

Abstract
Although the roles of mast cells (MCs) are essential in many inflammatory and fibrotic diseases, their role in Trypanosoma cruzi–induced cardiomyopathy is unexplored. In this study, we treated infected CBA mice with cromolyn, an MC stabilizer, and observed much greater parasitemia and interferon-γ levels, higher mortality, myocarditis, and cardiac damage. Although these data show that MCs are important in controlling acute infection, we observed MC apoptosis in the cardiac tissue and peritoneal cavity of untreated mice. In the heart, pericardial mucosal MC die, perhaps because of reduced amounts of local stem cell factor. Using RT-PCR in purified cardiac MCs, we observed that infection induced transcription of P2X7 receptor and Fas, two molecules reportedly involved in cell death and inflammatory regulation. In gld/gld mice (FasL−/−), apoptosis of cardiac, but not peritoneal, MCs was decreased. Conversely, infection of P2X7−/− mice led to reduced peritoneal, but not cardiac, MC death. These data illustrate the immunomodulatory role played by MCs in T. cruzi infection and the complexity of molecular interactions that control inflammatory pathways in different tissues and compartments.
Bone marrow–derived mast cell (MC) precursors circulate in the blood and lymphatic vessels and migrate to tissues such as muscle and mucosa and celomatic cavities, where they assume mature morphologic and functional characteristics under the influence of local microenvironmental factors.1 This may be one basis for the high heterogeneity of MCs, regarding the composition of cytoplasmic granules, agonist stimulation by growth factors, and secretion of cytokines and chemokines, exerting a central role in innate and adaptive immunity.2,3
MCs can be activated by aggregation of surface Fc receptors, such as FcεRI, FcγRI, and FcγRIII,4 and binding of pathogen-derived molecules to Toll-like receptors 1, 2, and 6.5 Moreover, a variety of molecules induce MC death in different pathologic conditions, such as tumor necrosis factor (TNF)–related apoptosis-inducing ligand, P2X7 receptor and Fas, low levels of stem cell factor (SCF or c-Kit) and IL-3, or high levels of glucocorticoids, illustrating the intricate complexity of MC responses.6–8 For example, the cell line MC/9 becomes resistant to Fas-L–induced cell death after activation through FcεRI and FcγR because of increased expression of FLICE-like inhibitory protein.9 Moreover, the binding of monomeric IgE to FcεRI promotes the survival of murine MCs, suppressing apoptosis induced by growth factor deprivation with no participation of Fas or Bcl-2 family members.10
Fas is a type II membrane protein belonging to the TNF family that is involved not only in apoptosis but also in the secretion of cytokines and chemokines, transcriptional control, proliferation,11 and inflammatory regulation.12 Another membrane receptor not only involved in apoptosis but also with a wider range of proinflammatory functions is the ATP-sensitive purinergic P2X7 receptor.13 P2X7 receptor activation through extracellular ATP renders cell membranes permeable to molecules up to 900 Da in macrophages and 400 Da in lymphocytes, leading to cell death.14 Several other effects have been proposed, including the activation and release of IL-1β,15 CD62-L shedding from lymphocytes,16 and maturation of T cells.17 Bone marrow–derived MCs and MC lines8 undergo apoptosis and Ca++ influx through P2X7 activation, recruiting caspases 8 and 3, with phosphorylation of ERK, JAK2, and STAT6 and enhanced secretion of IL-4, IL-13, IL-6, and TNF-α.8
Regarding the heart, MCs have been implicated in cardiovascular dysfunctions, such as ischemic heart disease, experimental myocardial infarction, myocarditis, heart failure, transplant-related fibrosis, and hypertensive heart disease.18–22 In the particular case of myocarditis induced by T. cruzi infection, inflammatory foci in humans and experimental models are mostly composed of CD8+ T cells, with cardiac dysfunction normally associated with fibrosis as the main cause of death.23–26 Many attempts have been made to clarify the relevance of T lymphocytes and secreted soluble factors produced by components of adaptive immunity, but components of innate immunity and their mechanisms are rarely addressed and less understood.27,28 For example, macrophages are important in the control of parasite replication, and, apparently, blood eosinophils are an important source of cytokines in patients with cardiodigestive forms of the disease.29 Regarding MCs, due to technical limitations, such as a reduced number of cardiac MCs, in situ identification, isolation from the tissue, and purification, most authors use histopathologic analysis and peritoneal MCs to study their relevance in T. cruzi infection. To date, acutely T. cruzi–infected rats present a decrease in cardiac MC numbers,30 whereas analysis of chronic human patients showed an increase in these cell numbers.31,32 The relevance of MCs to heart fibrosis is also a matter of debate31,33 because chronically infected rats and human patients show increased MC counts but not necessarily increased fibrosis.
Herein, we aimed to evaluate the functional role of MCs in acute T. cruzi infection by using cromolyn, an MC stabilizer that blocks histamine release, and to observe the aggravation of the disease. Because we observed MC apoptosis in untreated mice, we studied the mechanistic relevance of some membrane death receptors and soluble factors in the control of MC subpopulations after the infection. We observed that MCs control blood and tissue parasitemia, production of interferon-γ (IFN-γ), cardiac inflammation, and susceptibility to infection. Cardiac MCs trigger the transcription of Fas after infection and apparently die after Fas-L engagement. Although P2X7 receptor is expressed in cardiac MCs after infection, it seems to be involved in the death of only peritoneal MCs.
Materials and Methods
Mice and Infection
Ten- to 12-week-old specific pathogen–free male mice CBA/J, BALB/c, gld/gld (Fas-L−/−) (BALB/c background-susceptible mouse strain), and C57BL/6 (resistant) were obtained from the FIOCRUZ animal facility (CECAL, Rio de Janeiro, Brazil). P2X7−/− mice (C57BL/6 background), derived from Pfizer (Groton, CT) and generated by Solle et al,34 were a gift from Dr. Christopher A. Gabel and were bred at the Transgenic Mice Laboratory, Instituto de Biofísica Carlos Chagas Filho at the Universidade Federal do Rio de Janeiro.34 Mice were housed for at least 1 week before infection at the Animal Experimentation Division in conditions complying with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23, revised 1985). This project has protocol number 0308/06 at the FIOCRUZ Committee of Ethics in Research, according to resolution 196/96 of the National Health Council of the Brazilian Ministry of Health. For experimental infection, bloodstream trypomastigote forms of Trypanosoma cruzi CL strain were obtained from infected Swiss-Webster mice and were isolated as previously described.35 Mice were i.p. infected with 1 × 104 parasites in 200 μL of PBS (pH 7.2) (Sigma-Aldrich, St Louis, MO), and age-matched control mice received 200 μL of PBS.
Parasitemia, Mortality, and Blood Samples
Individual parasitemia was counted in 5 μL of blood collected from tail snips on day postinfection (dpi) 17, and mortality was determined daily. For creatine kinase–MB activity (cardiac isoform), plasma was collected on dpi 15 and was analyzed using a commercially available kit (Merck KGaA, Darmstadt, Germany) as described elsewhere.36 We used this marker of cardiomyocyte damage owing to its direct correlation with myocarditis.36 Data are expressed as a rate of NADPH increase (delta E/min) in five sequential readings using a spectrophotometer (Molecular Devices, Sunnyvale, CA) at 340 nm. To measure serum cytokines by flow cytometry, we used the Cytometric Bead Array kit (flex-inflammation) (BD Biosciences, San Jose, CA) for IL-6, IL-10, monocyte chemotactic protein-1, IFN-γ, TNF, and IL-12 p70, according to the manufacturer's instructions. For glucocorticoid quantification, serum levels of corticosterone were evaluated by means of radioimmunoassay (ICN Pharmaceuticals, Costa Mesa, CA) following the manufacturer's guidelines.
Cell Harvesting
Peritoneal cells were collected by washing the cavity of 10 control (noninfected) or 10 infected animals with 5 mL of ice-cold PBS/100 mmol/L cromolyn (MC stabilizing: cromolyn sodium ophthalmic solution; Alcon Laboratories, Fort Worth, TX). The cells were centrifuged at 50 × g for 10 minutes, resuspended in 10% of the individual collected volume, and counted in a Neubauer chamber.
For cardiac MC analysis, 20 control or infected mice were euthanized; the hearts were collected, rinsed with ice-cold PBS/cromolyn, and dissected in a Petri dish into 1- to 2-mm fragments. The samples were extensively washed again in cold PBS/cromolyn to remove contaminant blood cells and then were incubated in 6 to 7 mL of PBS/100 mmol/L cromolyn containing 0.2% type IV collagenase (2.6 U/mg; lot 51K8610, Sigma-Aldrich). Tissue dissociation occurred in 5 to 7 cycles of 20-minute incubations under very gentle circular agitation in a water bath at 37°C. After each cycle, isolated cells in the supernatant were centrifuged at 80 × g for 8 minutes at 4°C, washed in cold PBS/cromolyn, pooled for the last centrifugation, resuspended in complete medium [RPMI 1640 (Sigma-Aldrich)/100 mmol/L cromolyn, pH 7.4], and stored on ice.
To control the possible contamination of blood cells in tissue samples during enzymatic dissociation, especially for lymphocyte analysis, we labeled all the samples with anti-CD62-L (SouthernBiotech, Birmingham, AL) and proceeded to the purification of MCs (see later herein) only when CD62-L+ cells were <5% of events in flow cytometry (CyAn; DakoCytomation, Fort Collins, CO). Data were analyzed using Summit 4.3 software (DakoCytomation).
Cromolyn Treatment
CBA/J mice were daily treated with cromolyn diluted in PBS from dpi −3 until dpi 20, receiving 100 mg/kg in 200 μL by i.p. injection. Eight mice per group were divided as follows: infected and cromolyn-treated mice (G1), infected and PBS-treated mice (G2), infected mice with no treatment (G3), PBS injection and cromolyn-treated mice (G4), PBS injection and PBS-treated mice (G5), and PBS-injected mice with no treatment (G6). As a control for treatment efficacy, tracheal tissue was collected for histamine quantification as previously described.37 Briefly, this method consists of sample dilution with 0.1 N of HCl followed by 0.8 N of NaOH and further addition of the substrate o-phthaldialdehyde. After 4 minutes of incubation, the reaction was stopped with 3 N of HCl, and fluorescence was measured in a Shimadzu RF1501 spectrofluorophotometer (Shimadzu Corp., Kyoto, Japan) (excitation at 360 nm; emission at 450 nm).
Histopathologic Analysis
Control and infected mice were euthanized at the time points indicated in the figure legends, and the hearts were removed, sagittally divided, embedded in Tissue-Tek (OCT, Milles-Inc., Zoeterwoude, The Netherlands), and frozen at −70°C. The hearts were then cut into 16- to 18-μm-thick slices (unfixed samples) for MC identification as described later herein or into 5-μm-thick slices for H&E staining.
MC Staining
MCs (isolated or in tissue slices) were stained with either 1% toluidine blue (TB) or a mixed solution of 0.36% Alcian Blue (AB)/0.02% safranin (S)/0.01% TB (AB/S/TB) in acetate buffer (pH 1.42).38 AB was previously cleared39 by stirring for 1 hour in 90% acetone and then filtering through number 2 filter paper. The residue was recovered from the filter, air-dried, and immediately added to the mixed solution. This protocol identifies mucosal MCs (MMCs) as AB-positive cells, connective tissue MCs (CTMCs) as S- and/or TB-positive cells, and hybrid MCs.38 Note that quantification of MC numbers (average) was performed by scanning whole cardiac tissue sections, thus reflecting variations in MC numbers in different areas of the organ. These cells are mostly found in pericardium and perivascular areas, but very few MCs are observed in endocardium, reducing the final value. For qualitative results we show pericardial areas.
MC Purification
For peritoneal MCs, 5 mL of total peritoneal cells was overlaid on a 20-mL isotonic cromolyn-supplemented Percoll column (Sigma-Aldrich) at 72% and was centrifuged at 340 × g for 25 minutes. Pelleted cells were collected, washed twice with PBS/100 mmol/L cromolyn, and then resuspended in ice-cold complete medium until use.
For cardiac MCs, 5 mL of enzymatically dissociated total cells was overlaid on a 20-mL column of Percoll/cromolyn divided into two parts of 70% and 60% and centrifuged at 700 × g for 1 hour. Pelleted cells were also resuspended in ice-cold complete medium, and all the samples were used with at least 98% purity, as previously described.38
RT-PCR
Total mRNA was extracted from purified MCs using the RNeasy kit (Qiagen, Germantown, MD) and was converted to cDNA by reverse transcription using the iScript kit (Bio-Rad, Hercules, CA) as recommended by the manufacturer. Specific primers were used to amplify cDNA fragments by PCR as follows: 5′-CCAGGTTGTCTCCTGCGACT-3′ forward and 5′-ATACCAGGAAATGAGCTTGACAAAGT-3′ reverse to GAPDH; 5′-GTCCTGCCTCTGGTGCTTGCT-3′ forward and 5′-AGTGTCTGGGGTTGATTTTCC-3′ reverse to Fas, and 5′-CGAGTTGGTGCCAGTGTGGA-3′ forward and 5′-CCTGCTGTTGGTGGCCTCTT-3′ reverse to P2X7 receptor. The primers flanked conserved regions of genes and were used to amplify cDNA fragments by PCR under high stringency conditions. PCR amplification was performed in a final volume of 10 μL, with 1 ng of target cDNA, 5 pmol of each primer, 200 μmol/L of each deoxyribonucleotide triphosphate (Promega, Madison, WI), and 0.8 U of taqDNA polymerase (Cembiot, RS, Brazil) in a saline buffer (10 mmol/L Tris-HCl, 50 mmol/L KCl, and 1.5 mmol/L MgCl2; pH 8.5). All the samples were amplified in a Mastercycler thermocycler (Eppendorf AG, Hamburg, Germany) using 26 cycles as follows: denaturation step at 95°C for 3 minutes, annealing at 56°C for 1 minute, and extension at 72°C for 30 seconds, then 25 cycles of denaturation at 95°C for 45 seconds, annealing at 56°C for 30 seconds, and extension at 72°C for 1 minute, and the last extension cycle was changed to 5 minutes. A negative control (no cDNA) was included in all the experiments. PCR products were visualized in 6% silver–stained polyacrylamide gels (Sigma-Aldrich), and digital gel images were obtained using ImageScanner III (GE Healthcare Bio-Sciences Corp., Piscataway, NJ).
Real-Time Quantitative PCR
Hearts from control (G6) and infected (G3) mice on dpi 7 and 17 were extensively reperfused through the aorta with PBS. Total mRNA was extracted from fragments of left ventriculum using the RNeasy kit (Qiagen) and treated with DNase to exclude DNA contamination. RNA was converted to cDNA by reverse transcription using the high-capacity cDNA kit, as recommended by the manufacturer (Applied Biosystems, Foster City, CA). Real-time quantitative PCR was performed on an ABI Prism 7500 fast sequence detection system using SYBR Green PCR fast master mix (Applied Biosystems) following the manufacturer's protocols. The following primers and concentrations were used: chymase sense (200 nmol/L), 5′-TTGCCAGCCTGTGAGGAAA-3′; chymase antisense (200 nmol/L), 5′TACAGACAGGCCAGATCGCAT-3′; tryptase sense (200 nmol/L), 5′-CGACATTGATAATGACGAGCCTC-3′; tryptase antisense (200 nmol/L), 5′-ACAGGCTGTTTTCCACAATGG-3′; GAPDH sense (200 nmol/L), 5′-CCAGGTTGTCTCCTGCGACT-3′; and GAPDH antisense (200 nmol/L), 5′-ATACCAGGAAATGAGCTTGACAAAGT-3′. The conditions for the PCR were as follows: 95°C for 20 seconds, followed by 40 cycles at 95°C for 3 seconds and 60°C for 30 seconds. Each of these primer sets gave a unique product. PCR assays were tripled, and the data were pooled. Sample quantification was obtained by relative standard curve normalized by GAPDH.
Immunohistochemical Analysis
Hearts were collected and frozen as previously described,37 and 16-μm-thick sections were fixed in acetone at 4°C. Specimens were incubated with 3% H2O2 for 15 minutes to inactivate endogenous peroxidase activity, washed in PBS, and incubated with FcγR blocking solution (inactivated sheep serum 1:10 in PBS supplemented with 4% albumin) (Sigma-Aldrich) for 30 minutes. Heart tissue sections were then incubated for 12 hours at 4°C with antibodies against SCF or IL-3 (Santa Cruz Biotechnology, Santa Cruz, CA), washed with PBS/albumin, incubated with horseradish peroxidase–conjugated anti-goat IgG (R&D Systems, Minneapolis, MN) for 1 hour at room temperature, and revealed with 3-amino-9-ethyl-carbazol (Sigma-Aldrich) for 10 minutes protected from light. The slices were washed in PBS, stained with hematoxylin for 1 minute, and then analyzed by light microscopy using a BX50 Olympus microscope (Olympus, Center Valley, PA). In control experiments, no immunostaining was observed when primary antibody was omitted. For stromal labeling quantification, the microscope was coupled to a video camera (DEI-750; Optronics Engineering, Goleta, CA) with output processed and analyzed using Image-Pro Plus 4 image analyzer software (Media Cybernetics Inc., Bethesda, MD).
Cell Death Analysis
Hearts were collected and frozen as previously described,37 and 16-μm-thick sections were fixed in acetone at 4°C for the detection of apoptosis using the TUNEL-POD kit (Roche, Mannheim, Germany) according to the manufacturer's recommendations. After the reaction, the slices were stained with AB/S/TB solution for concomitant identification of MCs.
Statistical Analysis
Statistical analysis was performed using one-way analysis of variance followed by the Tukey posttest for nonparametric data, and results were considered significant at P < 0.05.
Results
Cromolyn Treatment of T. cruzi–Infected Mice
Parasitemia was detectable on dpi 13, peaking on dpi 17 and between dpi 26 and 29 (data not shown). This led us to plan the treatment with cromolyn and the dpi for sample collection, as described in Materials and Methods. In cromolyn-treated mice (G1), we observed an increase in parasitemia of approximately 10-fold on dpi 17 compared with in PBS-treated (G2) or untreated (G3) mice (Figure 1A). The stabilization of MCs also rendered the mice more susceptible to the infection, reaching 50% of mortality on dpi 20 only in this group (G1) (Figure 1B). Regarding serum cytokines, we found no alterations in IL-6, monocyte chemotactic protein-1, TNF, or IL-12 comparing treated and untreated groups (G1×G2/G3 and G4×G5/G6), but cromolyn-treated mice (G1) showed higher levels of IL-10 and a sixfold increase in IFN-γ on dpi 20 (Figure 1C). Cardiac damage (Figure 1D) and inflammatory infiltration (Figure 1, E–H) were more severe in the treated group (G1) (Figure 1G) than in untreated mice (G2/G3) on dpi 15 (Figure 1H), with diffuse inflammatory foci composed mostly of mononuclear cells. Flow cytometry analysis of CD8+ T cells harvested from the cardiac tissue showed no differences between the treated (G1) and untreated (G2/G3) groups regarding the expression of lymphocyte function–associated antigen-1, CD32, CD2, CD43, and CD44, with approximately 85% of CD8+ T cells in all groups (data not shown). To confirm the stabilization of MCs, we measured histamine levels in isolated tracheal tissue stimulated or not with the agonist compound 48/80 (all the groups were divided into compound-stimulated and compound-unstimulated tracheas) and found no stimulated histamine release after cromolyn treatment (G1 and G4), although there was always a basal release of histamine (see Supplemental Figure S1 at http://ajp.amjpathol.org). Although cromolyn is a well-known MC stabilizer and is extensively used in various allergic inflammatory responses, the mechanism of action of cromolyn is probably not restricted to inhibit MC degranulation, acting on monocytes and macrophages. Despite possible other functions, we decided to study in more detail other aspects of MCs, such as total number and subpopulations found in the heart and peritoneal cavity of acutely infected untreated mice. Therefore, all the results described later herein were determined in groups G3 and G6 (control).
Figure 1.
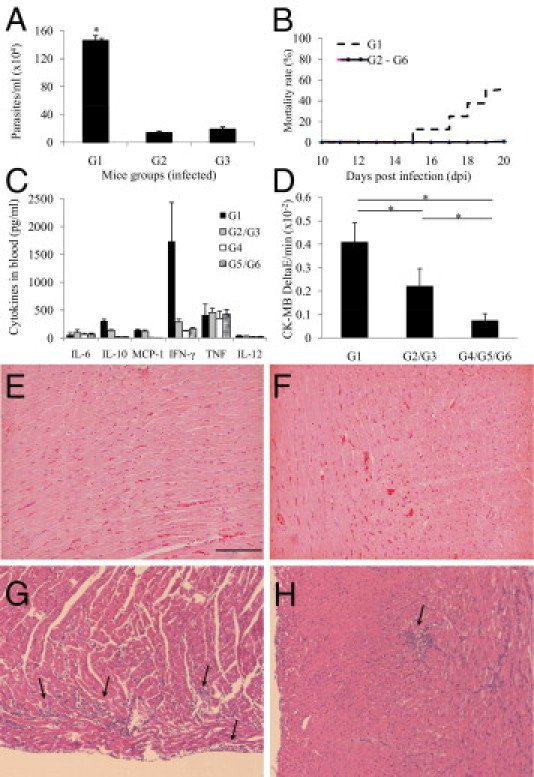
Cromolyn treatment. Mice were divided into T. cruzi–infected (G1-G3) and uninfected (G4-G6) groups, where G1 and G4 were cromolyn treated, G2 and G5 were PBS treated, and G3 and G6 were untreated. Parasitemia was counted in microscopic slides on dpi 17 (A), and mortality was checked daily (B). Plasma was collected for cytokine evaluation by flow cytometry (C) on dpi 20 and creatine kinas MB (CK-MB) activity (D) on dpi 15 from the indicated groups. MCP-1, monocyte chemotactic protein-1. Each bar represents the mean ± SD of three independent experiments with six mice per group. *P < 0.05 compared with the other groups. Representative cardiac tissue sections were collected from G4 (E), G5 (F), G1 (on dpi 15) (G), and G2 (on dpi 15) (H), all stained with H&E. Arrows indicate inflammatory foci. Scale bar = 100 μm.
MC Subpopulation Analysis
For most MC evaluations, we chose dpi 7 (no myocarditis) and dpi 17 (myocarditis similar to dpi 20), depending on the constraint of innate or adaptive immunity, respectively, and because previous data40 showed that the adaptive immune response takes over the control of infection from dpi 14. Regarding cardiac MC subpopulations,38 in control animals, AB/S/TB staining revealed mostly pericardial MMCs and some endocardial and perivascular CTMCs and MMCs (Figure 2A). However, after infection, there was a reduction in MC numbers, with a decrease in mainly pericardial MCs (Figure 2, B and C). Quantitative analysis of total cardiac MCs showed that this reduction was maintained at least until dpi 17 (Figure 3A) and was mostly due to the decrease in MMCs (Figure 3B). Note that depending on the MC stain used, this variation could be underestimated (Figure 3A; total number of MCs stained with TB compared with AB/S/TB stain for each time point). As found in the heart, total peritoneal MCs were also reduced on dpi 7, although the number of MCs increased to levels above those in control mice on dpi 17 (Figure 3C). CTMCs were predominantly found in the peritoneal cavity of control mice (Figure 3D), which is one of the richest compartments of MCs. On dpi 7, most of this subpopulation was reduced, followed by a decrease in hybrid MCs and MMCs (Figure 3D). All the subpopulations increased on dpi 17 (Figure 3D). The reduction of MCs in acutely infected cardiac tissue was confirmed by gene expression using real-time quantitative PCR with primers specific for tryptase and chymase (Figure 3E). These results indicate that different subpopulations of MCs are found in different anatomic compartments and are differently targeted by the infection.
Figure 2.
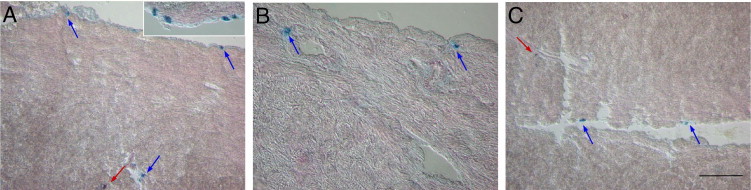
MC identification. Cardiac tissue sections were collected from uninfected and untreated mice (G6) (A and inset) and from T. cruzi–infected and untreated mice (G3) on dpi 7 (B) and dpi 17 (C) and were stained with AB/S/TB (A–C). Blue arrows indicate MMC subpopulations; red arrows, CTMC subpopulations. Data are representative of at least 15 mice per group in three independent experiments. Scale bar = 100 μm.
Figure 3.
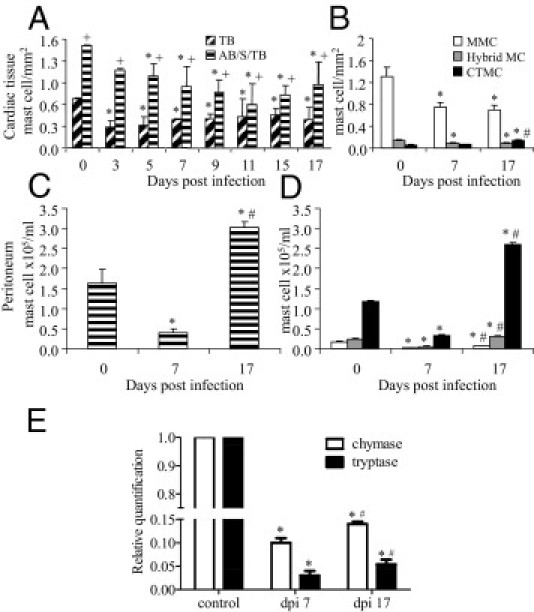
Cardiac and peritoneal MC density. MCs from control (G6) and T. cruzi–infected (G3) mice were collected at the indicated time points and were stained with AB/S/TB or TB for total cardiac MC quantification (A) and phenotypic separation as MMCs, CTMC, and hybrid MCs, as described in Materials and Methods (B). Each bar represents the mean ± SD of three experiments with six animals. Total peritoneal MCs were collected from control and infected mice on dpi 7 and 17 and were also stained with AB/S/TB (C) to reveal different MC subpopulations (D). E: Gene expression of chymase and tryptase in the left ventriculum of reperfused hearts was evaluated by real-time quantitative PCR. Each bar represents the mean ± SD of three independent experiments with seven animals per time point. *P < 0.05 compared with control; †P < 0.05 comparing AB/S/TB with TB; ‡P < 0.05 compared with dpi 7.
Mechanistic Pathways of MC Death in T. cruzi Infection
To test whether the reduction of cardiac MCs at least partially depended on apoptosis, a TUNEL-based analysis combined with MC staining was performed, and we observed no signs of apoptosis in control mice, either in the pericardium or endocardium (Figure 4A). However, on dpi 7, TUNEL+ MCs were detected mostly in the pericardium, the richest area of MCs in the heart (Figure 4B). On dpi 17, TUNEL+ MC were observed in the pericardium, endocardium, and perivascular areas but also in some mononuclear cells composing inflammatory foci (Figure 4, C and inset), as observed previously.25
Figure 4.
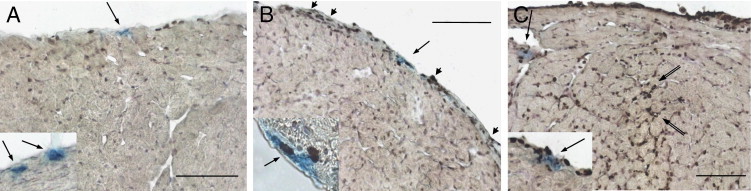
Cardiac MC apoptosis. All 16-μm-thick frozen sections were collected from control (G6) (A) and T. cruzi–infected (G3) mice on dpi 7 (B) and dpi 17 (C) for apoptosis detection by TUNEL and MC staining with AB/S/TB. Arrows indicate MCs; arrowheads, TUNEL+ pericardial cells; double arrows, TUNEL+ cells composing inflammatory foci. Two independent experiments were performed with six mice per group. Scale bars = 100 μm.
A variety of soluble and cell membrane stimuli are involved in the complex molecular network that determines MC survival or apoptosis. Soluble factors, such as SCF and IL-3, are important for MC survival, chemotaxis, maturation, and acquisition of effector functions.1,2,5,41 Therefore, infection-induced down-regulation of tissue factors could be directly triggering MC death or, in turn, leading to secondary expression of death receptors on MCs. Decreased expression of SCF was detected in the heart on dpi 7 and 17 compared with in control mice (Figure 5, A–D), although no alteration in IL-3 was observed (Figure 5, E–H). Because positive and negative modulations of SCF and other cytokines may affect the transcriptional regulation of MCs, we evaluated the production of Fas and P2X7 receptor mRNA in purified MCs for their apoptosis-inducing activity. Phenotypic heterogeneity of resident MCs is illustrated by the high levels of transcription of P2X7 receptor in peritoneal MCs but not in cardiac cells from control mice (Figure 6, A and B). P2X7 receptor transcription was sustained on dpi 7 in peritoneal MCs, and there was a down-regulation on dpi 17 (Figure 6A). In contrast, the infection induced P2X7 receptor transcription in cardiac MCs on dpi 7, with a decrease on dpi 17 (Figure 6B). Regarding Fas mRNA, we observed no transcription in peritoneal MCs at any time point studied (Figure 6C). However, as observed in the case of P2X7 receptor, the infection triggered the transcription of Fas in cardiac MCs on dpi 7, with a decrease on dpi 17 (Figure 6D). Although both molecules are not only implicated in cell death, at this point these results suggested that either molecule could be involved in the decrease in cardiac MC numbers. We, therefore, used P2X7−/− and gld/gld (Fas-L−/−) mice to assess the roles played by each molecule in MC regulation after the infection in vivo. As observed in infected CBA/J, BALB/c mice (gld/gld background) showed a reduced number of MCs in peritoneum (Table 1) and heart (Figure 7A) after infection, with a phenotypic profile (Figure 7B) similar to that found in CBA/J (Figure 3B). Conversely, gld/gld mice showed an increase in cardiac MCs after infection, also with a predominance of MMCs (Figure 7, C and D); these changes were not observed in peritoneal MCs (Table 1). Using the T. cruzi–resistant mouse lineage C57BL/6 (P2X7−/− background), we observed no infection-induced cardiac MC reduction on dpi 9 (Figure 7E), only on dpi 15, or on dpi 9 using a higher inoculum of parasites (1 × 105) (data not shown). However, MC subpopulation staining showed that there was a reduction in cardiac MMCs on dpi 9, which was compensated for by an enrichment of hybrid MCs (Figure 7F). With P2X7−/− mice, no MC reduction was detected in the heart (Figure 7G) after infection, a finding that could be due to resistance of this mouse strain with a slower rate of cardiac MC reduction or no role played by this receptor in cardiac MC death. The second alternative is more likely, as we found no reduction of MMCs in P2X7−/−-infected mice (Figure 7H). On the other hand, there was a decrease in peritoneal MC numbers in C57BL/6 but not in P2X7−/− mice after infection (Table 1). These data suggest that Fas/Fas-L plays a role in the control of cardiac MC death and that P2X7 receptor plays a role in the control of MC subpopulations of peritoneal MCs.
Figure 5.

SCF and IL-3 production in cardiac tissue. SCF (A–D) and IL-3 (E–H) levels were evaluated by immunohistochemical analysis in cardiac tissue from control (G6) mice (A and E) and from T. cruzi–infected (G3) mice on dpi 7 (B and F) and dpi 17 (C and G). Original magnification, ×200. SCF (D) and IL-3 (H) stromal labeling were quantified using Image-Pro Plus 4 image analyzer software in cardiac tissue from control (G6) and T. cruzi–infected (G3) mice on dpi 7 and 17. Each bar represents the mean ± SD of three independent experiments with five animals per time point. *P < 0.05 compared with control.
Figure 6.
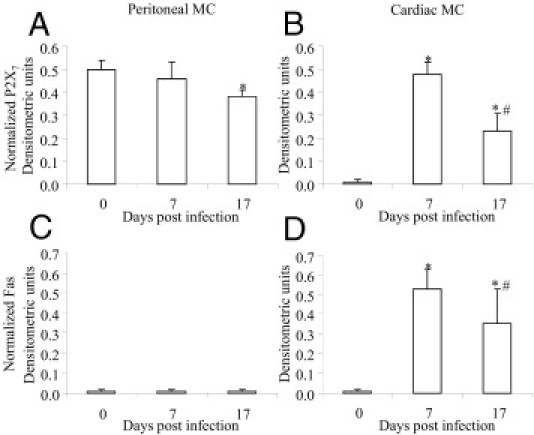
MC transcription of P2X7 receptor and Fas. Peritoneal (A and C) and cardiac (B and D) MCs were purified from control (G6) and T. cruzi–infected (G3) mice on dpi 7 and 17 using Percoll to evaluate the transcription of P2X7 receptor (A and B) and Fas molecule (C and D) by RT-PCR. Each bar represents the mean ± SD of densitometric units (normalized for the GAPDH) of three experiments per time point. The samples enriched in MCs (≥98%) were obtained from 20 animals. *P < 0.05 compared with control; †P < 0.05 compared with dpi 7.
Table 1.
Changes in MC Numbers in Peritoneum of Different Mouse Strains Infected with T. cruzi
| dpi | MCs (×105/mL) |
|||
|---|---|---|---|---|
| BALB/c | gld/gld | C57Bl/6 | P2X7−/− | |
| 0 | 2.5 ± 0.3 | 2.7 ± 0.3 | 1.9 ± 0.1 | 2.0 ± 0.3 |
| 9 | 0.7 ± 0.2⁎ | 0.9 ± 0.3⁎ | 0.9 ± 0.1⁎ | 1.9 ± 0.4⁎ |
Results are the mean ± SD of six mice per group.
P < 0.005.
Figure 7.
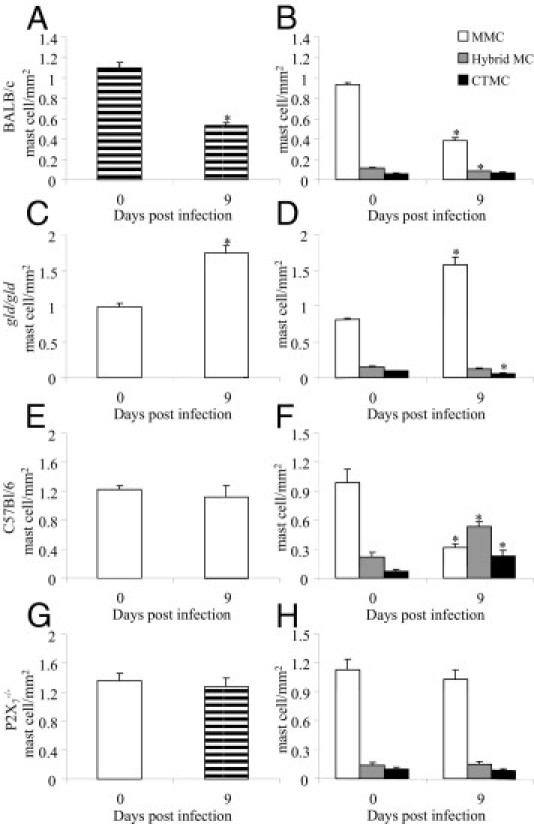
Fas-L and P2X7 receptors in cardiac MCs. Tissue sections were collected from control and T. cruzi–infected (as indicated in the panels) BALB/c (A and B), gld/gld (Fas-L−/−) (C and D), C57Bl/6 (E and F), and P2X7−/− (G and H) mice for total MC quantitation (left panels) and subpopulation identification (right panels). We used AB/S/TB staining solution. Each bar represents the mean ± SD of two independent experiments with six mice per group. *P < 0.05 compared with control.
Discussion
Murine infection with T. cruzi reproduces many characteristics of human infection, with electrical conductance disturbs, cellular inflammatory infiltration, cardiac damage, and fibrosis. In this study, we observed that an MC stabilizer (cromolyn) aggravates the infection, leading to increased myocarditis, parasitemia, and mortality. Although we cannot directly correlate the number of MCs with mortality rates, our data using cromolyn suggest that MCs are involved in resistance to the infection. For example, we observed no MC death when using a resistant mouse strain (C57Bl/6), except when the infection was induced using a higher parasite load. Accordingly, we observed previously that gld/gld mice have decreased myocarditis compared with BALB/c mice,12 and in the present study, we observed no death of MCs in gld/gld mice. This finding suggests that variations in the combination of hosts and parasites may lead to more or less death of MCs, where higher levels of MC death could account for more aggressive acute phases and higher mortality.
MCs are secretory cells strategically located in all vascularized tissues and cavities, able to initiate and modulate inflammatory processes.7,42–44 However, there are few studies evaluating MCs in Chagas' disease and/or experimental T. cruzi infection. Therefore, the goal was to evaluate the role(s) of MCs in T. cruzi infection and to elucidate the mechanistic pathways of variations in MC populations, as long as increase45–47 or decrease48 of MCs has been described in diverse pathologic conditions. Tissue MC density also seems to play a role in cardiac function because it was reported that the number of MCs increased after induced cardiac failure, and a reduction in MCs was associated with recovery of cardiac function after treatment with a chymase blocker.49 Moreover, cromolyn treatment reduces the number and local activation of MCs and inhibits the progression of atherosclerotic plaques.50
Previous studies evaluated the importance of some innate components in the control of T. cruzi infection.27,28,51,52 To date, IFN-γ released by natural killer cells and IL-12 and nitric oxide secreted by macrophages were associated with the control of parasitemia and mortality.24,40,53–55 In fact, it was demonstrated that IFN-γ and TNF-α, combined or separately, induce nitric oxide production by inducible nitric oxide synthase and, in turn, lead to parasite death.24,56,57 On the other hand, the present results using cromolyn showed that even in the presence of high levels of IFN-γ, blood parasite was much increased compared with untreated mice. This finding suggests that the IFN-γ–based control of parasite is more complex than previously described, probably involving alternative pathways in vivo, with a central role for MCs. In addition, high levels of IFN-γ itself can be cytotoxic and lead to host death.
Regarding numerical analysis of MCs, previous data revealed an increase in cardiac MCs in chronic chagasic patients, associated or not with severity of myocarditis and fibrosis.31,33 However, Chapadeiro et al30 observed a reduction in MC numbers in the heart of Wistar rats during the acute phase of T. cruzi infection and referred to a “scarcity” of MCs, although the possible causes of this reduction are still unclear. Similarly, the present data indicate a reduction of these cells in the heart and peritoneum during the early acute phase of the infection, and this could be due to a variety of factors, such as 1) reduced local secretion of MC survival factors, 2) expression of death receptors, and/or 3) reduced migration of blood MC precursors (Figure 8). Many soluble molecules can target these processes, and the glucocorticoid/SCF axis is probably also playing a role (Figure 8; see also Supplemental Figure S2 at http://ajp.amjpathol.org). For instance, increased levels of glucocorticoid hormones can down-modulate the secretion of SCF by tissue stromal cells, such as fibroblasts and epithelial cells.58 In turn, SCF is one of the most important secreted factors involved in MC survival, transcriptional regulation, chemotaxis, and maturation/differentiation.1,59 Finally, reduced SCF levels lead to MC apoptosis.41,60,61 Regarding T. cruzi infection, glucocorticoid levels were increased in a susceptible mouse strain (BALB/c) in the first week of infection,62 in accordance with the present results (see Supplemental Figure S2 at http://ajp.amjpathol.org). Moreover, in vivo blockade of glucocorticoid receptors62 increased mortality to 100%, although the authors did not measure the number or function of MCs in the heart. Therefore, we also evaluated the downstream mediator in the glucocorticoid/SCF system during the infection and observed a down-modulation of SCF in cardiac tissue. Another possibility to account for this reduction could be the death of pericardial SCF-producing cells due to the infection (Figure 8) because we observed intense pericardial apoptosis and myocarditis in this area rich in fibroblasts and epithelial cells. We propose that a reduction in SCF, possibly caused by higher levels of glucocorticoid hormones and/or infection-induced death of SCF-producing cells, leads directly or indirectly (through the expression of death receptors) to MC apoptosis (Figure 8). Moreover, according to the literature, a reduction in SCF leads to a significant decrease in histamine levels, MC and eosinophil infiltration, IL-4 production in vivo,63 and phenotypic modulation.1,64,65 All these responses can potentially affect the course and severity of the infection.
Figure 8.
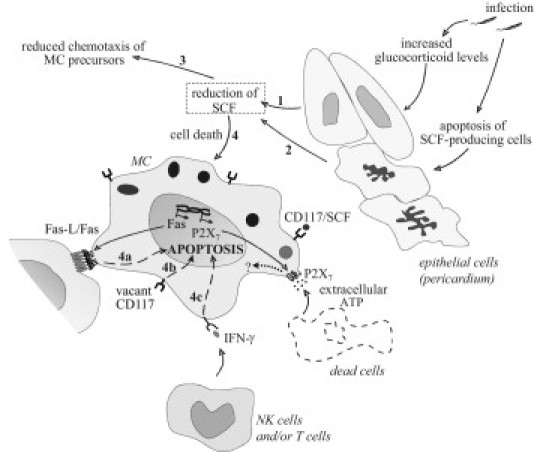
General scheme of pathways of pericardial MC death after T. cruzi infection. The increase in glucocorticoid levels induced by the infection (1) and/or possible death of pericardial SCF-producing cells (2) could lead to the reduction in cardiac stromal SCF. This down-modulation can trigger the decrease in the number of MCs by reduced chemotaxis of MC precursors to the heart (3) and/or apoptosis of mature cardiac MCs (4). Mechanistic pathways that could be playing a role in MC apoptosis include induction of Fas expression on MCs and interaction with Fas-L+ cells in the tissue (4a); predominance of vacant receptors of SCF (CD117) directly leading to MC death (4b); and increased levels of MC cytotoxic cytokines, such as IFN-γ, produced by natural killer (NK) cells in the early acute phase of the infection and later by T lymphocytes (4c). Although we observed no MC death through P2X7 receptor activation, using P2X7−/−-infected mice, the infection induced its transcription for still unexplored functions. Moreover, infected necrotic cells releasing extracellular ATP could be the source of receptor agonistic stimulus.
To explore mechanistic pathways that could further connect the reduction of SCF to MC death, we performed RT-PCR for two death receptors in peritoneal and cardiac MCs and observed that the infection induced the transcription of P2X7 receptor and Fas mRNA in the heart. Therefore, to evaluate whether these receptors were playing a role in the reduction of MC in vivo, we used the corresponding infected knockout mice. The reduction in cardiac MCs was reversed in the absence of Fas/Fas-L, suggesting that this pathway was critical for MC apoptosis. However, MC survival in infected gld/gld mice can be an indirect effect, due to reduced death of pericardial and endocardial SCF-producing cells in gld/gld mice, for example, sustaining high levels of SCF in these mice. Accordingly, we previously showed that T. cruzi–infected gld/gld mice have attenuated myocarditis and less tissue damage12 than infected BALB/c mice.
The P2X7 receptor plays many different inflammatory roles in MCs.8,66 However, although the infection induced P2X7 receptor transcription in cardiac MCs, P2X7−/− mice showed no changes in the numbers of MCs, indicating no involvement of this receptor in cardiac MC death. Regarding the phenotype of MCs in C57BL/6 mice, we observed, for the first time, an alteration in the relative frequency of subpopulations, with an enrichment of hybrid MCs and a reduction of CTMCs in the heart after T. cruzi infection. It is possible that this modulation is related to the natural resistance of the C57BL/6 strain to the infection, but this matter will be further investigated.
Regarding the number of peritoneal MCs, we observed in CBA/J and BALB/c mice a decrease of approximately fourfold in the first week of infection, which seems not to be induced by Fas activation because we observed no transcription of this molecule at any time. P2X7 receptor, on the other hand, was observed by RT-PCR in control and infected mice, and this could indicate that this receptor was not causal in the death of peritoneal MCs. However, the reduction in peritoneal MCs observed in infected C57BL/6 mice was not observed in infected P2X7−/− mice, indicating a role played by this molecule in cell death in vivo. Indeed, it could be possible that this mRNA was not being translated to produce a mature membrane molecule in noninfected peritoneal MCs but only after infection, as major histocompatibility complex class II in immature dendritic cells.67 Another possibility is that P2X7 receptor can be expressed on cell membranes of peritoneal MCs before infection but not be available for functional agonist stimuli, what could be triggered by the infection, as previously observed in thymocytes.68 This could be due to the expression of different P2X7 isotypes before and after infection, a conformational alteration induced by the inflammatory milieu, and/or coupling of the receptor to the intracellular signaling cascades after infection.
Besides the SCF importance in MC survival after infection, inflammatory cytokines can play an additional role in MC activation and death. It was described that IFN-γ can directly induce apoptosis in bone marrow MCs69 possibly by activation of caspases 8 and 9.70
In conclusion, we described some changes in cardiac and peritoneal MCs after experimental T. cruzi infection in a Fas- and P2X7 receptor–dependent manner. These data are useful to clarify the mechanisms of the infection that modulate MC populations and function, showing that MCs not only influence the inflammatory response but are constrained by the inflammatory milieu in different compartments.
Acknowledgments
We thank Drs. Emiliano Barreto, Marco Aurélio Martins, and Elen Melo for their invaluable suggestions and support in the experiments. We also thank the Program for Technological Development in Tools for Health-PDTISFIOCRUZ for use of its facilities.
Footnotes
Supported by the National Council for Scientific and Technological Development (CNPq), Instituto Oswaldo Cruz, and the Program for Centers of Excellence/Carlos Chagas Filho Foundation for Research Support of the State of Rio de Janeiro (PRONEX/FAPERJ).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.06.014.
Supplementary data
Histamine levels in tracheal tissue treatment control. Mice were divided into T. cruzi–infected (G1-G3) and uninfected (G4-G6) groups, where G1 and G4 were cromolyn treated, G2 and G5 were PBS treated, and G3 and G6 were untreated. Tracheal tissue was collected from every group on dpi 20 and was incubated in the presence or not of 0.05 μg/mL of compound 48/80 (MC agonist); histamine release was determined in supernatant through a radioimmunoassay. Each bar represents the mean ± SD of two independent experiments with six mice per group. *P < 0.05 comparing the same group with and without compound 48/80 stimulus.
Corticosterone levels in plasma. Mice were divided into T. cruzi–infected (G1-G3) and uninfected (G4-G6) groups, where G1 and G4 were cromolyn treated, G2 and G5 were PBS treated, and G3 and G6 were untreated. Plasma was collected on dpi 20, and corticosterone level was determined using radioimmunoassay. Each bar represents the mean ± SD of two independent experiments with six mice per group. *P < 0.05 compared with infected groups.
References
- 1.Metcalfe D.D., Baram D., Mekori Y.A. Mast cells. Physiol Rev. 1997;77:1033–1079. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 2.Lantz C.S., Boesiger J., Song C.H., Mach N., Kobayashi T., Mulligan R.C., Nawa Y., Dranoff G., Galli S.J. Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature. 1998;392:90–93. doi: 10.1038/32190. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson G., Mikovits J.A., Metcalfe D.D., Taub D.D. Mast cell migratory response to interleukin-8 is mediated through interaction with chemokine receptor CXCR2/interleukin-8RB. Blood. 1999;93:2791–2797. [PubMed] [Google Scholar]
- 4.Metz M., Maurer M. Mast cells: key effector cells in immune responses. Trends Immunol. 2007;28:234–241. doi: 10.1016/j.it.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Mekori Y.A., Metcalfe D.D. Mast cells in innate immunity. Immunol Rev. 2000;173:131–140. doi: 10.1034/j.1600-065x.2000.917305.x. [DOI] [PubMed] [Google Scholar]
- 6.e Silva P.M., Carvalho V.F., Cordeiro R.S., Martins M.A. Down-regulation of allergic responses in conditions of experimental diabetes: a role for glucocorticoids? Neuroimmunomodulation. 2009;16:13–18. doi: 10.1159/000179662. [DOI] [PubMed] [Google Scholar]
- 7.Galli S.J., Nakae S. Mast cells to the defense. Nat Immunol. 2003;4:1160–1162. doi: 10.1038/ni1203-1160. [DOI] [PubMed] [Google Scholar]
- 8.Bulanova E., Budagian V., Orinska Z., Hein M., Petersen F., Thon L., Adam D., Bulfone-Paus S. Extracellular ATP induces cytokine expression and apoptosis through P2X7 receptor in murine mast cells. J Immunol. 2005;174:3880–3890. doi: 10.4049/jimmunol.174.7.3880. [DOI] [PubMed] [Google Scholar]
- 9.Yoshikawa H., Nakajima Y., Tasaka K. Enhanced expression of Fas-associated death domain-like IL-1-converting enzyme (FLICE)-inhibitory protein induces resistance to Fas-mediated apoptosis in activated mast cells. J Immunol. 2000;165:6262–6269. doi: 10.4049/jimmunol.165.11.6262. [DOI] [PubMed] [Google Scholar]
- 10.Asai K., Kitaura J., Kawakami Y., Yamagata N., Tsai M., Carbone D.P., Liu F.T., Galli S.J., Kawakami T. Regulation of mast cell survival by IgE. Immunity. 2001;14:791–800. doi: 10.1016/s1074-7613(01)00157-1. [DOI] [PubMed] [Google Scholar]
- 11.Lambert C., Landau A.M., Desbarats J. Fas-beyond death: a regenerative role for Fas in the nervous system. Apoptosis. 2003;8:551–562. doi: 10.1023/A:1026113222478. [DOI] [PubMed] [Google Scholar]
- 12.Melo de Oliveira G., Lopes Diniz R., Batista W., Meuser Batista M., Bani Correa C., Cremonini de Araujo-Jorge T., Henriques-Pons A. Fas ligand-dependent Inflammatory regulation in acute myocarditis induced by Trypanosoma cruzi infection. Am J Pathol. 2007;171:79–86. doi: 10.2353/ajpath.2007.060643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.da Cruz C.M., Ventura A.L., Schachter J., Costa-Junior H.M., da Silva Souza H.A., Gomes F.R., Coutinho-Silva R., Ojcius D.M., Persechini P.M. Activation of ERK1/2 by extracellular nucleotides in macrophages is mediated by multiple P2 receptors independently of P2X7-associated pore or channel formation. Br J Pharmacol. 2006;147:324–334. doi: 10.1038/sj.bjp.0706559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mantuano-Barradas M., Henriques-Pons A., Araujo-Jorge T.C., Di Virgilio F., Coutinho-Silva R., Persechini P.M. Extracellular ATP induces cell death in CD4+/CD8+ double-positive thymocytes in mice infected with Trypanosoma cruzi. Microbes Infect. 2003;5:1363–1371. doi: 10.1016/j.micinf.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Ferrari D., Pizzirani C., Adinolfi E., Lemoli R.M., Curti A., Idzko M., Panther E., Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 16.Gu B., Bendall L.J., Wiley J.S. Adenosine triphosphate-induced shedding of CD23 and L-selectin (CD62L) from lymphocytes is mediated by the same receptor but different metalloproteases. Blood. 1998;92:946–951. [PubMed] [Google Scholar]
- 17.Tsukimoto M., Maehata M., Harada H., Ikari A., Takagi K., Degawa M. P2X7 receptor-dependent cell death is modulated during murine T cell maturation and mediated by dual signaling pathways. J Immunol. 2006;177:2842–2850. doi: 10.4049/jimmunol.177.5.2842. [DOI] [PubMed] [Google Scholar]
- 18.Kalesnikoff J., Galli S.J. New developments in mast cell biology. Nat Immunol. 2008;9:1215–1223. doi: 10.1038/ni.f.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palladini G., Tozzi R., Perlini S. Cardiac mast cells in the transition to heart failure: innocent bystanders or key actors? J Hypertens. 2003;21:1823–1825. doi: 10.1097/00004872-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Francis G.S., Tang W.H. Histamine, mast cells, and heart failure: is there a connection? J Am Coll Cardiol. 2006;48:1385–1386. doi: 10.1016/j.jacc.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Martin T.R., Ando A., Takeishi T., Katona I.M., Drazen J.M., Galli S.J. Mast cells contribute to the changes in heart rate, but not hypotension or death, associated with active anaphylaxis in mice. J Immunol. 1993;151:367–376. [PubMed] [Google Scholar]
- 22.Brower G.L., Janicki J.S. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J Card Fail. 2005;11:548–556. doi: 10.1016/j.cardfail.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Andrade Z.A., Andrade S.G., Sadigursky M., Wenthold R.J., Jr., Hilbert S.L., Ferrans V.J. The indeterminate phase of Chagas' disease: ultrastructural characterization of cardiac changes in the canine model. Am J Trop Med Hyg. 1997;57:328–336. doi: 10.4269/ajtmh.1997.57.328. [DOI] [PubMed] [Google Scholar]
- 24.Silva J.S., Machado F.S., Martins G.A. The role of nitric oxide in the pathogenesis of Chagas disease. Front Biosci. 2003;8:s314–s325. doi: 10.2741/1012. [DOI] [PubMed] [Google Scholar]
- 25.Henriques-Pons A., Oliveira G.M., Paiva M.M., Correa A.F., Batista M.M., Bisaggio R.C., Liu C.C., Cotta-De-Almeida V., Coutinho C.M., Persechini P.M., Araujo-Jorge T.C. Evidence for a perforin-mediated mechanism controlling cardiac inflammation in Trypanosoma cruzi infection. Int J Exp Pathol. 2002;83:67–79. doi: 10.1046/j.1365-2613.2002.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higuchi Mde L., Benvenuti L.A., Martins Reis M., Metzger M. Pathophysiology of the heart in Chagas' disease: current status and new developments. Cardiovasc Res. 2003;60:96–107. doi: 10.1016/s0008-6363(03)00361-4. [DOI] [PubMed] [Google Scholar]
- 27.Melo R.C., Machado C.R. Trypanosoma cruzi: peripheral blood monocytes and heart macrophages in the resistance to acute experimental infection in rats. Exp Parasitol. 2001;97:15–23. doi: 10.1006/expr.2000.4576. [DOI] [PubMed] [Google Scholar]
- 28.Teixeira M.M., Gazzinelli R.T., Silva J.S. Chemokines, inflammation and Trypanosoma cruzi infection. Trends Parasitol. 2002;18:262–265. doi: 10.1016/s1471-4922(02)02283-3. [DOI] [PubMed] [Google Scholar]
- 29.Cardoso G.M., Morato M.J., Gomes J.A., Rocha M.O., Bonfim I.P., Williams-Blangero S., VandeBerg J.L., Reis M.R., Magalhães E.F., Correa-Oliveira R. Comparative analysis of cell phenotypes in different severe clinical forms of Chagas' disease. Front Biosci. 2006;11:1158–1163. doi: 10.2741/1870. [DOI] [PubMed] [Google Scholar]
- 30.Chapadeiro E., Beraldo P.S., Jesus P.C., Oliveira Junior W.P., Junqueira Junior L.F. [Cardiac lesions in Wistar rats inoculated with various strains of Trypanosoma cruzi] Rev Soc Bras Med Trop. 1988;21:95–103. doi: 10.1590/s0037-86821988000300002. [DOI] [PubMed] [Google Scholar]
- 31.Almeida H.O., Pereira F.E., Tafuri W.L. [Mast cells in Chagas' chronic cardiopathy] Rev Inst Med Trop Sao Paulo. 1975;17:5–9. [PubMed] [Google Scholar]
- 32.Cabral H.R., Novak I.T., Glocker T.M., Castro Viera G.A. Chagas cardiopathy: identification and quantification of infiltrating cells in the hearts of cardiac death patients of different ages [in Spanish] Rev Fac Cien Med Univ Nac Cordoba. 2002;59:83–89. [PubMed] [Google Scholar]
- 33.Pinheiro M.C., Beraldo P.S., Junqueira Junior L.F., Lopes E.R., Chapadeiro E. A quantitative analysis of the mastocytes and eosinophilic granulocytes in the myocardium of Wistar rats chronically infected by Trypanosoma cruzi: a contribution to the knowledge of myocardial fibrosis [in Portuguese] Rev Soc Bras Med Trop. 1992;25:45–50. doi: 10.1590/s0037-86821992000100007. [DOI] [PubMed] [Google Scholar]
- 34.Solle M., Labasi J., Perregaux D.G., Stam E., Petrushova N., Koller B.H., Griffiths R.J., Gabel C.A. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–132. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- 35.de Araujo-Jorge T.C. The biology of Trypanosoma cruzi-macrophage interaction. Mem Inst Oswaldo Cruz. 1989;84:441–462. doi: 10.1590/s0074-02761989000400001. [DOI] [PubMed] [Google Scholar]
- 36.de Souza A.P., Olivieri B.P., de Castro S.L., Araujo-Jorge T.C. Enzymatic markers of heart lesion in mice infected with Trypanosoma cruzi and submitted to benznidazole chemotherapy. Parasitol Res. 2000;86:800–808. doi: 10.1007/s004360000262. [DOI] [PubMed] [Google Scholar]
- 37.Barreto E.O., Carvalho V.F., Lagente V., Lugnier C., Cordeiro R.S., Martins M.A., E Silva P.M. Increased levels of cyclic adenosine monophosphate contribute to the hyporesponsiveness of mast cells in alloxan diabetes. Int Immunopharmacol. 2004;4:755–762. doi: 10.1016/j.intimp.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Meuser-Batista M., Correa J.R., Soares M.J., Henriques-Pons A. Isolation of cardiac mast cells in experimental Trypanosoma cruzi infection. Tissue Cell. 2008;40:309–316. doi: 10.1016/j.tice.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 39.McAuliffe W.G. A note on the purification of Alcian blue. Stain Technol. 1983;58:374–376. doi: 10.3109/10520298309066816. [DOI] [PubMed] [Google Scholar]
- 40.Abrahamsohn I.A., Coffman R.L. Trypanosoma cruzi: IL-10, TNF, IFN-γ, and IL-12 regulate innate and acquired immunity to infection. Exp Parasitol. 1996;84:231–244. doi: 10.1006/expr.1996.0109. [DOI] [PubMed] [Google Scholar]
- 41.Iemura A., Tsai M., Ando A., Wershil B.K., Galli S.J. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am J Pathol. 1994;144:321–328. [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan J.J., Fernando J.F. Mast cell modulation of the immune response. Curr Allergy Asthma Rep. 2009;9:353–359. doi: 10.1007/s11882-009-0052-z. [DOI] [PubMed] [Google Scholar]
- 43.Lima M.C., Chagas M.S., Silva P.M., Calheiros A.S., Prouvost-Danon A., Fari-Neto H.C., Cordeiro R.S., Martins M.A. Histamine-potentiating activity in rat anaphylactic pleural fluid: role of serotonin. Braz J Med Biol Res. 1996;29:1049–1056. [PubMed] [Google Scholar]
- 44.Crivellato E., Beltrami C.A., Mallardi F., Ribatti D. The mast cell: an active participant or an innocent bystander? Histol Histopathol. 2004;19:259–270. doi: 10.14670/HH-19.259. [DOI] [PubMed] [Google Scholar]
- 45.Knight P.A., Wright S.H., Lawrence C.E., Paterson Y.Y., Miller H.R. Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell-specific granule chymase, mouse mast cell protease-1. J Exp Med. 2000;192:1849–1856. doi: 10.1084/jem.192.12.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobayashi Y., Okunishi H. Mast cells as a target of rheumatoid arthritis treatment. Jpn J Pharmacol. 2002;90:7–11. doi: 10.1254/jjp.90.7. [DOI] [PubMed] [Google Scholar]
- 47.He S.H., Chen H.Q., Zheng J. Inhibition of tryptase and chymase induced nucleated cell infiltration by proteinase inhibitors. Acta Pharmacol Sin. 2004;25:1677–1684. [PubMed] [Google Scholar]
- 48.Oliveira-Neto H.H., Leite A.F., Costa N.L., Alencar R.C., Lara V.S., Silva T.A., Leles C.R., Mendonca F.E., Batista A.C. Decrease in mast cells in oral squamous cell carcinoma: possible failure in the migration of these cells. Oral Oncol. 2007;43:484–490. doi: 10.1016/j.oraloncology.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 49.Matsumoto T., Wada A., Tsutamoto T., Ohnishi M., Isono T., Kinoshita M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation. 2003;107:2555–2558. doi: 10.1161/01.CIR.0000074041.81728.79. [DOI] [PubMed] [Google Scholar]
- 50.Bot I., de Jager S.C., Zernecke A., Lindstedt K.A., van Berkel T.J., Weber C., Biessen E.A. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation. 2007;115:2516–2525. doi: 10.1161/CIRCULATIONAHA.106.660472. [DOI] [PubMed] [Google Scholar]
- 51.Silva J.S., Aliberti J.C., Martins G.A., Souza M.A., Souto J.T., Padua M.A. The role of IL-12 in experimental Trypanosoma cruzi infection. Braz J Med Biol Res. 1998;31:111–115. doi: 10.1590/s0100-879x1998000100014. [DOI] [PubMed] [Google Scholar]
- 52.Umekita L.F., Mota I. How are antibodies involved in the protective mechanism of susceptible mice infected with T. cruzi? Braz J Med Biol Res. 2000;33:253–258. doi: 10.1590/s0100-879x2000000300001. [DOI] [PubMed] [Google Scholar]
- 53.Cardillo F., Voltarelli J.C., Reed S.G., Silva J.S. Regulation of Trypanosoma cruzi infection in mice by gamma interferon and interleukin 10: role of NK cells. Infect Immun. 1996;64:128–134. doi: 10.1128/iai.64.1.128-134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Norris K.A., Schrimpf J.E., Flynn J.L., Morris S.M., Jr Enhancement of macrophage microbicidal activity: supplemental arginine and citrulline augment nitric oxide production in murine peritoneal macrophages and promote intracellular killing of Trypanosoma cruzi. Infect Immun. 1995;63:2793–2796. doi: 10.1128/iai.63.7.2793-2796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodrigues M.M., Ribeirao M., Boscardin S.B. CD4 Th1 but not Th2 clones efficiently activate macrophages to eliminate Trypanosoma cruzi through a nitric oxide dependent mechanism. Immunol Lett. 2000;73:43–50. doi: 10.1016/s0165-2478(00)00205-4. [DOI] [PubMed] [Google Scholar]
- 56.Machado F.S., Martins G.A., Aliberti J.C., Mestriner F.L., Cunha F.Q., Silva J.S. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation. 2000;102:3003–3008. doi: 10.1161/01.cir.102.24.3003. [DOI] [PubMed] [Google Scholar]
- 57.Martins G.A., Cardoso M.A., Aliberti J.C., Silva J.S. Nitric oxide-induced apoptotic cell death in the acute phase of Trypanosoma cruzi infection in mice. Immunol Lett. 1998;63:113–120. doi: 10.1016/s0165-2478(98)00066-2. [DOI] [PubMed] [Google Scholar]
- 58.Finotto S., Mekori Y.A., Metcalfe D.D. Glucocorticoids decrease tissue mast cell number by reducing the production of the c-kit ligand, stem cell factor, by resident cells: in vitro and in vivo evidence in murine systems. J Clin Invest. 1997;99:1721–1728. doi: 10.1172/JCI119336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okayama Y., Kawakami T. Development, migration, and survival of mast cells. Immunol Res. 2006;34:97–115. doi: 10.1385/IR:34:2:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piliponsky A.M., Levi-Schaffer F. Regulation of apoptosis in mast cells. Apoptosis. 2000;5:435–441. doi: 10.1023/a:1009680500988. [DOI] [PubMed] [Google Scholar]
- 61.Mekori Y.A., Oh C.K., Metcalfe D.D. The role of c-Kit and its ligand, stem cell factor, in mast cell apoptosis. Int Arch Allergy Immunol. 1995;107:136–138. doi: 10.1159/000236955. [DOI] [PubMed] [Google Scholar]
- 62.Roggero E., Perez A.R., Tamae-Kakazu M., Piazzon I., Nepomnaschy I., Besedovsky H.O., Bottasso O.A., del Rey A. Endogenous glucocorticoids cause thymus atrophy but are protective during acute Trypanosoma cruzi infection. J Endocrinol. 2006;190:495–503. doi: 10.1677/joe.1.06642. [DOI] [PubMed] [Google Scholar]
- 63.Reber L., Da Silva C.A., Frossard N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur J Pharmacol. 2006;533:327–340. doi: 10.1016/j.ejphar.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 64.Berent-Maoz B., Piliponsky A.M., Daigle I., Simon H.U., Levi-Schaffer F. Human mast cells undergo TRAIL-induced apoptosis. J Immunol. 2006;176:2272–2278. doi: 10.4049/jimmunol.176.4.2272. [DOI] [PubMed] [Google Scholar]
- 65.Hartmann K., Wagelie-Steffen A.L., von Stebut E., Metcalfe D.D. Fas (CD95. APO-1) antigen expression and function in murine mast cells. J Immunol. 1997;159:4006–4014. [PubMed] [Google Scholar]
- 66.Nakamura H., Saito H., Ikura Y. The stimuli releasing histamine from murine bone marrow-derived mast cells, 1: the presence of P2-purinoceptors. Arerugi. 1989;38:1359–1363. [PubMed] [Google Scholar]
- 67.Villadangos J.A., Schnorrer P., Wilson N.S. Control of MHC class II antigen presentation in dendritic cells: a balance between creative and destructive forces. Immunol Rev. 2005;207:191–205. doi: 10.1111/j.0105-2896.2005.00317.x. [DOI] [PubMed] [Google Scholar]
- 68.Cascabulho C.M., Menna-Barreto R.F., Coutinho-Silva R., Persechini P.M., Henriques-Pons A. P2X7 modulatory web in Trypanosoma cruzi infection. Parasitol Res. 2008;103:829–838. doi: 10.1007/s00436-008-1063-8. [DOI] [PubMed] [Google Scholar]
- 69.Mann-Chandler M.N., Kashyap M., Wright H.V., Norozian F., Barnstein B.O., Gingras S., Parganas E., Ryan J.J. IFN-γ induces apoptosis in developing mast cells. J Immunol. 2005;175:3000–3005. doi: 10.4049/jimmunol.175.5.3000. [DOI] [PubMed] [Google Scholar]
- 70.Kulkarni K., Selesniemi K., Brown T.L. Interferon-γ sensitizes the human salivary gland cell line, HSG, to tumor necrosis factor-α induced activation of dual apoptotic pathways. Apoptosis. 2006;11:2205–2215. doi: 10.1007/s10495-006-0281-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histamine levels in tracheal tissue treatment control. Mice were divided into T. cruzi–infected (G1-G3) and uninfected (G4-G6) groups, where G1 and G4 were cromolyn treated, G2 and G5 were PBS treated, and G3 and G6 were untreated. Tracheal tissue was collected from every group on dpi 20 and was incubated in the presence or not of 0.05 μg/mL of compound 48/80 (MC agonist); histamine release was determined in supernatant through a radioimmunoassay. Each bar represents the mean ± SD of two independent experiments with six mice per group. *P < 0.05 comparing the same group with and without compound 48/80 stimulus.
Corticosterone levels in plasma. Mice were divided into T. cruzi–infected (G1-G3) and uninfected (G4-G6) groups, where G1 and G4 were cromolyn treated, G2 and G5 were PBS treated, and G3 and G6 were untreated. Plasma was collected on dpi 20, and corticosterone level was determined using radioimmunoassay. Each bar represents the mean ± SD of two independent experiments with six mice per group. *P < 0.05 compared with infected groups.