK due to metformin, another AMPK activator, was observed after 24 hours of treatment (see Supplemental Figure S5 at http://ajp.amjpathol.org), which clearly implies an indirect mechanism of activation, in agreement with the findings of others,39,40 and contribution of different pathways.
Figure 6.

AMPK activation partially rescues defective aged myofibroblast function. A: AMPK activation enhances collagen lattice contraction. Cells were cultured for 30 days in the presence of 0.5 mmol/L AICAR. At 72 hours before assay, cells were passaged, and medium was supplemented with 10 ng/mL TGF-β1 and 20 μmol/L Compound C, as indicated. Free-floating collagen matrices were incubated in serum-free medium for 24 hours. B: AICAR and TGF-β1 treatment increased α-SMA expression in aged fibroblasts. Cells were cultured for 30 days in the presence of 0.5 mmol/L AICAR. At 72 hours before assay, cells were passaged, and medium was supplemented with 10 ng/mL TGF-β1. Lower panel, Ponceau S staining of proteins transferred into nitrocellulose membrane. C: Cells were treated with 0.5 mmol/L AICAR for 1 hour, which promoted phosphorylation of AMPKα subunit at Thr172. D: TGF-β1 enhanced AICAR-dependent AMPKα phosphorylation (Thr172). Cells were exposed to 0.5 mmol/L AICAR for 30 days, then treated with 10 ng/mL TGF-β1 for 30 minutes. E: AICAR had no effect on Smad3 phosphorylation. Cells were treated with 0.5 mmol/L AICAR for 30 days, and 10 ng/mL TGF-β1 was applied for 30 minutes. F: TGF-β1–dependent AMPK activation relies on activation of FAK, Tak1, and JNK but not p38MAPK. Fibroblasts were serum-starved for 48 hours, then pretreated for 30 minutes with various kinase inhibitors. Subsequently, 10 ng/mL TGF-β1 was applied for 30 minutes, and cells were harvested. CC, Compound C (20 μmol/L); PP2 (10 μmol/L); SB, SB203580 (20 μmol/L); SP6, SP6000125 (25 μmol/L); T, TGF-β1 (10 ng/mL); 5Z, (5Z)–7-oxozeaenol (100 nmol/L).
α-SMA is transcriptionally regulated by Smads.32 It was hypothesized that AMPK activation increased phosphorylation of Smad3 and, therefore, contributed to its up-regulated expression. To address that issue, we examined the level of pSmad3 and Smad3 using Western blot analysis on samples from the aged fibroblast cultures treated with and without AICAR in the presence of TGF-β1 (Figure 6E). No substantial changes in Smad3 phosphorylation due to AICAR treatment were observed, which suggests that the AMPK-dependent increase in α-SMA expression does not rely on Smad3 phosphorylation.
Inasmuch as it has been confirmed that TGF-β1 activates AMPK in cardiac fibroblasts, we examined phosphorylation of AMPK in response to TGF-β1 in the presence of various kinase inhibitors. Cardiac fibroblasts were pretreated with PP2 (inhibitor of Src kinase that acts upstream of FAK), SB203580 (p38MAPK inhibitor), SP600125 (JNK inhibitor), and (5Z)–7-oxozeaenol (Tak1 inhibitor) before TGF-β1 stimulation. AMPK phosphorylation was evaluated using Western blot analysis (Figure 6F). Inhibition of FAK, Tak1, or JNK resulted in reduced activation of AMPK. p38MAPK inhibition had no effect on TGF-β1–dependent AMPK phosphorylation.
Several reports have linked AMPK and p38MAPK as downstream signaling molecules.41 p38MAPK is involved in controlling α-SMA expression.42 We evaluated TGF-β1–dependent phosphorylation of p38MAPK in fibroblasts isolated from 4- and 30-month-old mice. Twofold increase of p38MAPK phosphorylation on Thr180/Thr182 residues in fibroblasts from 4-month-old animals in response to TGF-β1 was observed. TGF-β1 failed to activate p38MAPK in fibroblasts isolated from aged mice (Figure 7A). However, when subjected to TGF-β1/AICAR treatment, fibroblasts derived from aged animals demonstrated up-regulated p38MAPK phosphorylation (Figure 7B). Compound C prevented AICAR-induced increased phosphorylation of p38MAPK, which indicates that p38MAPK activation is dependent on AMPK.
Figure 7.
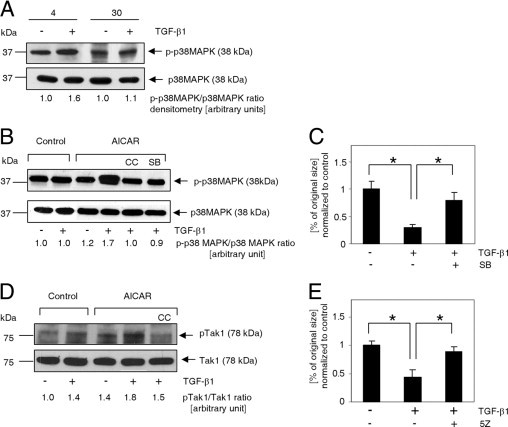
p38MAPK and Tak1 kinases are involved in TGF-β1/AICAR-dependent signal amplification. A: Immunoblot analysis of p38MAPK phosphorylation (Thr180/Thr182) in response to 30 minutes of activation with 10 ng/mL TGF-β1 in cardiac fibroblasts isolated from young (4 months old) and aged (30 months old) mice. B: p38MAPK activation depends on synergistic action of TGF-β1 and AICAR. Cells were exposed to AICAR (0.5 mmol/L) or TGF-β1 (10 ng/mL) for 1 hour. C: p38MAPK inhibition reduces collagen lattice contraction (free-floating collagen gel model). Cells were cultured in the presence of 0.5 mmol/L AICAR. At 72 hours before assay, cells were passaged, and the medium was supplemented with 10 ng/mL TGF-β1 and 20 μmol/L SB203580, as indicated. Collagen disks were incubated in serum-free medium for 24 hours. D: Tak1 phosphorylation (Thr184/Thr187) was enhanced via simultaneous application of AICAR and TGF-β1. E: Tak1 inhibition eradicated the ability of myofibroblasts to contract a collagen pad. Cells were cultured in the presence of 0.5 mmol/L AICAR. At 72 hours before assay, cells were passaged, and medium was supplemented with 10 ng/mL TGF-β1 and 100 nmol/L (5Z)−7-oxozeaenol, as indicated. Collagen matrices were incubated for 24 hours under serum-free conditions. B–E: Cells were derived only from 30-month-old animals. For Western blot analysis, cells were pretreated with inhibitors for 30 minutes before application of AICAR or TGF-β1. For collagen pad contraction, the medium was supplemented with inhibitors every 24 hours. CC, Compound C; SB, SB203580; 5Z, (5Z)−7-oxozeaenol.
To further corroborate the role of p38MAPK in the TGF-β1/AMPK axis, collagen lattices were populated with cells incubated with the p38MAPK inhibitor (SB203580). Blockade of p38MAPK phosphorylation resulted in substantially reduced TGF-β1/AICAR-dependent contraction of free-floating collagen matrices (Figure 7C).
To further elucidate this important signaling pathway, we wished to identify the relevant AMPK kinase. AMPK activation was originally proposed to be dependent on Thr172 phosphorylation via action of upstream kinases including LKB1,43,44 calmodulin-dependent protein kinase kinase IIβ,45 and Tak1.46 However, Xie et al47 suggested that LKB1 actually acts downstream of Tak1 in mouse fibroblasts. Because TGF-β1 seems to be crucial in AMPK-dependent p38MAPK activation and because it is part of the FAK/Tak1/JNK pathway crucial for myofibroblast activation,35 we chose to analyze Tak1 activation in aged fibroblasts treated with TGF-β1 and AICAR. Tak1 phosphorylation on Thr184 and Thr187 residues has been linked to its activation.48 Tak1 phosphorylation was slightly increased by TGF-β1 or AICAR. However, AICAR/TGF-β1 combined treatment nearly doubled the phosphorylation of Tak1 when compared with the untreated control (Figure 7D). Cells pretreated with an AMPK inhibitor and then stimulated with AICAR/TGF-β1 demonstrated reduced Tak1 phosphorylation (a level comparable with that with TGF-β1 stimulation alone), which is evidence that AMPK is involved in Tak1 activation, possibly via a positive feedback loop, which is consistent with the data of Xie et al.47
To further corroborate involvement of Tak1 in AICAR/TGF-β1–mediated signal amplification, we pretreated aged fibroblasts with (5Z)-7-oxozeaenol, a Tak1 inhibitor, and stimulated the cells with AICAR and TGF-β1 for 3 days. A free-floating collagen pad contraction assay revealed almost complete inhibition of contraction in pads populated with cells treated with (5Z)-7-oxozeaenol (Figure 7E).
Discussion
We have previously demonstrated that aged mice form inadequate scars after MI.6,7 Mesenchymal progenitor cells have been identified as a potential fibroblast source,8,10 and recent evidence suggests they may contribute to cardiac tissue repair by differentiating to scar-forming myofibroblasts.49 As demonstrated herein, those cardiac MSCs are positive for CD44, Nanog, and Oct3/4, and are multipotential. A decrease in Nanog expression was observed in cells isolated from aged mice. The mechanism by which Nanog maintains stemness remains unclear; however, it has been hypothesized that Nanog regulates the expression of genes critical to stem cell renewal and differentiation. Nanog and Oct3/4 associate with histone deacetylase in repression complexes by which they control gene transcription and prevent differentiation.50 In contrast, methylation (down-regulation) of Oct3/4 and Nanog promoters was demonstrated in embryonic stem cells on differentiation.51
Bmp2 expression is increased in aged MSCs, consistent with the role of this protein in adipogenesis and with the preadipocytic phenotype of the aged cells. In contrast, Bmp4 levels decreased, which may indicate that it has no role in preadipocytic commitment by the MSCs. Conversely, Bmp4 suppresses neuronal differentiation and sustains murine embryonic stem cell self-renewal via a mechanism involving increased expression of ld and Nanog proteins.26 Bmp4 is a secreted protein, and, therefore, it may act via a paracrine mechanism on neighboring cells within the stem cell niche. Reduced expression of Bmp4 mRNA in the aged stem cells may contribute to escape from their normal self-renewal fate and the promotion of lineage commitment, which results in diminished Nanog and perhaps increased Dlk1 expression. Dlk1 is a modulator of adipogenesis.52,53 Different isoforms of Dlk1 may have opposite roles in the control of adipogenesis; however, it is agreed that Dlk1 is elevated in the preadipocytic state. With age progression, an adipogenic program overcomes osteogenesis54 (rat marrow cells) and myogenesis55 (murine myoblasts). Herein, we have presented evidence that the stem cells derived from aged hearts have an enhanced tendency to undergo adipocytic differentiation when subjected to a relatively low dosage of insulin (see Materials and Methods, Differentiation). Higher insulin concentrations forced cells derived from young mice into adipogenesis (data not shown), which suggests that aged stem cells might be more prone to insulin signaling due to changes in metabolism or activatory pathways. Recently, a new population of adipocyte and fibroblast progenitor cells residing in muscle has been identified.56 Those cells proliferate and differentiate in response to muscle damage, and their existence may explain the sarcopenia observed in elderly and obese individuals.57 Perhaps the altered lineage choice observed in the cardiac MSCs isolated from aged animals results from a similar mechanism, a common fibroblast or adipocyte progenitor that preferentially gives rise to adipocytes in older age and exhibits compromised maturation toward the myofibroblast phenotype. Because AICAR reduces adipogenesis in preadipocytes,19,37 it was assumed that by applying AICAR together with adipocytic differentiation medium we would reduce the formation of adipocytes in the aged stem cell culture. However, AICAR did not reduce the adipocytic potential of the aged stem cells (Figure 5C). Perhaps AICAR exerts its action by promoting the lineage choice of already committed cells. AICAR-enhanced differentiation of progenitor cells placed in specific differentiation medium has been reported.58,59 Some percentage of stem cells isolated from aged hearts, when subjected to AICAR in serum-free medium, differentiated into myofibroblasts (Figure 5B), which suggests that AICAR activation can bypass serum-dependent pathways necessary for myofibroblast maturation.
In cardiac wound healing, myofibroblasts migrate toward the site of injury and contract and stabilize the scar under the influence of TGF-β. We have modeled these two functions in vitro using two assays: directed migration in response to TGF-β1 and contraction of a collagen pad under the influence of TGF-β1. Cells from aged animals were defective in both functions.
Signaling of TGF-β1 is mediated by a complex of two types of TβRs, both of which possess serine and threonine kinase activity. Binding of TGF-β1 to the receptor complex can promote several scenarios. Ligand activation of TβRII receptor kinase leads to phosphorylation and, thereby, to activation of TβRI, which then activates and phosphorylates Smad2 and Smad3 proteins, which are subsequently moved into the nucleus, where they associate with other transcription factors and activate transcription of target genes60,61 (Smad-dependent pathway), and activates Smad-independent signals including GTPase Ras and ERK62,63, promotes JNK phosphorylation via FAK and Tak1,34,35 and p38MAPK phosphorylation mediated by Fyn.42 Up-regulation of all of these pathways has been associated with fibroblast and myofibroblast activation.32,34,35,42,63 There seems to be signal redundancy, in which both Smad-dependent and Smad-independent pathways are involved in TGF-β1–mediated responses in cardiac fibroblasts; however, the key signal transducers are TβRs. As shown in Figure 2E, when compared with fibroblasts isolated from young mice, the fibroblasts generated from aged cardiac MSCs exhibit reduced TβRI and TβRII expression and, therefore, demonstrate reduced responsiveness to TGF-β1, which translates into decreased phosphorylation of Smad3, FAK, Tak1, JNK, and p38MAPK (Figure 3, D–G, and Figure 7A). Consequently, myofibroblasts from aged heart express less α-SMA (Figure 3C), in agreement with the findings of Bujak et al,7 who demonstrated decreased myofibroblast accumulation in the aged infarcted myocardium. Even with a noticeable lack of FAK, Tak1, p38MAPK, and JNK phosphorylation in response to 30 minutes of stimulation with TGF-β1 observed in fibroblasts isolated from aged mice (Figure 3, E–G, and Figure 7A), the pharmacologic inhibition of Tak1, p38MAPK, or JNK resulted in abolished TGF-β1–mediated contraction of free-floating collagen pads (Figure 7, C and E; see also Supplemental Figure S6 at http://ajp.amjpathol.org), which suggests that these pathways are still important for fibroblasts isolated from aged mice, and perhaps their activation kinetics is different from that in fibroblasts isolated from young mice. It is important to note that small differences observed in response to TGF-β1 in aged cells (Figure 3F versus Figure 7D) and lower basal phosphorylation of JNK (Figure 3G) in cells isolated from young versus aged mice were probably caused by culturing these cells under low-adhesion conditions. It was hypothesized that fibroblasts isolated from aged animals are more sensitive to surface changes due to reduced integrin expression (unpublished observation).
It became obvious that one of the reasons for the dysfunction of fibroblasts isolated from aged mice was insufficient TGF-β1–dependent signal transduction caused by reduced expression of TβRs. To remedy that defect, we overexpressed constitutively active TβRI. Overexpression of TβRs is efficacious in colon and pancreatic cancer cells.64,65 Herein, we have demonstrated that overexpression of a constitutively active TβRI mutant improved the motility of aged cardiac fibroblasts but not their ability to contract collagen pads (Figure 4). Myofibroblast maturation and migration are the opposite spectra of TβR-mediated responses; expression of α-SMA increases fibroblast contractile activity66 and decreases fibroblast motility.67 Clearly, overexpression of TβRI promoted the actin cytoskeleton reorganization required for directed migration68 and, therefore, partially rescued the functioning of aged fibroblasts. However, it was not sufficient to reconstitute defective myofibroblast functions and matrix contraction.
Thus, we amplified the existing signals downstream of TβRs, which was achieved by up-regulating AMPK. AICAR had a profound effect on TGF-β1–induced maturation in aged myofibroblasts but did not rescue directed migration (see Supplemental Figure S7 at http://ajp.amjpathol.org). Our results demonstrate that application of AICAR increased phosphorylation of AMPK (Figure 6C), although it did not substantially improve myofibroblast maturation (Figure 6, A and B). However, when AICAR was combined with TGF-β1, α-SMA expression was clearly up-regulated and pad contraction was improved. The data revealed that AICAR/TGF-β1 treatment did not affect either Smad3 phosphorylation (Figure 6F) or expression of TβRs (data not shown).
On the basis of the experiments presented herein, we propose a unique mechanism that sensitizes the aged fibroblasts to TGF-β1 through a non-canonical pathway (Figure 8, schema), which has a direct clinical implication for older patients experiencing MI, because the MI rate increases in the aged population but the reparative capacity of the aged heart is severely compromised.69 AMPK protects against post-ischemic cardiac dysfunction due to its role in glucose uptake70; however, we are not aware of any study that evaluated the role of AMPK in post-MI scar formation. Therefore, this in vitro study sets the foundation for possible new therapeutic interventions for recovery after MI.
Figure 8.

Rescue of the aged fibroblast to myofibroblast differentiation by amplification of TGF-β signaling via the Tak1/AMPK/p38MAPK pathway. Stimulation of cardiac fibroblasts derived from young animals (Young) with TGF-β1 resulted in phosphorylation of Smad3 and increased expression of α-SMA (left panel). Cardiac fibroblasts derived from 30-month-old animals (Aged) demonstrated reduced expression of both TβRI and TβRII, which resulted in decreased signal transduction and diminished α-SMA expression (middle panel). AICAR/TGF-β1 synergistically activated Tak1, AMPK, and p38MAPK, resulting in up-regulation of α-SMA expression (right panel). Large and small font sizes indicate, respectively, up-regulation or reduction of expression.
Acknowledgment
We thank Dorellyn Lee for technical assistance.
Footnotes
Supported by grants R01HL076661 and R01HL089792 from the National Institutes of Health and by the Medallion Foundation and the Hankamer Foundation.
Supplemental material for this article can be found on http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.06.022.
Supplementary data
Cultured cardiac MSC characteristics. A: Brightfield image. B: CD44 staining using immunofluorescence (red). Nuclei were counterstained using DAPI (blue). Expression of Nanog (C) and Oct3/4 (D) in undifferentiated stem cells was analyzed using immunohistochemistry (IHC). Right panels are appropriate IgG controls. Scale bars: 50 μm (A, C, and D); 25 μm (B).
Progenitor cells differentiate into fibroblasts regardless of age. A: Brightfield image of young (4 months old) and aged (30 months old) fibroblasts derived from progenitor cells cultured in DMEM/F12 supplemented with 10% FBS. Immunohistochemical staining of fibroblast markers collagen type l (B) and DDR2 (C). Each image is representative of three independent experiments. Scale bar = 25μm.
Fibroblasts derived from aged animals exhibit impaired chemokinesis in a wound-healing assay. Photomicrographs demonstrate the process of wound repair by cardiac fibroblasts isolated from animals aged 4, 14, 24, or 30 months. Cells were grown to confluence in DMEM containing 10% FBS. Then the cell layer was wounded (example represents 0 hour). Wound closure was monitored every 24 hours. All data are representative of four experiments.
TGF-β1–induced α-SMA expression in young fibroblasts cultured under low-adhesion conditions in Petri dishes. Cells were serum-starved for 48 hours, then incubated with 10 ng/mL TGF-β1 for 72 hours, as indicated. Right panel, Ponceau S staining.
Effect of metformin on AMPK activation. Serum-starved fibroblast cultures were incubated with 2.5 mmol/L metformin for 1 and 24 hours, as indicated.
JNK inhibition eradicates TGF-β1–dependent collagen pad contraction. Fibroblasts isolated from 30-month-old mice were incubated for 3 days in the presence of 10 ng/mL TGF-β1 (T) with or without 25 μmol/L SP600125 (SP6), and then were seeded in collagen pads. Contraction of free-floating collagen matrices was evaluated 24 hours later.
AICAR does not enhance migration capability of aged fibroblasts. Cells were incubated with AICAR for 3 days, then serum-starved in the presence of AICAR. Cells were placed in Boyden's chambers and allowed to migrate in response to 10 ng/mL TGF-β1. As a positive control, cells isolated from 4-month-old animals were used. Data are given as mean ± SD of three independent experiments. *P < 0.05.
References
- 1.Camelliti P., Borg T.K., Kohl P. Structural and functional characterization of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 2.Carlson S., Trial J., Soeller C., Entman M.L. Cardiac mesenchymal stem cells contribute to scar formation after myocardial infarction. Cardiovasc Res. 2011;91:99–107. doi: 10.1093/cvr/cvr061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jugdutt B.I. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1395–1403. doi: 10.1161/01.CIR.0000085658.98621.49. [DOI] [PubMed] [Google Scholar]
- 4.Dobaczewski M., Bujak M., Zymek P., Ren G., Entman M.L., Frangogiannis N.G. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324:475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 5.Dewald O., Ren G., Duerr G.D., Zoerlein M., Klemm C., Gersch C., Tincey S., Michael L.H., Entman M.L., Frangogiannis N.G. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–677. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gould K.E., Taffet G.E., Michael L.H., Christie R.M., Konkol D.L., Pocius J.S., Zachariah J.P., Chaupin D.F., Daniel S.L., Sandusky G.E., Jr, Hartley C.J., Entman M.L. Heart failure and greater infarct expansion in middle-aged mice: a relevant model for postinfarction failure. Am J Physiol Heart Circ Physiol. 2002;282:H615–H621. doi: 10.1152/ajpheart.00206.2001. [DOI] [PubMed] [Google Scholar]
- 7.Bujak M., Kweon H.J., Chatila K., Li N., Taffet G., Frangogiannis N.G. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–1392. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beltrami A.P., Cesselli D., Bergamin N., Marcon P., Rigo S., Puppato E., D'Aurizio F., Verardo R., Piazza S., Pignatelli A., Poz A., Baccarani U., Damiani D., Fanin R., Mariuzzi L., Finato N., Masolini P., Burelli S., Belluzzi O., Schneider C., Beltrami C.A. Multipotent cells can be generated in vitro from several adult human organs (heart, liver, and bone marrow) Blood. 2007;110:3438–3446. doi: 10.1182/blood-2006-11-055566. [DOI] [PubMed] [Google Scholar]
- 9.Pittenger M.F., Mackay A.M., Beck S.C., Jaiswal R.K., Douglas R., Mosca J.D., Moorman M.A., Simonetti D.W., Craig S., Marshak D.R. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 10.da Silva Meirelles L., Chagastelles P.C., Nardi N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. 2006;119:2204–2213. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 11.Sarugaser R., Hanoun L., Keating A., Stanford W.L., Davies J.E. Human mesenchymal stem cells self-renew and differentiate according to a deterministic hierarchy. PLoS One. 2009;4:e6498. doi: 10.1371/journal.pone.0006498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Ippolito G., Schiller P.C., Ricordi C., Roos B.A., Howard G.A. Age-related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J Bone Miner Res. 1999;14:1115–1122. doi: 10.1359/jbmr.1999.14.7.1115. [DOI] [PubMed] [Google Scholar]
- 13.Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R., Slukvin I.I., Thomson J.A. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 15.Silva J., Nichols J., Theunissen T.W., Guo G., van Oosten A.L., Barrandon O., Wray J., Yamanaka S., Chambers I., Smith A. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138:722–737. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choy L., Derynck R. Transforming growth factor-beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J Biol Chem. 2003;278:9609–9619. doi: 10.1074/jbc.M212259200. [DOI] [PubMed] [Google Scholar]
- 17.Sabina R.L., Patterson D., Holmes E.W. 5-Amino-4-imidazolecarboxamide riboside (Z-riboside) metabolism in eukaryotic cells. J Biol Chem. 1985;260:6107–6114. [PubMed] [Google Scholar]
- 18.Adamo L., Zhang Y., Garcia-Cardena G. AICAR activates the pluripotency transcriptional network in embryonic stem cells and induces KLF4 and KLF2 expression in fibroblasts. BMC Pharmacol. 2009;9:2. doi: 10.1186/1471-2210-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Habinowski S.A., Witters L.A. The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem Biophys Res Commun. 2001;286:852–856. doi: 10.1006/bbrc.2001.5484. [DOI] [PubMed] [Google Scholar]
- 20.Shih D.T., Lee D.C., Chen S.C., Tsai R.Y., Huang C.T., Tsai C.C., Shen E.Y., Chiu W.T. Isolation and characterization of neurogenic mesenchymal stem cells in human scalp tissue. Stem Cells. 2005;23:1012–1020. doi: 10.1634/stemcells.2004-0125. [DOI] [PubMed] [Google Scholar]
- 21.Song L., Webb N.E., Song Y., Tuan R.S. Identification and functional analysis of candidate genes regulating mesenchymal stem cell self-renewal and multipotency. Stem Cells. 2006;24:1707–1718. doi: 10.1634/stemcells.2005-0604. [DOI] [PubMed] [Google Scholar]
- 22.Rosenfeldt H., Lee D.J., Grinnell F. Increased c-fos mRNA expression by human fibroblasts contracting stressed collagen matrices. Mol Cell Biol. 1998;18:2659–2667. doi: 10.1128/mcb.18.5.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyden S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J Exp Med. 1962;115:453–466. doi: 10.1084/jem.115.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng X.H., Derynck R. Ligand-independent activation of transforming growth factor (TGF) beta signaling pathways by heteromeric cytoplasmic domains of TGF-beta receptors. J Biol Chem. 1996;271:13123–13129. doi: 10.1074/jbc.271.22.13123. [DOI] [PubMed] [Google Scholar]
- 25.Huang H., Song T.J., Li X., Hu L., He Q., Liu M., Lane M.D., Tang Q.Q. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci USA. 2009;106:12670–12675. doi: 10.1073/pnas.0906266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ying Q.L., Nichols J., Chambers I., Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- 27.Kuroda T., Tada M., Kubota H., Kimura H., Hatano S.Y., Suemori H., Nakatsuji N., Tada T. Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol. 2005;25:2475–2485. doi: 10.1128/MCB.25.6.2475-2485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodda D.J., Chew J.L., Lim L.H., Loh Y.H., Wang B., Ng H.H., Robson P. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem. 2005;280:24731–24737. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- 29.Eghbali M., Tomek R., Sukhatme V.P., Woods C., Bhambi B. Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts: regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res. 1991;69:483–490. doi: 10.1161/01.res.69.2.483. [DOI] [PubMed] [Google Scholar]
- 30.Chen M.M., Lam A., Abraham J.A., Schreiner G.F., Joly A.H. CTGF expression is induced by TGF-beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32:1805–1819. doi: 10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- 31.Delvoye P., Wiliquet P., Leveque J.L., Nusgens B.V., Lapiere C.M. Measurement of mechanical forces generated by skin fibroblasts embedded in a three-dimensional collagen gel. J Invest Dermatol. 1991;97:898–902. doi: 10.1111/1523-1747.ep12491651. [DOI] [PubMed] [Google Scholar]
- 32.Hu B., Wu Z., Phan S.H. Smad3 mediates transforming growth factor-beta–induced alpha–smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- 33.Yu L., Hebert M.C., Zhang Y.E. TGF-beta receptor–activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S., Xu S.W., Kennedy L., Pala D., Chen Y., Eastwood M., Carter D.E., Black C.M., Abraham D.J., Leask A. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell. 2007;18:2169–2178. doi: 10.1091/mbc.E06-12-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi-wen X., Parapuram S.K., Pala D., Chen Y., Carter D.E., Eastwood M., Denton C.P., Abraham D.J., Leask A. Requirement of transforming growth factor beta–activated kinase 1 for transforming growth factor beta–induced alpha–smooth muscle actin expression and extracellular matrix contraction in fibroblasts. Arthritis Rheum. 2009;60:234–241. doi: 10.1002/art.24223. [DOI] [PubMed] [Google Scholar]
- 36.Park S.K., Prolla T.A. Gene expression profiling studies of aging in cardiac and skeletal muscles. Cardiovasc Res. 2005;66:205–212. doi: 10.1016/j.cardiores.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Giri S., Rattan R., Haq E., Khan M., Yasmin R., Won J.S., Key L., Singh A.K., Singh I. AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr Metab (Lond) 2006;3:31. doi: 10.1186/1743-7075-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ku C.H., Johnson P.H., Batten P., Sarathchandra P., Chambers R.C., Taylor P.M., Yacoub M.H., Chester A.H. Collagen synthesis by mesenchymal stem cells and aortic valve interstitial cells in response to mechanical stretch. Cardiovasc Res. 2006;71:548–556. doi: 10.1016/j.cardiores.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 39.Fryer L.G., Parbu-Patel A., Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 40.Hawley S.A., Gadalla A.E., Olsen G.S., Hardie D.G. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 41.Du J.H., Xu N., Song Y., Xu M., Lu Z.Z., Han C., Zhang Y.Y. AICAR stimulates IL-6 production via p38 MAPK in cardiac fibroblasts in adult mice: a possible role for AMPK. Biochem Biophys Res Commun. 2005;337:1139–1144. doi: 10.1016/j.bbrc.2005.09.174. [DOI] [PubMed] [Google Scholar]
- 42.Vepachedu R., Gorska M.M., Singhania N., Cosgrove G.P., Brown K.K., Alam R. Unc119 regulates myofibroblast differentiation through the activation of Fyn and the p38 MAPK pathway. J Immunol. 2007;179:682–690. doi: 10.4049/jimmunol.179.1.682. [DOI] [PubMed] [Google Scholar]
- 43.Shaw R.J., Kosmatka M., Bardeesy N., Hurley R.L., Witters L.A., DePinho R.A., Cantley L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakamoto K., Zarrinpashneh E., Budas G.R., Pouleur A.C., Dutta A., Prescott A.R., Vanoverschelde J.L., Ashworth A., Jovanovic A., Alessi D.R., Bertrand L. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab. 2006;290:E780–E788. doi: 10.1152/ajpendo.00443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hurley R.L., Anderson K.A., Franzone J.M., Kemp B.E., Means A.R., Witters L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 46.Momcilovic M., Hong S.P., Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 47.Xie M., Zhang D., Dyck J.R., Li Y., Zhang H., Morishima M., Mann D.L., Taffet G.E., Baldini A., Khoury D.S., Schneider M.D. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci USA. 2006;103:17378–17383. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim S.I., Kwak J.H., Na H.J., Kim J.K., Ding Y., Choi M.E. Transforming growth factor-beta (TGF-beta1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-beta receptor kinase activity in mesangial cells. J Biol Chem. 2009;284:22285–22296. doi: 10.1074/jbc.M109.007146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu G.D., Bowdish M.E., Jin Y.S., Zhu H., Mitsuhashi N., Barsky L.W., Barr M.L. Contribution of mesenchymal progenitor cells to tissue repair in rat cardiac allografts undergoing chronic rejection. J Heart Lung Transplant. 2005;24:2160–2169. doi: 10.1016/j.healun.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 50.Liang J., Wan M., Zhang Y., Gu P., Xin H., Jung S.Y., Qin J., Wong J., Cooney A.J., Liu D., Songyang Z. Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat Cell Biol. 2008;10:731–739. doi: 10.1038/ncb1736. [DOI] [PubMed] [Google Scholar]
- 51.Li J.Y., Pu M.T., Hirasawa R., Li B.Z., Huang Y.N., Zeng R., Jing N.H., Chen T., Li E., Sasaki H., Xu G.L. Synergistic function of DNA methyltransferases Dnmt3a and Dnmt3b in the methylation of Oct4 and Nanog. Mol Cell Biol. 2007;27:8748–8759. doi: 10.1128/MCB.01380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smas C.M., Sul H.S. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell. 1993;73:725–734. doi: 10.1016/0092-8674(93)90252-l. [DOI] [PubMed] [Google Scholar]
- 53.Nueda M.L., Baladron V., Sanchez-Solana B., Ballesteros M.A., Laborda J. The EGF-like protein dlk1 inhibits notch signaling and potentiates adipogenesis of mesenchymal cells. J Mol Biol. 2007;367:1281–1293. doi: 10.1016/j.jmb.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 54.Beresford J.N., Bennett J.H., Devlin C., Leboy P.S., Owen M.E. Evidence for an inverse relationship between the differentiation of adipocytic and osteogenic cells in rat marrow stromal cell cultures. J Cell Sci. 1992;102(Pt 2):341–351. doi: 10.1242/jcs.102.2.341. [DOI] [PubMed] [Google Scholar]
- 55.Taylor-Jones J.M., McGehee R.E., Rando T.A., Lecka-Czernik B., Lipschitz D.A., Peterson C.A. Activation of an adipogenic program in adult myoblasts with age. Mech Ageing Dev. 2002;123:649–661. doi: 10.1016/s0047-6374(01)00411-0. [DOI] [PubMed] [Google Scholar]
- 56.Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM: Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12:153–163 [DOI] [PMC free article] [PubMed]
- 57.Vettor R., Milan G., Franzin C., Sanna M., De Coppi P., Rizzuto R., Federspil G. The origin of intermuscular adipose tissue and its pathophysiological implications. Am J Physiol Endocrinol Metab. 2009;297:E987–E998. doi: 10.1152/ajpendo.00229.2009. [DOI] [PubMed] [Google Scholar]
- 58.Zang Y., Yu L.F., Pang T., Fang L.P., Feng X., Wen T.Q., Nan F.J., Feng L.Y., Li J. AICAR induces astroglial differentiation of neural stem cells via activating the JAK/STAT3 pathway independently of AMP-activated protein kinase. J Biol Chem. 2008;283:6201–6208. doi: 10.1074/jbc.M708619200. [DOI] [PubMed] [Google Scholar]
- 59.Li X., Han Y., Pang W., Li C., Xie X., Shyy J.Y., Zhu Y. AMP-activated protein kinase promotes the differentiation of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1789–1795. doi: 10.1161/ATVBAHA.108.172452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y., Derynck R. Regulation of Smad signalling by protein associations and signalling crosstalk. Trends Cell Biol. 1999;9:274–279. doi: 10.1016/s0962-8924(99)01579-2. [DOI] [PubMed] [Google Scholar]
- 61.Massague J., Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yue J., Mulder K.M. Requirement of Ras/MAPK pathway activation by transforming growth factor beta for transforming growth factor beta 1 production in a Smad-dependent pathway. J Biol Chem. 2000;275:30765–30773. doi: 10.1074/jbc.M000039200. [DOI] [PubMed] [Google Scholar]
- 63.Stratton R., Rajkumar V., Ponticos M., Nichols B., Shiwen X., Black C.M., Abraham D.J., Leask A. Prostacyclin derivatives prevent the fibrotic response to TGF-beta by inhibiting the Ras/MEK/ERK pathway. FASEB J. 2002;16:1949–1951. doi: 10.1096/fj.02-0204fje. [DOI] [PubMed] [Google Scholar]
- 64.MacKay S.L., Auffenberg T., Tannahill C.L., Ksontini R., Josephs M.D., Nowak M., Moldawer L.L., Copeland E.M., III Transfection of the type II TGF-beta receptor into colon cancer cells increases receptor expression, inhibits cell growth, and reduces the malignant phenotype. Ann Surg. 1998;227:781–789. doi: 10.1097/00000658-199806000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wagner M., Kleeff J., Lopez M.E., Bockman I., Massaque J., Korc M. Transfection of the type I TGF-beta receptor restores TGF-beta responsiveness in pancreatic cancer. Int J Cancer. 1998;78:255–260. doi: 10.1002/(sici)1097-0215(19981005)78:2<255::aid-ijc21>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 66.Hinz B., Celetta G., Tomasek J.J., Gabbiani G., Chaponnier C. Alpha–smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ronnov-Jessen L., Petersen O.W. A function for filamentous alpha–smooth muscle actin: retardation of motility in fibroblasts. J Cell Biol. 1996;134:67–80. doi: 10.1083/jcb.134.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nobes C.D., Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lloyd-Jones D., Adams R.J., Brown T.M., Carnethon M., Dai S., De Simone G., American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 70.Russell R.R., III, Li J., Coven D.L., Pypaert M., Zechner C., Palmeri M., Giordano F.J., Mu J., Birnbaum M.J., Young L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cultured cardiac MSC characteristics. A: Brightfield image. B: CD44 staining using immunofluorescence (red). Nuclei were counterstained using DAPI (blue). Expression of Nanog (C) and Oct3/4 (D) in undifferentiated stem cells was analyzed using immunohistochemistry (IHC). Right panels are appropriate IgG controls. Scale bars: 50 μm (A, C, and D); 25 μm (B).
Progenitor cells differentiate into fibroblasts regardless of age. A: Brightfield image of young (4 months old) and aged (30 months old) fibroblasts derived from progenitor cells cultured in DMEM/F12 supplemented with 10% FBS. Immunohistochemical staining of fibroblast markers collagen type l (B) and DDR2 (C). Each image is representative of three independent experiments. Scale bar = 25μm.
Fibroblasts derived from aged animals exhibit impaired chemokinesis in a wound-healing assay. Photomicrographs demonstrate the process of wound repair by cardiac fibroblasts isolated from animals aged 4, 14, 24, or 30 months. Cells were grown to confluence in DMEM containing 10% FBS. Then the cell layer was wounded (example represents 0 hour). Wound closure was monitored every 24 hours. All data are representative of four experiments.
TGF-β1–induced α-SMA expression in young fibroblasts cultured under low-adhesion conditions in Petri dishes. Cells were serum-starved for 48 hours, then incubated with 10 ng/mL TGF-β1 for 72 hours, as indicated. Right panel, Ponceau S staining.
Effect of metformin on AMPK activation. Serum-starved fibroblast cultures were incubated with 2.5 mmol/L metformin for 1 and 24 hours, as indicated.
JNK inhibition eradicates TGF-β1–dependent collagen pad contraction. Fibroblasts isolated from 30-month-old mice were incubated for 3 days in the presence of 10 ng/mL TGF-β1 (T) with or without 25 μmol/L SP600125 (SP6), and then were seeded in collagen pads. Contraction of free-floating collagen matrices was evaluated 24 hours later.
AICAR does not enhance migration capability of aged fibroblasts. Cells were incubated with AICAR for 3 days, then serum-starved in the presence of AICAR. Cells were placed in Boyden's chambers and allowed to migrate in response to 10 ng/mL TGF-β1. As a positive control, cells isolated from 4-month-old animals were used. Data are given as mean ± SD of three independent experiments. *P < 0.05.