

Abstract
Experimental nonhuman primate models of asthma exhibit multiple features that are characteristic of an eosinophilic/T helper 2 (Th2)-high asthma subtype, characterized by the increased expression of Th2 cytokines and responsive genes, in humans. Here, we determine the molecular pathways that are present in a house dust mite–induced rhesus asthma model by analyzing the genomewide lung gene expression profile of the rhesus model and comparing it with that of human Th2-high asthma. We find that a prespecified human Th2 inflammation gene set from human Th2-high asthma is also present in rhesus asthma and that the expression of the genes comprising this gene set is positively correlated in human and rhesus asthma. In addition, as in human Th2-high asthma, the Th2 gene set correlates with physiologic markers of allergic inflammation and disease in rhesus asthma. Comparison of lung gene expression profiles from human Th2-high asthma, the rhesus asthma model, and a common mouse asthma model indicates that genes associated with Th2 inflammation are shared by all three species. However, some pathophysiologic aspects of human asthma (ie, subepithelial fibrosis, angiogenesis, neural biology, and immune host defense biology) are better represented in the gene expression profile of the rhesus model than in the mouse model. Further study of the rhesus asthma model may yield novel insights into the pathogenesis of human Th2-high asthma.
Asthma is characterized by variable airflow obstruction, airway hyperreactivity, and chronic airway inflammation. Despite these common clinical features, asthma is a heterogeneous disease that can be subclassified by a number of different measures, including the nature of the airway inflammation.1,2 Airway eosinophilia, a hallmark of T helper 2 (Th2) inflammation, defines a major subtype of severe asthma, with other major subtypes defined by neutrophilic and paucigranulocytic inflammation.1–4 Recent analyses of airway gene expression in mild-to-moderate asthmatics not being treated with corticosteroids defines a molecular signature of Th2 inflammation that correlates with airway and peripheral eosinophilia, airway hyperreactivity, subepithelial fibrosis, distinct patterns of mucin expression, and elevated IgE.5,6 The Th2-high subtype of asthma in mild-to-moderate asthmatics is associated with the cytokines IL-5 and IL-13 and is correlated with a clinical response to inhaled corticosteroid treatment.
Nonhuman primate models of asthma have been used to study pathogenic mechanisms and the efficacy of therapies, given the close similarities between monkeys and humans.7–13 Several features of lung and immune biology in nonhuman primates more accurately model human biology than the mouse, which is the most commonly used animal species for preclinical asthma studies.8 For example, in both humans and rhesus monkeys the primary distal airway is the respiratory bronchiole, whereas in rodents it is the nonalveolarized bronchiole. Basal cells are found throughout the tracheobronchial airways of both humans and rhesus monkeys but only in the tracheas of mice. In addition, Clara cells are found only in the bronchioles of both humans and rhesus monkeys but throughout the tracheobronchial airways of mice. Some unique features of asthma found in humans and rhesus monkeys but not mice include intrinsic airway hyperreactivity (except for the A/J mouse strain), smooth muscle hypertrophy in the more distal bronchi and respiratory bronchioles, exfoliation of epithelial sheets, and mast cell infiltration of airway smooth muscle.11,14,15 Thus, the study of nonhuman primate models of asthma is an important complement and supplement to studies of mouse asthma models in preclinical asthma research.
Gene expression profiling of disease tissues enables the genomewide assessment of molecular pathways that are dysregulated in disease. Although several groups have performed genomewide transcriptional profiling of preclinical mouse asthma models,16–24 and a few publications have assessed gene expression in nonhuman primate asthma models,10,25,26 data are limited on genomewide gene expression in nonhuman primate asthma models, and we are not aware of any publications that have directly compared the genomewide transcriptional profiles of human asthma with those of any preclinical asthma model, regardless of species.
A model of allergic asthma induced in rhesus monkeys by house dust mites (HDMs) exhibits multiple pathophysiologic features of human allergic asthma and has been used to assess the efficacy of immunomodulatory therapies.9–12 Here, we have conducted gene expression profiling of lung airway samples from this rhesus asthma model. We assess the relation between rhesus lung gene expression and physiologic parameters of inflammation and disease. In addition, we compare lung gene expression of human Th2-high asthma with that of the rhesus asthma model, as well as that of a commonly used mouse asthma model, to assess similarities and differences in lung gene expression between human Th2-high asthma and these two animal models of asthma.
Materials and Methods
Human Subjects
Bronchial biopsy RNA from 27 nonsmoking patients with mild-to-moderate asthma and healthy nonsmoking subjects were obtained from the University of California San Francisco Airway Tissue Bank, a specimen biorepository approved by the University of California San Francisco Committee on Human Research. Endobronchial biopsies had been collected from a subset of patients for whom we have previously described gene expression profiles of bronchial epithelial brushings.6,27 Three to six endobronchial biopsies were collected from the carinae of the second- to fourth-order bronchi. Informed consent was obtained from all human subjects.
Rhesus Model of Allergic Asthma
An HDM-induced allergic asthma model in young adult rhesus monkeys (Macaca mulatta) has been described previously.12 The model consisted of an 18-month disease development phase, followed by a treatment phase. Data from the treatment phase was published previously.12 Data from the 18-month disease development phase is presented in this article. The animal protocol was approved by the Institutional Animal Care and Use Committee Ethical Review Board at the University of California Davis. All monkeys were selected from the California National Primate Research Center's breeding colony on the basis of social rank, treated with ivermectin subcutaneously at 0.2 mg/kg for potential parasites, and isolated indoors for 1 month. Briefly, 12 adult rhesus monkeys were sensitized with a subcutaneous injection of HDM allergen extract followed by 3 biweekly intranasal instillations of HDM and 6 weekly aerosol challenges of HDM. Ten adult rhesus monkeys were subjected to control treatment with PBS injections and mock aerosol challenges. After the sensitization procedure, sensitized monkeys were regularly exposed to aerosolized HDM for 2 to 3 hours twice a week, for a total of 18 months, and also received additional subcutaneous and intranasal HDM boosts at weeks 56, 71, and 75. Although all 12 sensitized animals were enrolled and characterized for the duration of the study, only 4 of the 10 control animals were kept and characterized beyond the postsensitization time point, because of cost constraints. The six control animals that were dropped from the study were randomly selected for exclusion such that there were no differences in the mean group values of measured parameters in the control group between the 4-animal subset and the entire group of 10 control animals. Data and samples were collected over a 2-week period, starting at weeks −4 to −8 for the presensitization time point, week 15 for the postsensitization time point, and week 75 for the 18-month time point, whereby week 0 denotes the beginning of the sensitization protocol. In the first week of data and sample collection, peripheral blood and serum samples were obtained just before an aerosol challenge for determination of complete blood counts, flow cytometric analysis, and serum ELISAs. Blood leukocyte values and differential counts were determined as described previously.11 Forty-eight hours after the aerosol challenge, pulmonary mechanics and airway hyperreactivity to methacholine were determined as described previously.11 In the second week of data and sample collection, bronchoalveolar lavage fluid (BALF) samples were collected 48 hours after an aerosol challenge for determination of leukocyte differentials and flow cytometric analysis, as described previously.11 Immediately after the BALF collection, five lung biopsies were obtained from the subcarina at the lower or middle lobes of the lung by flexible bronchoscopy with the use of a 1-mm biopsy forceps, after intubation with an appropriate-sized, cuffed endotracheal tube. Animals were deprived of food 8 hours before the bronchoscopy to minimize the risk of aspiration during anesthesia. During the bronchoscopy procedure, oxygen saturation and heart rate monitoring were provided continuously, and supplemental oxygen was routinely given. The bronchoscope (2.7 to 3.6 mm in diameter; Olympus, Center Valley, PA) was passed through the nose, 5 mg of lidocaine was instilled on the larynx for topical anesthesia, and the bronchoscope was directed into the trachea. A second dose of 5 mg of lidocaine was administered through the bronchoscope for topical anesthesia and to reduce the cough reflex, and the bronchoscope was directed to the thin shelf of tissues dividing segmental or subsegmental airways. Three biopsies were preserved in RNAlater RNA stabilization reagent (Qiagen, Valencia, CA) for RNA extraction and gene expression profiling, and two biopsies were reserved for histopathology analyses. Intradermal skin testing for reactivity to HDM antigen was performed as described previously11 at approximately 5 months before the start of sensitization for the presensitization time point and at week 5 for the postsensitization time point, whereby week 0 denotes the beginning of the sensitization protocol. During all data and sample collection, animals were sedated with ketamine (10 mg/kg, i.m.) and then anesthetized with propofol (0.1 to 0.2 mg/kg/min, i.v.), with the dose adjusted as deemed necessary by the attending veterinarian.
Flow Cytometry
Peripheral blood mononuclear cells and BALF cells were prepared for immunofluoresence staining and analyzed by flow cytometry on a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ).
Total and HDM-Specific IgE ELISAs
Total IgE in rhesus monkey sera was measured by ELISA with the use of monoclonal anti-IgE MAE11 (Genentech, South San Francisco, CA) for capture and peroxidase-conjugated goat anti-human IgE antibody (Kirkegaard and Perry Laboratories, Gaithersburg, MD) for detection. The standard range was 0.098 to 12.5 ng/mL for human IgE. The minimum sample dilution was 1:10 to avoid any interference from sera in the assay. Rhesus monkey IgE concentrations were calculated by dividing the concentrations obtained on the basis of a human IgE standard curve by a correlation factor of 0.029, which was determined with purified cynomolgus monkey IgE (Genentech). HDM-specific IgE titers were measured by ELISA with the use of monoclonal anti-IgE MAE11 for capture and biotinylated HDM allergen Der f1 (Indoor Biotechnologies, Charlottesville, VA) for detection, followed by horseradish peroxidase–conjugated streptavidin (GE Healthcare, Little Chalfont, Buckinghamshire, UK). For calculation of titers, a cut point was set at twice the absorbance of a 1:100 diluted blank rhesus monkey serum (Bioreclamation, Westbury, NY). The dilution factor at which an absorbance value equaled the cut point was calculated from a linear interpolation of absorbance values obtained from serial dilutions of samples. Titer is reported as the log10 of the dilution factor. Titers for negative samples are reported as <1.52 because a minimum sample dilution factor of 33.3 was used.
Histopathology
Biopsies were placed individually in cryomolds and submerged in optimal cutting temperature compound media (Sakura Finetek, Torrance, CA). Cryomolds were then chilled to set optimal cutting temperature compound and stored at −80°C. All sections were cut 5-μm thick and stained with H&E for histologic analysis. All biopsies were scored in a blinded manner at the time of collection.
Microarray Gene Expression Profiling
Human lung airway biopsy RNA was isolated from homogenized bronchial biopsies as described previously.5,6 Total RNA was extracted from individual biopsy samples with the use of RNeasy Mini Kits (Qiagen), following the manufacturer's guidelines, and all biopsy RNA from each individual subject was pooled for further processing and analysis. RNA samples were quantified with a Nanodrop ND-1000 UV-spectrophotometer (Thermo Scientific, West Palm Beach, FL), and RNA quality was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). The quantity of total RNA used in a two-round amplification protocol ranged from 10 ng to 50 ng per sample. First-round amplification and second-round cDNA syntheses were done with the Message Amp II aRNA Amplification Kit (Applied Biosystems, Foster City, CA). Cyanine-5 dye was incorporated with the Quick Amp Labeling kit (Agilent Technologies). Each cyanine-5–labeled test sample (750 ng) was pooled with cyanine-3–labeled Universal Human Reference RNA (Stratagene, La Jolla, CA) and hybridized onto Agilent Whole Human Genome 4 × 44K arrays as described in the manufacturer's protocol. Arrays were washed, dried, and scanned on the Agilent scanner according to the manufacturer's protocol. Microarray image files were analyzed with Feature Extraction software 9.5 (Agilent Technologies). Human lung airway biopsy microarray data have been deposited in Gene Expression Omnibus with the accession code GSE23611.
Rhesus lung airway biopsies were recovered from RNAlater and homogenized in RLT buffer (Qiagen) with the use of an MM300 mixer mill (Retsch, Haan, Germany). RNA was isolated from homogenized tissue with the use of RNeasy Micro Kits (Qiagen) with on-column DNase treatment, following the manufacturer's guidelines, and concentrated by ethanol precipitation. RNA samples were quantified with a Nanodrop ND-1000 UV-spectrophotometer (Thermo Scientific), and RNA quality was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies). Subsequently, RNA was amplified with the Low RNA Input Fluorescent Linear Amplification protocol (Agilent Technologies). A T7 RNA polymerase single round of linear amplification was performed to incorporate Cyanine-3 and Cyanine-5 label into cRNA. Each Universal Human Reference (Stratagene) cRNA labeled with Cyanine-3 and test sample cRNA labeled with Cyanine-5 (750 ng) was fragmented for 30 minutes at 60°C before loading onto Agilent Whole Human Genome microarrays (Agilent Technologies). Samples were hybridized for 18 hours at 60°C with constant rotation. Microarrays were washed, dried, and scanned on the Agilent scanner according to the manufacturer's protocol. Microarray image files were analyzed with Feature Extraction software version 7.5 with default parameters and Lowess normalization to yield summary ratio data (Agilent Technologies). Rhesus lung airway biopsy microarray data have been deposited in Gene Expression Omnibus with the accession code GSE23327.
Statistical and Bioinformatic Analyses
Rhesus model data were expressed as mean ± SD. P values were calculated with JMP version 8.0.1 (SAS Institute, Cary, NC). A Wilcoxon/Kruskal–Wallis Test (rank sums) was used to compare the sensitized and nonsensitized groups. P values < 0.05 were considered significant.
For analysis of human gene expression microarray data, all statistical calculations were performed with R for Windows version 2.9.2, JMP version 8.0.1 (SAS Institute), and Partek Genomics Suite 6.5 (Partek Incorporated, St Louis, MO). Probes with features <50% present were removed before each statistical test. Differential gene expression analysis between [Th2-high asthma] and [Th2-low asthma AND healthy control] was conducted by Welch's t-test, because of unequal sample sizes (14 and 26, respectively), and unequal variances were detected by Bartlett's test among array features.
For analysis of rhesus gene expression microarray data, all statistical calculations were performed with the R Project software package, version 2.10.0. Linear modeling was performed with the Limma package of Bioconductor. Control probes, probes exhibiting CVs in the lowest quartile, and probes without Entrez Gene annotation were excluded. In cases in which multiple probes mapped to the same Entrez Gene record, the probe with the highest CV was retained, and the others were excluded. Moderated t-statistics from modeling were used to calculate adjusted P values with the use of the method of Benjamini–Hochberg. Probes were defined as differentially expressed when the false discovery rate was <20%. Correlation coefficients between gene expression and physiologic parameters were performed with Spearman's method. Nominal correlation P values were calculated with algorithm AS 89 and adjusted with Storey's q-value method. For comparison of human, rhesus, and mouse gene expression microarray data, expression data from murine studies were linked to orthologous human genes with the use of National Center for Biotechnology Information annotation: first, GenBank transcript annotation for each human or murine probe was linked to a gene of the same species by GenBank annotation; second, human and mouse genes were linked by Homologene cluster data. Statistical thresholds for determining differential expression in the animal models were P value was <0.05 and fold change was >1.2.
Results
A Rhesus Asthma Model Exhibits Features of Allergic/Th2 Inflammation
We developed a model of allergic asthma in rhesus monkeys, as described previously,12 in which 12 adult rhesus monkeys were sensitized to HDM allergen and 10 adult rhesus monkeys were subjected to mock sensitization. During 18 months of disease development and progression, we assessed lung physiologic parameters (pulmonary mechanics and airway hyperreactivity to methacholine), lung inflammatory parameters (BALF cell composition and histology of lung airway biopsies), and systemic parameters (total and HDM-specific serum IgE, complete peripheral blood count, and skin test reactivity to HDM) at presensitization, 15-week postsensitization, and 18-month time points. Overall, animals in the sensitized group developed several physiologic features of asthma, including airway hyperreactivity, lung inflammation, and systemic allergic sensitization, with some heterogeneity in disease parameters on an animal-to-animal basis, as would be expected in an outbred population of rhesus monkeys. These physiologic features of disease are comparable to those observed in previous studies of the rhesus asthma model9–11 and are also similar in magnitude to those observed in human asthma.
As shown in Figure 1, A and B, BALF eosinophils and HDM-specific serum IgE titers increased in sensitized animals during the disease development phase at both the postsensitization and 18-month time points. Sensitized animals also exhibited a statistically significant increase in BALF neutrophils and in the extent of activated CD4 T cells in the BALF (see Supplemental Figure S1 at http://ajp.amjpathol.org). Peripheral blood eosinophils (Figure 1C), total BALF lymphocytes (Figure 1D), and total serum IgE (Figure 1E) were not significantly different between the sensitized and nonsensitized animals and did not change during the disease development period. Blinded scoring of histologic analysis of airway biopsies was consistent with the development of airway inflammation in the sensitized group. Although the small size of the rhesus monkeys created challenges for biopsy collection and quality, mild-to-moderate airway inflammation was apparent in most of the sensitized animals from which data could be obtained (Figure 2, A and B; see also Supplemental Table S1 at http://ajp.amjpathol.org). In addition, all sensitized animals developed skin test reactivity to HDM that was apparent at both the postsensitization and the 18-month time points (see Supplemental Table S2 at http://ajp.amjpathol.org).
Figure 1.
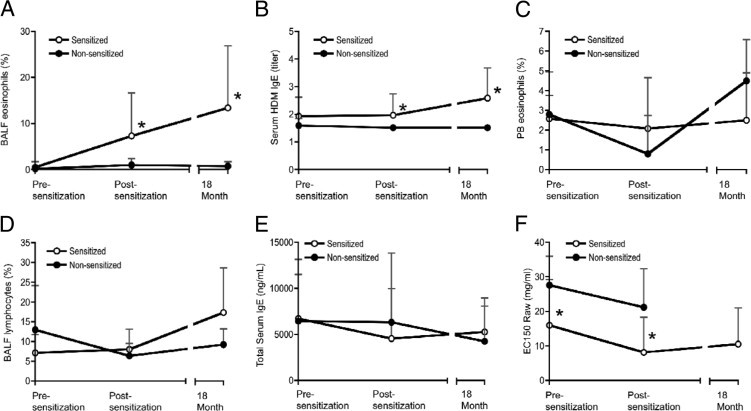
Sensitization to HDM antigen induces allergic inflammation and airway hyperreactivity in rhesus monkeys. Twelve rhesus monkeys were sensitized with a subcutaneous injection of HDM antigen extract and intranasal HDM, followed by regular exposure to aerosolized HDM for 2 to 3 hours twice a week, for a total of 18 months. Sensitized rhesus monkeys also received additional subcutaneous and intranasal HDM boosts at weeks 56, 71, and 75. Ten rhesus monkeys were subjected to control treatment with PBS injections and mock aerosol challenges. Data were collected at week −4 to −8 for the presensitization time point, week 15 for the postsensitization time point, and week 75 for the 18-month time point, whereby week 0 denotes the beginning of the sensitization protocol, for all 12 sensitized and all 10 nonsensitized monkeys, except for the 18-month time point, whereby data were collected for all 12 sensitized and only 4 of the 10 nonsensitized monkeys. BALF eosinophils (A), serum HDM IgE titers (B), peripheral blood (PB) eosinophils (C), BALF lymphocytes (D), total serum IgE (E), and airway hyperreactivity to methacholine (F) in sensitized and nonsensitized groups at presensitization, postsensitization, and 18-month time points. Airway hyperreactivity is expressed as EC150, the effective concentration of methacholine required to increase airway resistance (Raw) to 150% of baseline, whereby a lower EC150 indicates greater airway hyperreactivity. Data are expressed as mean ± SD. *P < 0.05 versus nonsensitized animals, Wilcoxon/Kruskal-Wallis Test (rank sums).
Figure 2.
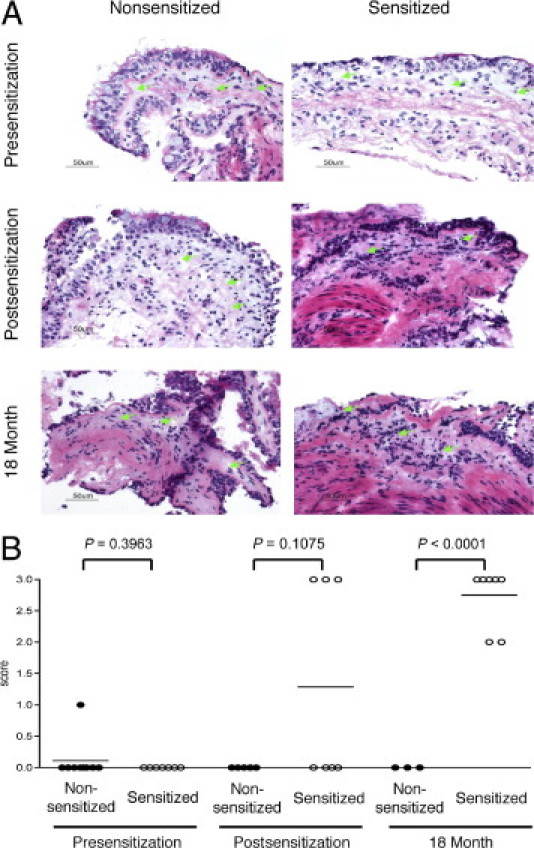
Sensitization to HDM antigen induces airway inflammation in rhesus monkeys. A: Inflammatory cell infiltration of the airway lamina propria was assessed in lung biopsies from sensitized and nonsensitized monkeys at presensitization, postsensitization, and 18-month time points. Original magnification, ×40; H&E. Green arrows indicate lamina propria. No difference is seen in lamina propria cellularity between groups in the presensitization biopsies. Increased lamina propria cellularity is seen in sensitized animals at the postsensitization and 18-month time points. B: An increase in inflammation is observed in the sensitized versus nonsensitized group biopsies at the postsensitization and 18-month time points. Inflammation was scored in the lamina propria underlying airway epithelium, when assessable; 0 indicates no inflammatory infiltrate; 1, minimal inflammation/rare scattered immune cells; 2, mild inflammation/scattered immune cells; 3, moderate inflammation/immune cell clusters; and 4, severe inflammation/diffuse immune cell infiltrate. Individual animal scores are shown as individual circles, with the mean inflammation score for a group of animals indicated as a line. *P < 0.0001 (mean inflammation score) versus nonsensitized animals, Dunnett's test.
The sensitized group was selected to have a greater average intrinsic airway hyperreactivity to methacholine than the nonsensitized group at the presensitization time point, to model the intrinsic airway hyperreactivity observed in human asthma. With the use of a definition of airway hyperreactivity of a methacholine EC150 value of <8 mg/mL, which is similar to the definition of airway hyperreactivity in human asthmatics,28 4 of the 12 animals in the sensitized group and none of the animals in the nonsensitized group were hyperreactive before sensitization. Both sensitized and nonsensitized groups had increased airway hyperreactivity at the postsensitization time point, compared with the presensitization time point (Figure 1F). However, many more animals in the sensitized group were hyperreactive at the postsensitization time point, compared with the nonsensitized group (9 of the 12 animals in the sensitized group were hyperreactive, whereas only 1 of the 10 animals in the nonsensitized group was hyperreactive). When we compared all 12 animals in the sensitized group with all 10 animals in the nonsensitized group at the presensitization and postsensitization time points, we found no statistically significant difference in the change in airway hyperreactivity between the groups, because of minimal changes or slight decreases in the hyperreactivity of the 4 animals in the sensitized group that were already hyperreactive at the presensitization time point. However, when we compared the 8 animals in the sensitized group and the 10 animals in the nonsensitized group that were not hyperreactive before sensitization and which had comparable levels of airway hyperreactivity at the presensitization time point, we found that the sensitized group showed a greater increase in airway hyperreactivity at the postsensitization time point than the nonsensitized group, indicating that the sensitization protocol induced airway hyperreactivity in the sensitized group (see Supplemental Figure S2 at http://ajp.amjpathol.org).
The Rhesus Asthma Model Exhibits a Th2 Inflammation Gene Expression Signature in the Lung Airways
To compare the molecular pathways and mediators in the rhesus asthma model with those of human asthma, we analyzed lung airway biopsies from all 12 sensitized and all 10 nonsensitized animals at the postsensitization time point. RNA from these biopsies was prepared and analyzed on Agilent Whole Human Genome expression microarray chips, and genes were assessed for differential expression by considering pairwise comparisons between sensitized and control animals. We assessed the genes that were differentially expressed in the postsensitization time point samples from sensitized rhesus monkeys (versus nonsensitized) and compared them with the genes that were differentially expressed in lung airway biopsies from a previously described cohort of mild-to-moderate asthmatic patients who were not being treated with steroids (versus healthy controls).5,6
Our previous studies identified a set of three genes that are specifically induced in human asthmatic epithelium, are directly regulated by the Th2 cytokine IL-13 in vitro, and can serve as a surrogate marker of Th2 inflammation in the lung.6,27 These three genes (periostin, chloride channel regulator 1/CLCA1, and serpin peptidase inhibitor clade B member 2/serpinB2) are increased not only in human asthmatic epithelial cell brushings but also in matched lung biopsies and define a Th2-high subset of patients who have airway eosinophilia, increased serum IgE, airway hyperreactivity, and subepithelial fibrosis.6 Additional analysis of lung biopsies from Th2-high asthmatics identified a set of 93 probes corresponding to 79 uniquely annotated genes that are highly intercorrelated and that define a larger Th2 inflammation gene set in lung biopsies, compared with the 3-gene set in lung epithelium.5 Several other groups have also defined Th2 inflammation gene sets, and many of the genes comprising our epithelial 3-gene set and our biopsy 79-gene set have also been identified by other groups in their Th2 inflammation gene sets and/or have been previously shown to be induced by the Th2 cytokines IL-4 and IL-13.5,19,20,25,26,29–34
We found that periostin, CLCA1, and serpinB2 are increased in rhesus asthma lung biopsies (Figure 3A). Moreover, the human biopsy 79-gene set is correlated between human asthma and rhesus asthma (Figure 3B). Taken together, these data indicate that there is a strong Th2 inflammation gene signature in rhesus asthma and that the expression of Th2 inflammation genes is similar between human Th2-high asthma and rhesus asthma.
Figure 3.
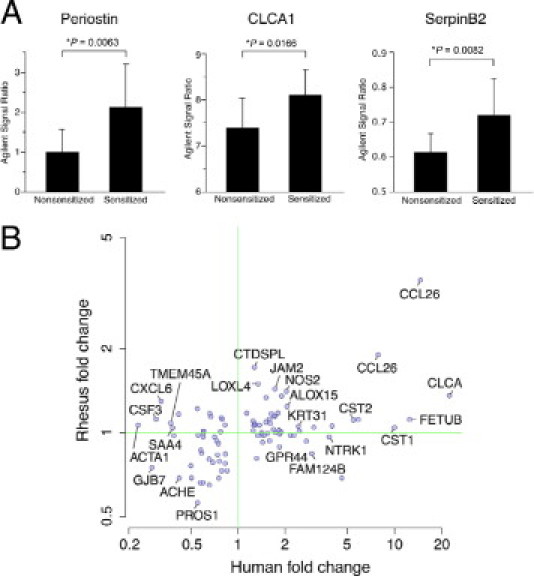
A Th2 inflammation gene set is shared and correlated in lung airway biopsies of human asthma and a rhesus asthma model. A: Each gene in a previously defined human asthma epithelial three-gene set is increased in rhesus asthma biopsies. Microarray signal ratios for periostin (Agilent probe ID A_24_P347411), CLCA1 (Agilent probe ID A23_P153185), and serpinB2 (Agilent probe ID A_23_P51217) are expressed as mean ± SD. *P < 0.05 versus nonsensitized animals, t-test. B: A previously defined human asthma biopsy 79-gene set is correlated in human and rhesus asthma (Pearson's correlation coefficient ρ = 0.495; P = 4.5 × 10−7). The fold change for each of 93 Agilent probes corresponding to 79 uniquely annotated genes is compared for human Th2-high asthma (versus Th2-low asthma and healthy controls) and rhesus asthma (versus nonsensitized animals). Each point corresponds to one probe, but for clarity only a subset of the 93 probes is labeled.
Th2 Inflammation Genes Correlate with Physiologic Measures of Th2 Inflammation and Disease in the Rhesus Asthma Model
Because our Th2 inflammation genes correlated with physiologic parameters of Th2 inflammation and disease in human asthma,5,6 we investigated whether this was also true for the rhesus asthma model. We assessed the correlation of our Th2 inflammation gene set at the postsensitization time point in the rhesus asthma model with BALF eosinophils, BALF lymphocytes, peripheral blood eosinophils, total serum IgE, HDM-specific serum IgE, and airway hyperreactivity to methacholine. To reduce the statistical penalty for multiple testing, we limited the genes tested to a subset of 25 genes from the previously defined 79-gene set in human asthmatics. This 25-gene set was generated by first applying filters of a fold change > 1.2 and a q-value < 0.05 to whole-genome expression profiles of lung airway biopsies from the Th2-high subset of asthmatic patients (comparing the Th2-high asthma subset with both the Th2-low asthma subset and the healthy control subjects) to generate a 35-gene set, followed by an additional filter of increased expression (fold change > 1) in rhesus asthma lung airway biopsies compared with nonsensitized control rhesus lung airway biopsies to generate the final 25-gene set. Many of the genes in the 25-gene set were modestly up-regulated in rhesus lung airway biopsies with respect to the mean fold change between sensitized and nonsensitized groups, because of the heterogeneity of the animals in each group. Importantly, however, there was sufficient variability in gene expression and physiologic parameters in individual animals to support testing for correlations.
With the use of a cutoff threshold corresponding to a false discovery rate of 20%, we found that 10 of the 25 genes were correlated with BALF eosinophils, 6 of the 25 genes were correlated with HDM-specific serum IgE levels and peripheral blood eosinophils, and 5 of the 25 genes were correlated with airway hyperreactivity. This correlation between Th2 inflammation genes and disease parameters on an animal-to-animal basis in the sensitized group is similar to that observed in human asthma.5,6 There was no correlation between the set of 25 genes and total serum IgE and very little correlation between the set of 25 genes and BALF lymphocytes, because only 1 of the 25 genes was correlated with BALF lymphocytes. A summary of the correlation of the 25 genes with the various physiologic parameters is shown in Table 1, and a plot of the correlation of each gene in the 25-gene set with BALF eosinophils is shown in Supplemental Figure S3 at http://ajp.amjpathol.org.
Table 1.
Correlation of Th2 Inflammation Signature Genes with Physiological Parameters of Th2 Inflammation and Disease in the Rhesus Asthma Model
| Entrez gene ID | Gene symbol | Gene Name | Human biopsy fold change⁎ | Rhesus biopsy fold change† | BALF eosinophil |
BALF lymphocyte |
PB eosinophil |
Total IgE |
Serum HDM IgE |
AHR (EC150)¶ |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ρ‡ | P§ | ρ‡ | P§ | ρ‡ | P§ | ρ‡ | P§ | ρ‡ | P§ | ρ‡ | P§ | |||||
| 10344 | CCL26 | Chemokine (C-C motif) ligand 26 | 4.06 | 3.22 | 0.736∥ | 0.001∥ | 0.225 | 0.37 | 0.283 | 0.292 | −0.075 | 0.767 | 0.628∥ | 0.005∥ | −0.604∥ | 0.008∥ |
| 84171 | LOXL4 | Lysyl oxidase-like 4 | 1.27 | 1.53 | 0.653∥ | 0.005∥ | −0.022 | 0.966 | 0.405 | 0.128 | −0.027 | 0.936 | 0.559∥ | 0.02∥ | −0.292 | 0.263 |
| 4843 | NOS2A | Nitric oxide synthase 2A (inducible, hepatocytes) | 1.39 | 1.42 | 0.245 | 0.328 | 0.389 | 0.11 | 0.615∥ | 0.007∥ | 0.112 | 0.656 | 0.238 | 0.341 | −0.568∥ | 0.014∥ |
| 948 | CD36 | CD36 molecule (thrombospondin receptor) | 1.66 | 1.33 | 0.211 | 0.401 | −0.302 | 0.224 | 0.468∥ | 0.062∥ | 0.195 | 0.436 | 0.465∥ | 0.052∥ | −0.464∥ | 0.052∥ |
| 1012 | CDH13 | Cadherin 13, H-cadherin (heart) | 1.24 | 1.30 | 0.433 | 0.073 | 0.003 | 0.99 | 0.643∥ | 0.005∥ | −0.096 | 0.705 | 0.418 | 0.084 | −0.322 | 0.192 |
| 58494 | JAM2 | Junctional adhesion molecule 2 | 1.40 | 1.29 | 0.513∥ | 0.029∥ | −0.313 | 0.206 | 0.509∥ | 0.037∥ | 0.061 | 0.811 | 0.458∥ | 0.056∥ | −0.22 | 0.38 |
| 51751 | HIGD1B | HIG1 domain family, member 1B | 1.54 | 1.18 | 0.159 | 0.528 | −0.1 | 0.693 | 0.176 | 0.5 | −0.015 | 0.954 | 0.326 | 0.187 | −0.587∥ | 0.01∥ |
| 1179 | CLCA1 | Chloride channel, calcium activated, family member 1 | 5.76 | 1.17 | 0.311 | 0.209 | 0.457∥ | 0.057∥ | 0.046 | 0.843 | 0.088 | 0.729 | 0.231 | 0.356 | −0.22 | 0.38 |
| 7855 | FZD5 | Frizzled homolog 5 (Drosophila) | 1.42 | 1.16 | 0.384 | 0.116 | 0.062 | 0.806 | 0.659∥ | 0.004∥ | 0.007 | 0.98 | 0.392 | 0.107 | −0.224 | 0.371 |
| 1470 | CST2 | Cystatin SA | 2.78 | 1.13 | 0.449∥ | 0.062∥ | 0.098 | 0.699 | −0.156 | 0.561 | −0.106 | 0.674 | 0.088 | 0.729 | −0.066 | 0.796 |
| 3497 | IGHE | Immunoglobulin heavy chain epsilon | 2.57 | 1.12 | 0.441∥ | 0.067∥ | 0.165 | 0.512 | −0.148 | 0.572 | −0.228 | 0.361 | 0.057 | 0.823 | −0.172 | 0.495 |
| 522 | ATP5J | ATP synthase, H+ transporting, subunit F6 | 1.42 | 1.12 | 0.382 | 0.118 | −0.065 | 0.799 | 0.681∥ | 0.003∥ | −0.026 | 0.921 | 0.351 | 0.153 | −0.208 | 0.409 |
| 246 | ALOX15 | Arachidonate 15-lipoxygenase | 1.44 | 1.12 | −0.132 | 0.602 | −0.104 | 0.681 | 0.256 | 0.31 | 0.24 | 0.335 | 0.187 | 0.457 | −0.462∥ | 0.054∥ |
| 26998 | FETUB | Fetuin B | 3.34 | 1.12 | 0.602∥ | 0.008∥ | 0.369 | 0.131 | 0.145 | 0.579 | 0.059 | 0.818 | 0.268 | 0.283 | −0.22 | 0.38 |
| 13 | AADAC | Arylacetamide deacetylase (esterase) | 1.48 | 1.10 | −0.138 | 0.585 | 0.154 | 0.542 | 0.347 | 0.187 | −0.148 | 0.558 | −0.072 | 0.776 | −0.195 | 0.438 |
| 84969 | C20orf100 | TOX high mobility group box family member 2 | 1.44 | 1.08 | 0.518∥ | 0.028∥ | −0.085 | 0.736 | 0.115 | 0.662 | −0.146 | 0.563 | 0.282 | 0.257 | −0.245 | 0.327 |
| 116372 | LYPD1 | LY6/PLAUR domain containing 1 | 1.44 | 1.08 | 0.583∥ | 0.011∥ | −0.095 | 0.709 | 0.327 | 0.222 | 0.051 | 0.843 | 0.441∥ | 0.067∥ | 0.103 | 0.684 |
| 116159 | CYYR1 | Cysteine/tyrosine-rich 1 | 1.24 | 1.07 | 0.306 | 0.444 | −0.229 | 0.569 | 0.575 | 0.132 | 0.015 | 0.88 | 0.664∥ | 0.042∥ | −0.208 | 0.417 |
| 9951 | HS3ST4 | Heparan sulfate (glucosamine) 3-O-sulfotransferase 4 | 2.78 | 1.06 | 0.5∥ | 0.035∥ | −0.019 | 0.941 | 0.141 | 0.622 | 0.187 | 0.456 | 0.377 | 0.123 | −0.306 | 0.218 |
| 1469 | CST1 | Cystatin SN | 3.21 | 1.06 | 0.195 | 0.438 | 0.018 | 0.944 | −0.251 | 0.339 | −0.226 | 0.366 | 0.062 | 0.806 | 0.041 | 0.873 |
| 3881 | KRT31 | Keratin 31 | 1.41 | 1.05 | 0.449∥ | 0.062∥ | −0.056 | 0.825 | −0.173 | 0.497 | 0.053 | 0.837 | 0.36 | 0.142 | −0.268 | 0.282 |
| 7850 | IL1R2 | Interleukin 1 receptor, type II | 1.33 | 1.02 | 0.315 | 0.203 | −0.313 | 0.206 | 0.092 | 0.688 | 0.154 | 0.541 | 0.404 | 0.097 | −0.233 | 0.353 |
| 390010 | NKX1-2 | NK1 homeobox 2 | 1.52 | 1.01 | −0.008 | 0.974 | 0.304 | 0.22 | 0.251 | 0.273 | −0.373 | 0.129 | 0.133 | 0.598 | 0.118 | 0.641 |
| 79861 | TUBAL3 | Tubulin, alpha-like 3 | 1.49 | 1.01 | −0.057 | 0.823 | 0.145 | 0.567 | −0.251 | 0.349 | −0.071 | 0.78 | 0.023 | 0.929 | −0.274 | 0.271 |
| 5587 | PRKD1 | Protein kinase D1 | 1.29 | 1.00 | −0.011 | 0.967 | 0.149 | 0.556 | −0.175 | 0.514 | −0.148 | 0.558 | −0.152 | 0.548 | 0.191 | 0.448 |
PB, peripheral blood; AHR, airway hyperreactivity; EC150, the effective concentration of methacholine required to increase airway resistance to 150% of baseline.
Mean fold increase in expression of a gene in the Th2-high subset of human asthma (comparing the Th2-high asthma subset with both the Th2-low asthma subset and the healthy control subjects).
Mean fold-increase in expression of a gene in sensitized rhesus monkeys compared with nonsensitized rhesus monkeys.
Correlation coefficient.
Nominal P value from the Spearman's correlation test.
Airway hyperreactivity is expressed as EC150, whereby a lower EC150 indicates greater hyperreactivity, such that negative correlations with EC150 indicate a positive correlation with airway hyperreactivity.
Data have passed threshold for significance (P = 0.069) at a false discovery rate of 20%.
Comparison of Human, Rhesus, and Mouse Asthma Gene Expression Profiles Shows Common and Distinct Pathophysiologies
The most commonly used species in preclinical animal models of asthma is the mouse. To assess which aspects of human Th2 asthma are recapitulated in rhesus and mouse asthma models, we compared a published lung gene expression profile in a standard mouse asthma model with the airway biopsy gene expression profiles in rhesus asthma and human Th2 asthma. We reanalyzed the lung gene expression microarray data from a published study of an acute ovalbumin-induced asthma model in BALB/c mice (BALB/c OVA)21 with the use of the same analytic methods that we applied to our rhesus and human data sets, and compared the results with the use of homology links between human and mouse genes defined by Homologene.35 Although there are multiple variations of mouse asthma models consisting of different sensitization and challenge protocols, as well as different antigens, the BALB/c OVA mouse asthma model whose gene expression we analyzed is the most commonly used version of mouse asthma in the literature. As expected, our analysis of the BALB/c OVA gene expression data set produced results similar to those originally published.
When comparing genes that are commonly up-regulated in human, rhesus, and BALB/c OVA mouse asthma, we found that a dominant gene signature that is shared among all three species contains genes that are associated with Th2 inflammation. Of the 35-gene set that represents the Th2 inflammation signature in the Th2-high subset of human asthma, as described above, many of the genes are also up-regulated in either sensitized rhesus or sensitized mice, although few of the genes are up-regulated in both species (Figure 4).
Figure 4.
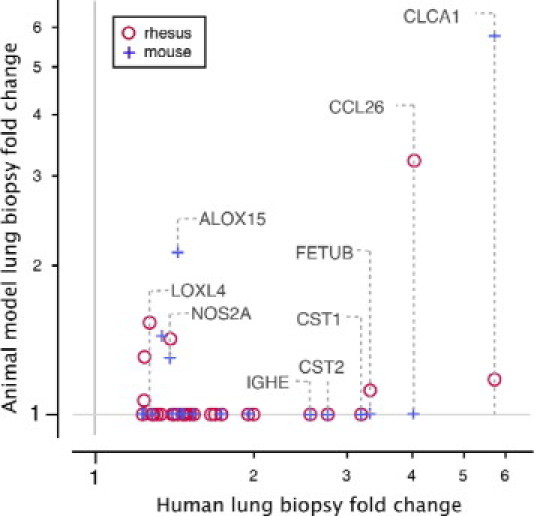
Human asthma Th2 inflammation genes are also up-regulated in rhesus and mouse models of asthma. Thirty-five genes that represent a Th2 inflammation signature in the Th2-high subset of human asthma, defined by applying filters of a fold change >1.2 and a q-value of <0.05 to whole-genome expression profiles of lung airway biopsies from the Th2-high subset of asthmatic patients (comparing the Th2-high asthma subset with both the Th2-low asthma subset and the healthy control subjects), were tested for differential expression in the rhesus asthma model and an OVA-induced mouse asthma model. The fold change of each gene in each model species that meets a false discovery rate cutoff of 20% is plotted against the fold change of that gene in human Th2-high asthma. The fold change of rhesus and mouse asthma model genes that do not meet the false discovery rate cutoff is plotted as 1.0.
To more broadly assess the similarities and differences between human, rhesus, and BALB/c OVA mouse asthma, all genes differentially expressed in human Th2-high asthma biopsies (comparing the Th2-high asthma subset with both the Th2-low asthma subset and the healthy control subjects) were examined for significant differential expression in rhesus and BALB/c OVA asthma. Most genes that were differentially expressed in disease were specific to a single species, with 309 genes in common between human and rhesus asthma and 217 genes in common between human and BALB/c OVA asthma (Figure 5). Only 11 genes were up-regulated in all three species (see Supplemental Table S3 at http://ajp.amjpathol.org). Of these 11 genes, periostin has been previously described to be associated with Th2 inflammation and is produced downstream of IL-4 and IL-13 signaling.6,27
Figure 5.
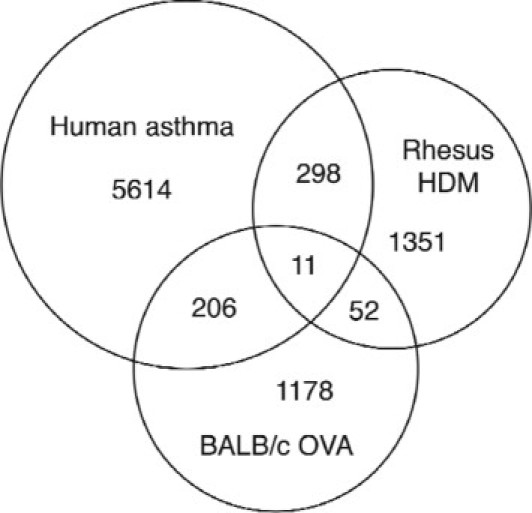
A subset of genes is commonly up-regulated among human Th2-high asthma, rhesus asthma, and/or mouse asthma. The Venn diagram depicts the total number of genes that are up-regulated in human Th2-high asthma, HDM-sensitized rhesus monkeys, and OVA-sensitized BALB/c mice. Intersections of the Venn diagram depict the total number of genes that are shared by two or three species.
To determine which biological pathways in human Th2-high asthma are specifically represented in either rhesus or BALB/c OVA asthma, we used the Database for Annotation, Visualization, and Integrated Discovery36 to identify sets of genes from the Gene Ontology, SwissProt/Protein Information Resource, KEGG, INTERPRO, and SMART databases that are enriched among the genes shared by human and rhesus asthma or human and mouse asthma (last accessed May 13, 2010). A total of 30 groups with an adjusted P value of <10−1 are specific to human Th2-high asthma and rhesus asthma and describe biological processes (Table 2). These groups are enriched for genes associated with extracellular matrix biology, cell morphogenesis, nervous system biology, immune host defense biology, and angiogenesis (Table 3). In contrast, only four groups with an adjusted P value of <10−1 are specific to human Th2-high asthma and BALB/c OVA asthma and describe biological processes (Table 4). These groups include extracellular matrix biology, sequence-specific DNA-binding proteins, and proteins associated with developmental biology.
Table 2.
Increased Expression of Biological Gene Sets in Both Human Th2-High Asthma and the Rhesus Asthma Model, as Identified by DAVID Analysis
| DAVID term (biology) | P⁎ |
|---|---|
| Extracellular region part | 4.19 × 10−5 |
| Extracellular matrix | 8.08 × 10−5 |
| Proteinaceous extracellular matrix | 1.90 × 10−4 |
| Cellular component morphogenesis | 3.77 × 10−4 |
| Cell morphogenesis involved in differentiation | 5.73 × 10−4 |
| Cell morphogenesis | 0.00107 |
| Cell projection organization | 0.00126 |
| Axonogenesis | 0.00156 |
| Cell projection morphogenesis | 0.00253 |
| Neuron projection morphogenesis | 0.00267 |
| Cell morphogenesis involved in neuron differentiation | 0.00294 |
| Neuron projection development | 0.00313 |
| Cell part morphogenesis | 0.00313 |
| Extracellular matrix | 0.00415 |
| Extracellular region | 0.0193 |
| Biological adhesion | 0.0410 |
| Cell motion | 0.0423 |
| Axon guidance | 0.0426 |
| Cell adhesion | 0.0429 |
| Neuron development | 0.0429 |
| Defense response | 0.0452 |
| Response to extracellular stimulus | 0.0453 |
| Neuron differentiation | 0.0460 |
| Regulation of nervous system development | 0.0577 |
| Angiogenesis | 0.0619 |
| Extracellular space | 0.0643 |
| Cell adhesion | 0.0753 |
| Osteogenesis | 0.0865 |
| Developmental protein | 0.0911 |
| Cell junction | 0.0920 |
DAVID, Database for Annotation, Visualization, and Integrated Discovery.
Corrected by the method of Benjamini-Hochberg for multiple comparisons.
Table 3.
Genes that Comprise the Extracellular Matrix, Axonogenesis, Defense Response, and Angiogenesis Gene Sets Identified by DAVID Analysis of Human Th2-High Asthma and the Rhesus Asthma Model
| DAVID term (biology) | Agilent probe ID | Entrez gene ID | Gene symbol | Gene name |
|---|---|---|---|---|
| Extracellular matrix | A_23_P211212 | 80781 | COL18A1 | Collagen, type XVIII, alpha 1 |
| A_23_P42322 | 1302 | COL11A2 | Collagen, type XI, alpha 2 | |
| A_24_P334130 | 2335 | FN1 | Fibronectin 1 | |
| A_23_P44291 | 100134403 | CRTAP | Cartilage associated protein | |
| A_24_P179225 | 4147 | MATN2 | Matrilin 2 | |
| A_23_P24870 | 960 | CD44 | CD44 molecule (Indian blood group) | |
| A_23_P112554 | 1306 | COL15A1 | Collagen, type XV, alpha 1 | |
| A_23_P7642 | 6678 | SPARC | Secreted protein, acidic, cysteine-rich (osteonectin) | |
| A_24_P315821 | 339366 | ADAMTSL5 | ADAMTS-like 5 | |
| A_23_P124084 | 4016 | LOXL1 | Lysyl oxidase-like 1 | |
| A_23_P7727 | 1404 | HAPLN1 | Hyaluronan and proteoglycan link protein 1 | |
| A_23_P159325 | 51129 | ANGPTL4 | Angiopoietin-like 4 | |
| A_23_P59388 | 667 | DST | Dystonin | |
| A_23_P204286 | 4256 | MGP | Matrix Gla protein | |
| A_23_P20697 | 9719 | ADAMTSL2 | Similar to ADAMTS-like 2; ADAMTS-like 2 | |
| A_24_P331918 | 1291 | COL6A1 | Collagen, type VI, alpha 1 | |
| A_23_P68487 | 655 | BMP7 | Bone morphogenetic protein 7 | |
| A_32_P32254 | 1291 | COL6A1 | Collagen, type VI, alpha 1 | |
| A_23_P102117 | 80326 | WNT10A | Wingless-type MMTV integration site family, member 10A | |
| A_23_P56578 | 5212 | VIT | Vitrin | |
| A_23_P121533 | 10417 | SPON2 | Spondin 2, extracellular matrix protein | |
| Axonogenesis | A_23_P7752 | 57556 | SEMA6A | Sema domain, transmembrane domain (TM), and cytoplasmic domain, (semaphorin) 6A |
| A_32_P100974 | 6152 | RPL24 | Ribosomal protein L24; ribosomal protein L24 pseudogene 6 | |
| A_23_P65307 | 84189 | SLITRK6 | SLIT and NTRK-like family, member 6 | |
| A_24_P365807 | 1947 | EFNB1 | Ephrin-B1 | |
| A_23_P59388 | 667 | DST | Dystonin | |
| A_23_P55099 | 5578 | PRKCA | Protein kinase C, alpha | |
| A_32_P152586 | 7080 | NKX2-1 | NK2 homeobox 1 | |
| A_24_P519638 | 89884 | LHX4 | LIM homeobox 4 | |
| A_23_P210756 | 6616 | SNAP25 | Synaptosomal-associated protein, 25kDa | |
| A_23_P68487 | 655 | BMP7 | Bone morphogenetic protein 7 | |
| A_23_P132175 | 65078 | RTN4R | Reticulon 4 receptor | |
| A_24_P42624 | 8633 | UNC5C | Unc-5 homolog C (C. elegans) | |
| A_23_P121533 | 10417 | SPON2 | Spondin 2, extracellular matrix protein | |
| Defense response | A_23_P16384 | 199713 | NALP7 | NLR family, pyrin domain containing 7 |
| A_23_P502464 | 4843 | NOS2A | Nitric oxide synthase 2, inducible | |
| A_23_P26965 | 6357 | CCL13 | Chemokine (C-C motif) ligand 13 | |
| A_23_P215484 | 10344 | CCL26 | Chemokine (C-C motif) ligand 26 | |
| A_24_P334130 | 2335 | FN1 | Fibronectin 1 | |
| A_23_P24870 | 960 | CD44 | CD44 molecule (Indian blood group) | |
| A_23_P22660 | 10800 | CYSLTR1 | Cysteinyl leukotriene receptor 1 | |
| A_24_P12573 | 10344 | CCL26 | Chemokine (C-C motif) ligand 26 | |
| A_23_P25354 | 23676 | P2RX7 | Purinergic receptor P2X, ligand-gated ion channel, 7 | |
| A_23_P143331 | 650 | BMP2 | Bone morphogenetic protein 2 | |
| A_23_P209954 | 10578 | GNLY | Granulysin | |
| A_23_P119295 | 51295 | ECSIT | ECSIT homolog (Drosophila) | |
| Angiogenesis | A_23_P159325 | 51129 | ANGPTL4 | Angiopoietin-like 4 |
| A_24_P7950 | 10451 | VAV3 | Vav 3 guanine nucleotide exchange factor | |
| A_23_P48217 | 81575 | APOLD1 | Apolipoprotein L domain containing 1 | |
| A_24_P334130 | 2335 | FN1 | Fibronectin 1 | |
| A_32_P210642 | 51162 | EGFL7 | EGF-like-domain, multiple 7 | |
| A_23_P112554 | 1306 | COL15A1 | Collagen, type XV, alpha 1 |
DAVID, Database for Annotation, Visualization, and Integrated Discovery.
Table 4.
Increased Expression of Biological Gene Sets in Both Human Th2-High Asthma and the BALB/c OVA Mouse Asthma Model, as Identified by DAVID Analysis
| DAVID term (biology) | P⁎ |
|---|---|
| Extracellular structure organization | 0.00210 |
| Sequence-specific DNA binding | 0.0160 |
| Developmental protein | 0.0540 |
| Embryonic organ development | 0.0720 |
DAVID, Database for Annotation, Visualization, and Integrated Discovery.
Corrected by the method of Benjamini-Hochberg for multiple comparisons.
Discussion
It is increasingly recognized that asthma is a heterogeneous disease1,2 and that a diversity of pathogenic mechanisms can give rise to the common clinical manifestations of variable airflow obstruction, airway hyperreactivity, and chronic airway inflammation that are observed in human asthmatics. The composition of the airway inflammation in the asthmatic lung has been studied by several groups and has been found to be heterogeneous in disease,1–4 with one common subtype of asthma defined by eosinophilic lung inflammation that is associated with Th2 inflammatory gene expression.5,6 A better understanding of the pathophysiology of eosinophilic/Th2-high asthma and other asthma subtypes will facilitate the identification of novel asthma therapeutics, as well as the identification of the appropriate patient populations for treatment with these therapies.
Studies of nonhuman primate models of asthma have yielded important insights into human asthma pathogenesis, given the strong similarities between nonhuman primates and humans. Although mice are the most commonly used species in preclinical asthma research, studies of nonhuman primate models of asthma can complement and supplement studies of mouse models of asthma, given the limitations of mouse asthma models in modeling human disease.14,37–40 Here, we have assessed the genomewide lung gene expression profile of an allergic asthma model induced in rhesus monkeys by HDM and have compared it with the lung gene expression profiles of the eosinophilic/Th2-high subtype of human asthma and a commonly used mouse asthma model. We find that a dominant gene expression signature in all three species is a Th2 inflammation gene signature. Significantly, there is a strong correlation between a prespecified set of human Th2 inflammation genes in human Th2-high asthma and in the rhesus asthma model. In addition, similar to studies of this lung Th2 inflammation gene set in human asthma,5,6 we find that the Th2 inflammation gene set correlates with physiologic measures of Th2 inflammation and disease in rhesus asthma, including BALF eosinophils, peripheral blood eosinophils, HDM-specific serum IgE, and airway hyperreactivity. Thus, consistent with previous reports,11 the rhesus asthma model exhibits many features of human Th2-high asthma. In support of our observations, others have also reported gene expression that is associated with Th2 inflammation in the lungs or BALF of nonhuman primate models of asthma, including a previous study of the HDM-induced rhesus asthma model studied here.10,25,26 Differences in the specific genes detected in our study versus previous studies of nonhuman primate asthma models may be due to i) differences in assessed genes (our study used a prespecified gene set that was generated by applying statistical cutoffs to a human asthma biopsy microarray gene expression data set, whereas previously published studies assessed hand-curated gene lists), ii) differences in assay sensitivity (we assessed gene expression levels by microarray, whereas some previously published studies assessed gene expression levels by quantitative PCR), and/or iii) differences in animal model samples (we assessed airway biopsy gene expression, whereas previously published studies assessed either whole lung or BALF cell gene expression).
Additional analysis of the lung gene expression profiles of human, rhesus, and BALB/c OVA mouse asthma showed that only a small subset of genes is shared among the three different species. The lack of significant gene overlap among the three species may be due to several reasons, including i) imperfect cross-hybridization between rhesus mRNA transcripts and human microarray probes, which would reduce the number of rhesus genes that can be assessed by microarray; ii) incomplete orthology between human and mouse genes, which would reduce the number of mouse genes that can be compared with human genes; and iii) limitations in the ability of the animal models to fully mimic human disease biology. Imperfect cross-hybridization between rhesus mRNA transcripts and human microarray probes because of differences in homology between rhesus and human gene sequences would lead to false-negative signals. We calculate that 99% of human genes lie in regions possessing synteny to a sequenced portion of the rhesus macaque genome, and that 96% of the human probes on the Agilent microarray platform are perfect matches in sequence to the rhesus genes. In addition, analysis of our human and rhesus microarray expression data indicates that the average expression intensity of each gene is similar between the two species. Thus, imperfect cross-hybridization between rhesus mRNA transcripts and human microarray probes is unlikely to be a main cause of the lack of significant gene overlap between rhesus and human gene expression. Ultimately, the main effect of microarray platform and species orthology limitations on our conclusions is probably a potential underestimation of the similarities between human asthma and the rhesus and mouse asthma models.
When we compared the genes that are shared by human Th2-high asthma and rhesus asthma with the genes that are shared by human Th2-high asthma and BALB/c OVA mouse asthma, we found that some of the biological pathways that are present in the gene expression profile of human Th2-high asthma are better represented in the gene expression profile of the rhesus asthma model than that of the BALB/c OVA model. These include extracellular matrix biology, cell morphogenesis, nervous system biology, immune host defense biology, and angiogenesis. One limitation of our approach, which may have influenced the outcomes of our analyses, is that we assessed airway biopsy gene expression from human and rhesus and total lung homogenate gene expression from mouse. Thus, the composition of the tissues being compared among human, rhesus, and mouse are different. However, a previously published study of gene expression in total lung homogenate from an allergic asthma model induced in cynomolgus monkeys by Ascaris suum antigen also identified Th2 inflammation and remodeling genes, although not nervous system or angiogenesis biology.26 Moreover, it should be noted that the pattern of immune cell accumulation in the lung after allergen challenge differs between rodents and primates. Whereas in mice the immune cell accumulation is primarily perivascular with diffuse peribronchial accumulation, in humans and rhesus the immune cell accumulation is primarily in the larger conducting airways. As such, the biological pathways present in the areas of the mouse airways that are comparable to the areas sampled by biopsies in humans and rhesus monkeys are probably more divergent than those present in total mouse lung homogenate.
Subepithelial fibrosis of the lung, which consists of extracellular matrix deposition in the basement membrane, is a pathological characteristic of asthma and may contribute to irreversible lung obstruction.41,42 A previous study of the rhesus asthma model showed increased thickening of the basement membrane, as assessed by histology.11 In contrast, commonly used short-term mouse models of asthma, such as the BALB/c OVA model that we analyzed in this study,21 do not develop subepithelial fibrosis and lung remodeling, although more chronic mouse models of asthma can develop some features of fibrosis and remodeling.39 Neuronal dysfunction and dysregulation, encompassing activation of both the parasympathetic and sympathetic nervous systems, may contribute to the pathogenesis of asthma.43–45 Neural mediators can have direct effects on smooth muscle, airway glands, and alveolar walls; there is also substantial interaction between the nervous system and the immune system in the lungs of asthmatic persons. The lung vasculature is increased in asthmatics, and the degree of vascular abnormalities in asthma is associated with asthma severity.46–48 Vascularization is not increased in nontransgenic allergen-induced mouse asthma models,49,50 although overexpression of vascular endothelial growth factor in the mouse lung results in increased vascularization, Th2 inflammation, and remodeling.49,51 In contrast, in the rhesus monkey model of asthma, bronchial vascular density was increased at the mid-to-lower airway generations and was independent of changes in the interstitial compartment.7
Given the multiple variations of mouse asthma models in the literature, comprising differences in sensitization and challenge protocols, differences in allergens, and differences in mouse strains, it was not possible for us to compare human Th2-high asthma gene expression with that of every mouse asthma model. We therefore chose to compare lung gene expression in human asthma, rhesus asthma, and the most commonly used version of mouse asthma in the literature, which is an acute OVA-induced mouse asthma model in the BALB/c strain. Some of the gene expression features of human Th2-high asthma that are observed in the rhesus asthma model, but not in the mouse asthma model, may be present in other versions of mouse asthma that incorporate a more chronic sensitization and challenge protocol or that use HDM antigen. To address this, we performed additional comparisons of the gene expression profile of human Th2-high asthma with those of a chronic OVA-induced mouse asthma model16 and an acute HDM-induced mouse asthma model,18 both of which are publically available. The chronic OVA-induced mouse asthma models share features of cell cycle/cell division, cellular substructures, lymphocyte/T-cell activation and differentiation, immune response/inflammation, extracellular region, chemotaxis, and chitinase activity with human Th2-high asthma (see Supplemental Table S4 at http://ajp.amjpathol.org). The acute HDM-induced mouse asthma model shares features of cell cycle/cell division, chemotaxis, extracellular region/extracellular matrix, immune response/inflammation, and chitinase activity with human Th2-high asthma (see Supplemental Table S5 at http://ajp.amjpathol.org). However, similar to the acute OVA-induced mouse asthma model, neither the chronic OVA-induced mouse asthma model nor the acute HDM-induced mouse asthma model exhibits features of neural biology or angiogenesis, features that are shared between the rhesus asthma model and human Th2-high asthma.
Our study is the first to our knowledge to directly compare genomewide human asthma lung gene expression with that of any preclinical asthma model, regardless of species. We find that a lung Th2 inflammation gene signature that is present in the eosinophilic/Th2-high subtype of human asthma is modeled by a HDM antigen-induced allergic asthma model in rhesus monkeys, as well as by a BALB/c OVA mouse asthma model. In addition, some gene profiles that are present in human Th2-high asthma are better represented in the gene expression profile of the rhesus asthma model, compared with that of the BALB/c OVA mouse asthma model. These include some inflammatory pathways, subepithelial fibrosis and remodeling, angiogenesis, and neural biology that may be associated with neurogenic inflammation. Further study of the rhesus asthma model may complement and supplement studies of mouse asthma models, yield novel insights into the pathogenesis of human Th2-high asthma, and aid in the identification of new therapeutic targets and disease biomarkers for asthma, including potentially some of the genes reported in this study.
Acknowledgment
We thank the Primate Services Unit at the California National Primate Research Center for animal handling, care, and coordination, which were critical to this study.
Footnotes
Supported by Genentech Inc. and NCRR grant RR00169.
A.R.A., J.K.J., S.L.B., D.F.C., G.F., M.T., B.-T.T., Y.G.M., L.D., H.F.C., Z.M., J.R.A., and L.C.W. are employed by Genentech, Inc.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.06.009.
Current address of S.L.B., Department of Translational Biology, BioMarin Pharmaceutical Inc., Novato, CA.
Supplementary data
Sensitization to HDM antigen induces inflammation in the bronchoalveolar lavage (BAL) of rhesus monkeys. Twelve rhesus monkeys were sensitized with a subcutaneous injection of HDM antigen extract and intranasal HDM, followed by regular exposure to aerosolized HDM for 2 to 3 hours twice a week, for a total of 18 months. Sensitized rhesus monkeys also received additional subcutaneous and intranasal HDM boosts at weeks 56, 71, and 75. Ten rhesus monkeys were subjected to control treatment with PBS injections and mock aerosol challenges. Data were collected at week −4 to −8 for the presensitization time point, week 15 for the postsensitization time point, and week 75 for the 18-month time point, whereby week 0 denotes the beginning of the sensitization protocol, for all 12 sensitized and all 10 nonsensitized monkeys, except for the 18-month time point, whereby data was collected for all 12 sensitized and only 4 of the 10 nonsensitized monkeys. Percentage of BALF neutrophils (A), number of BALF neutrophils (B), and percentage of BALF CD4 lymphocytes that are activated (C) in the sensitized and control groups at presensitization, postsensitization, and 18-month time points. Data are expressed as mean ± SD. *P < 0.05 versus nonsensitized animals, Wilcoxon/Kruskal-Wallis Test (rank sums).
Sensitized rhesus monkeys show an increase in airway hyperreactivity at the postsensitization time point, compared with nonsensitized rhesus monkeys. Eight animals in the sensitized group and 10 animals in the nonsensitized group lacked airway hyperreactivity at the presensitization time point, using a definition of airway hyperreactivity of a methacholine EC150 value of < 8 mg/mL. A: Baseline levels of airway reactivity at the presensitization time point are comparable between the 8 sensitized and 10 nonsensitized animals that were not hyperreactive before sensitization. B: The 8 sensitized animals showed a greater increase in airway reactivity at the postsensitization time point than did the 10 nonsensitized animals. Airway hyperreactivity is expressed as EC150, the effective concentration of methacholine required to increase airway resistance (Raw) to 150% of baseline, whereby a lower EC150 indicates greater airway hyperreactivity. The ratio of EC150 at the postsensitization time point to the EC150 at the presensitization time point was calculated for each animal. Data are expressed as mean ± SD. *P < 0.05 versus nonsensitized animals, Dunnett's test.
Th2 inflammation genes are correlated with the percentage of eosinophils in the BALF of sensitized rhesus monkeys. Twenty-five genes that represent a Th2 inflammation signature were identified by first applying filters of a fold change of >1.2 and a q-value of <0.05 to whole-genome expression profiles of lung airway biopsies from the Th2-high subset of asthmatic patients (comparing the Th2-high asthma subset with both the Th2-low asthma subset and the healthy control subjects), followed by an additional filter of increased expression (fold change > 1) in rhesus asthma lung airway biopsies compared with nonsensitized control rhesus lung airway biopsies. Expression levels for these 25 Th2 inflammation genes were compared with measures of BALF eosinophil relative abundance in the rhesus asthma model with the use of Spearman's correlation; ρ is the correlation coefficient. The dotted line is the significance threshold (corresponding to q-value = 0.2).
References
- 1.Green R.H., Brightling C.E., Bradding P. The reclassification of asthma based on subphenotypes. Curr Opin Allergy Clin Immunol. 2007;7:43–50. doi: 10.1097/ACI.0b013e3280118a32. [DOI] [PubMed] [Google Scholar]
- 2.Wenzel S.E. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;368:804–813. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- 3.Douwes J., Gibson P., Pekkanen J., Pearce N. Non-eosinophilic asthma: importance and possible mechanisms. Thorax. 2002;57:643–648. doi: 10.1136/thorax.57.7.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haldar P., Pavord I.D. Noneosinophilic asthma: a distinct clinical and pathologic phenotype. J Allergy Clin Immunol. 2007;119:1043–1052. doi: 10.1016/j.jaci.2007.02.042. quiz 1053–1044. [DOI] [PubMed] [Google Scholar]
- 5.Choy D.F., Modrek B., Abbas A.R., Kummerfeld S., Clark H.F., Wu L.C., Fedorowicz G., Modrusan Z., Fahy J.V., Woodruff P.G., Arron J.R. Gene expression patterns of Th2 inflammation and intercellular communication in asthmatic airways. J Immunol. 2011;186:1861–1869. doi: 10.4049/jimmunol.1002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodruff P.G., Modrek B., Choy D.F., Jia G., Abbas A.R., Ellwanger A., Koth L.L., Arron J.R., Fahy J.V. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avdalovic M.V., Putney L.F., Schelegle E.S., Miller L., Usachenko J.L., Tyler N.K., Plopper C.G., Gershwin L.J., Hyde D.M. Vascular remodeling is airway generation-specific in a primate model of chronic asthma. Am J Respir Crit Care Med. 2006;174:1069–1076. doi: 10.1164/rccm.200506-848OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coffman R.L., Hessel E.M. Nonhuman primate models of asthma. J Exp Med. 2005;201:1875–1879. doi: 10.1084/jem.20050901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fanucchi M.V., Schelegle E.S., Baker G.L., Evans M.J., McDonald R.J., Gershwin L.J., Raz E., Hyde D.M., Plopper C.G., Miller L.A. Immunostimulatory oligonucleotides attenuate airways remodeling in allergic monkeys. Am J Respir Crit Care Med. 2004;170:1153–1157. doi: 10.1164/rccm.200404-533OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller L.A., Hurst S.D., Coffman R.L., Tyler N.K., Stovall M.Y., Chou D.L., Putney L.F., Gershwin L.J., Schelegle E.S., Plopper C.G., Hyde D.M. Airway generation-specific differences in the spatial distribution of immune cells and cytokines in allergen-challenged rhesus monkeys. Clin Exp Allergy. 2005;35:894–906. doi: 10.1111/j.1365-2222.2005.02271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schelegle E.S., Gershwin L.J., Miller L.A., Fanucchi M.V., Van Winkle L.S., Gerriets J.P., Walby W.F., Omlor A.M., Buckpitt A.R., Tarkington B.K., Wong V.J., Joad J.P., Pinkerton K.B., Wu R., Evans M.J., Hyde D.M., Plopper C.G. Allergic asthma induced in rhesus monkeys by house dust mite (Dermatophagoides farinae) Am J Pathol. 2001;158:333–341. doi: 10.1016/S0002-9440(10)63973-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seshasayee D., Lee W.P., Zhou M., Shu J., Suto E., Zhang J., Diehl L., Austin C.D., Meng Y.G., Tan M., Bullens S.L., Seeber S., Fuentes M.E., Labrijn A.F., Graus Y.M., Miller L.A., Schelegle E.S., Hyde D.M., Wu L.C., Hymowitz S.G., Martin F. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest. 2007;117:3868–3878. doi: 10.1172/JCI33559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Scott M.R., Hooker J.L., Ehrmann D., Shibata Y., Kukoly C., Salleng K., Westergaard G., Sandrasagra A., Nyce J. Dust mite-induced asthma in cynomolgus monkeys. J Appl Physiol. 2004;96:1433–1444. doi: 10.1152/japplphysiol.01128.2003. [DOI] [PubMed] [Google Scholar]
- 14.Boyce J.A., Austen K.F. No audible wheezing: nuggets and conundrums from mouse asthma models. J Exp Med. 2005;201:1869–1873. doi: 10.1084/jem.20050584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Winkle L.S., Baker G.L., Chan J.K., Schelegle E.S., Plopper C.G. Airway mast cells in a rhesus model of childhood allergic airways disease. Toxicol Sci. 2010;116:313–322. doi: 10.1093/toxsci/kfq119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Valentin E., Crahay C., Garbacki N., Hennuy B., Gueders M., Noel A., Foidart J.M., Grooten J., Colige A., Piette J., Cataldo D. New asthma biomarkers: lessons from murine models of acute and chronic asthma. Am J Physiol Lung Cell Mol Physiol. 2009;296:L185–L197. doi: 10.1152/ajplung.90367.2008. [DOI] [PubMed] [Google Scholar]
- 17.Fulkerson P.C., Zimmermann N., Hassman L.M., Finkelman F.D., Rothenberg M.E. Pulmonary chemokine expression is coordinately regulated by STAT1. STAT6, and IFN-gamma. J Immunol. 2004;173:7565–7574. doi: 10.4049/jimmunol.173.12.7565. [DOI] [PubMed] [Google Scholar]
- 18.Kelada S.N., Wilson M.S., Tavarez U., Kubalanza K., Borate B., Whitehead G., Maruoka S., Roy M.G., Olive M., Carpenter D.E., Brass D.M., Wynn T.A., Cook D.A., Evans C.M., Schwartz D.A., Collins F.S. Strain-dependent genomic factors affect allergen-induced airway hyper-responsiveness in mice. Am J Respir Cell Mol Biol. 2011 doi: 10.1165/rcmb.2010-0315OC. [Epub ahead of press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuperman D.A., Lewis C.C., Woodruff P.G., Rodriguez M.W., Yang Y.H., Dolganov G.M., Fahy J.V., Erle D.J. Dissecting asthma using focused transgenic modeling and functional genomics. J Allergy Clin Immunol. 2005;116:305–311. doi: 10.1016/j.jaci.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 20.Lewis C.C., Aronow B., Hutton J., Santeliz J., Dienger K., Herman N., Finkelman F.D., Wills-Karp M. Unique and overlapping gene expression patterns driven by IL-4 and IL-13 in the mouse lung. J Allergy Clin Immunol. 2009;123 doi: 10.1016/j.jaci.2009.01.003. 795–804e798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis C.C., Yang J.Y., Huang X., Banerjee S.K., Blackburn M.R., Baluk P., McDonald D.M., Blackwell T.S., Nagabhushanam V., Peters W., Voehringer D., Erle D.J. Disease-specific gene expression profiling in multiple models of lung disease. Am J Respir Crit Care Med. 2008;177:376–387. doi: 10.1164/rccm.200702-333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park S.G., Choi J.W., Kim H., Roh G.S., Bok J., Go M.J., Kwack K., Oh B., Kim Y. Genome-wide profiling of antigen-induced time course expression using murine models for acute and chronic asthma. Int Arch Allergy Immunol. 2008;146:44–56. doi: 10.1159/000112502. [DOI] [PubMed] [Google Scholar]
- 23.Rolph M.S., Sisavanh M., Liu S.M., Mackay C.R. Clues to asthma pathogenesis from microarray expression studies. Pharmacol Ther. 2006;109:284–294. doi: 10.1016/j.pharmthera.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Zimmermann N., King N.E., Laporte J., Yang M., Mishra A., Pope S.M., Muntel E.E., Witte D.P., Pegg A.A., Foster P.S., Hamid Q., Rothenberg M.E. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–1874. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayanoglu G., Desai B., Fick R.B., Jr, Grein J., de Waal Malefyt R., Mattson J., McClanahan T., Olmstead S., Reece S.P., Van Scott M.R., Wardle R.L. Modelling asthma in macaques: longitudinal changes in cellular and molecular markers. Eur Respir J. 2011;37:541–552. doi: 10.1183/09031936.00047410. [DOI] [PubMed] [Google Scholar]
- 26.Zou J., Young S., Zhu F., Gheyas F., Skeans S., Wan Y., Wang L., Ding W., Billah M., McClanahan T., Coffman R.L., Egan R., Umland S. Microarray profile of differentially expressed genes in a monkey model of allergic asthma. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-5-research0020. research0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woodruff P.G., Boushey H.A., Dolganov G.M., Barker C.S., Yang Y.H., Donnelly S., Ellwanger A., Sidhu S.S., Dao-Pick T.P., Pantoja C., Erle D.J., Yamamoto K.R., Fahy J.V. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104:15858–15863. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crapo R.O., Casaburi R., Coates A.L., Enright P.L., Hankinson J.L., Irvin C.G., MacIntyre N.R., McKay R.T., Wanger J.S., Anderson S.D., Cockcroft D.W., Fish J.E., Sterk P.J. Guidelines for methacholine and exercise challenge testing-1999: This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med. 2000;161:309–329. doi: 10.1164/ajrccm.161.1.ats11-99. [DOI] [PubMed] [Google Scholar]
- 29.Balzar S., Westcott J.Y., Trudeau J.B., Sun Y., Conrad D.J., Wenzel S.E. Expression and activation of 15-lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and collagen deposition. Clin Exp Allergy. 2002;32:1558–1565. doi: 10.1046/j.1365-2222.2002.01477.x. ChuHW. [DOI] [PubMed] [Google Scholar]
- 30.Gould H.J., Sutton B.J. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8:205–217. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 31.Komiya A., Nagase H., Yamada H., Sekiya T., Yamaguchi M., Sano Y., Hanai N., Furuya A., Ohta K., Matsushima K., Yoshie O., Yamamoto K., Hirai K. Concerted expression of eotaxin-1, eotaxin-2, and eotaxin-3 in human bronchial epithelial cells. Cell Immunol. 2003;225:91–100. doi: 10.1016/j.cellimm.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Suresh V., Mih J.D., George S.C. Measurement of IL-13-induced iNOS-derived gas phase nitric oxide in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2007;37:97–104. doi: 10.1165/rcmb.2006-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurmond R.L., Gelfand E.W., Dunford P.J. The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat Rev Drug Discov. 2008;7:41–53. doi: 10.1038/nrd2465. [DOI] [PubMed] [Google Scholar]
- 34.Zuo L., Fulkerson P.C., Finkelman F.D., Mingler M., Fischetti C.A., Blanchard C., Rothenberg M.E. IL-13 induces esophageal remodeling and gene expression by an eosinophil-independent: IL-13R alpha 2-inhibited pathway. J Immunol. 2010;185:660–669. doi: 10.4049/jimmunol.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, et al: Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 38:D5-D16 [DOI] [PMC free article] [PubMed]
- 36.Dennis G., Jr, Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 37.Finkelman F.D., Wills-Karp M. Usefulness and optimization of mouse models of allergic airway disease. J Allergy Clin Immunol. 2008;121:603–606. doi: 10.1016/j.jaci.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mestas J., Hughes C.C. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 39.Taube C., Dakhama A., Gelfand E.W. Insights into the pathogenesis of asthma utilizing murine models. Int Arch Allergy Immunol. 2004;135:173–186. doi: 10.1159/000080899. [DOI] [PubMed] [Google Scholar]
- 40.Wenzel S., Holgate S.T. The mouse trap: It still yields few answers in asthma. Am J Respir Crit Care Med. 2006;174:1173–1178. doi: 10.1164/rccm.2609002. [DOI] [PubMed] [Google Scholar]
- 41.Jeffery P.K. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164:S28–S38. doi: 10.1164/ajrccm.164.supplement_2.2106061. [DOI] [PubMed] [Google Scholar]
- 42.Pascual R.M., Peters S.P. Airway remodeling contributes to the progressive loss of lung function in asthma: an overview. J Allergy Clin Immunol. 2005;116:477–486. doi: 10.1016/j.jaci.2005.07.011. quiz 487. [DOI] [PubMed] [Google Scholar]
- 43.Nockher W.A., Renz H. Neurotrophins and asthma: novel insight into neuroimmune interaction. J Allergy Clin Immunol. 2006;117:67–71. doi: 10.1016/j.jaci.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 44.Pisi G., Olivieri D., Chetta A. The airway neurogenic inflammation: clinical and pharmacological implications. Inflamm Allergy Drug Targets. 2009;8:176–181. doi: 10.2174/187152809788681047. [DOI] [PubMed] [Google Scholar]
- 45.Undem B.J., Carr M.J. The role of nerves in asthma. Curr Allergy Asthma Rep. 2002;2:159–165. doi: 10.1007/s11882-002-0011-4. [DOI] [PubMed] [Google Scholar]
- 46.Hoshino M., Nakamura Y., Hamid Q.A. Gene expression of vascular endothelial growth factor and its receptors and angiogenesis in bronchial asthma. J Allergy Clin Immunol. 2001;107:1034–1038. doi: 10.1067/mai.2001.115626. [DOI] [PubMed] [Google Scholar]
- 47.Li X., Wilson J.W. Increased vascularity of the bronchial mucosa in mild asthma. Am J Respir Crit Care Med. 1997;156:229–233. doi: 10.1164/ajrccm.156.1.9607066. [DOI] [PubMed] [Google Scholar]
- 48.Vrugt B., Wilson S., Bron A., Holgate S.T., Djukanovic R., Aalbers R. Bronchial angiogenesis in severe glucocorticoid-dependent asthma. Eur Respir J. 2000;15:1014–1021. doi: 10.1034/j.1399-3003.2000.01507.x. [DOI] [PubMed] [Google Scholar]
- 49.Lee C.G., Link H., Baluk P., Homer R.J., Chapoval S., Bhandari V., Kang M.J., Cohn L., Kim Y.K., McDonald D.M., Elias J.A. Vascular endothelial growth factor (VEGF) induces remodeling and enhances TH2-mediated sensitization and inflammation in the lung. Nat Med. 2004;10:1095–1103. doi: 10.1038/nm1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzaki Y., Hamada K., Sho M., Ito T., Miyamoto K., Akashi S., Kashizuka H., Ikeda N., Nakajima Y., Iwase M., Homma I., Kobzik L., Kimura H. A potent antiangiogenic factor, endostatin prevents the development of asthma in a murine model. J Allergy Clin Immunol. 2005;116:1220–1227. doi: 10.1016/j.jaci.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 51.Baluk P., Lee C.G., Link H., Ator E., Haskell A., Elias J.A., McDonald D.M. Regulated angiogenesis and vascular regression in mice overexpressing vascular endothelial growth factor in airways. Am J Pathol. 2004;165:1071–1085. doi: 10.1016/S0002-9440(10)63369-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sensitization to HDM antigen induces inflammation in the bronchoalveolar lavage (BAL) of rhesus monkeys. Twelve rhesus monkeys were sensitized with a subcutaneous injection of HDM antigen extract and intranasal HDM, followed by regular exposure to aerosolized HDM for 2 to 3 hours twice a week, for a total of 18 months. Sensitized rhesus monkeys also received additional subcutaneous and intranasal HDM boosts at weeks 56, 71, and 75. Ten rhesus monkeys were subjected to control treatment with PBS injections and mock aerosol challenges. Data were collected at week −4 to −8 for the presensitization time point, week 15 for the postsensitization time point, and week 75 for the 18-month time point, whereby week 0 denotes the beginning of the sensitization protocol, for all 12 sensitized and all 10 nonsensitized monkeys, except for the 18-month time point, whereby data was collected for all 12 sensitized and only 4 of the 10 nonsensitized monkeys. Percentage of BALF neutrophils (A), number of BALF neutrophils (B), and percentage of BALF CD4 lymphocytes that are activated (C) in the sensitized and control groups at presensitization, postsensitization, and 18-month time points. Data are expressed as mean ± SD. *P < 0.05 versus nonsensitized animals, Wilcoxon/Kruskal-Wallis Test (rank sums).
Sensitized rhesus monkeys show an increase in airway hyperreactivity at the postsensitization time point, compared with nonsensitized rhesus monkeys. Eight animals in the sensitized group and 10 animals in the nonsensitized group lacked airway hyperreactivity at the presensitization time point, using a definition of airway hyperreactivity of a methacholine EC150 value of < 8 mg/mL. A: Baseline levels of airway reactivity at the presensitization time point are comparable between the 8 sensitized and 10 nonsensitized animals that were not hyperreactive before sensitization. B: The 8 sensitized animals showed a greater increase in airway reactivity at the postsensitization time point than did the 10 nonsensitized animals. Airway hyperreactivity is expressed as EC150, the effective concentration of methacholine required to increase airway resistance (Raw) to 150% of baseline, whereby a lower EC150 indicates greater airway hyperreactivity. The ratio of EC150 at the postsensitization time point to the EC150 at the presensitization time point was calculated for each animal. Data are expressed as mean ± SD. *P < 0.05 versus nonsensitized animals, Dunnett's test.
Th2 inflammation genes are correlated with the percentage of eosinophils in the BALF of sensitized rhesus monkeys. Twenty-five genes that represent a Th2 inflammation signature were identified by first applying filters of a fold change of >1.2 and a q-value of <0.05 to whole-genome expression profiles of lung airway biopsies from the Th2-high subset of asthmatic patients (comparing the Th2-high asthma subset with both the Th2-low asthma subset and the healthy control subjects), followed by an additional filter of increased expression (fold change > 1) in rhesus asthma lung airway biopsies compared with nonsensitized control rhesus lung airway biopsies. Expression levels for these 25 Th2 inflammation genes were compared with measures of BALF eosinophil relative abundance in the rhesus asthma model with the use of Spearman's correlation; ρ is the correlation coefficient. The dotted line is the significance threshold (corresponding to q-value = 0.2).