

Abstract
A cluster of six microRNAs (miRNAs), miR-17-92, is processed from the transcript of C13orf25, a gene amplified in some lymphomas and solid tumors. We find that levels of the miRNAs in the cluster do not vary entirely in parallel with each other or with the primary RNA in B-cell lines or normal cells, suggesting that processing or stability of the miRNAs is differentially regulated. Using luciferase reporter assays, we identified the region required for maximum promoter activity. Additional deletions and mutations indicated that the promoter is regulated by the collaborative activity of several transcription factors, most of which individually have only a moderate effect; mutation of a cluster of putative SP1-binding sites, however, reduces promoter activity by 70%. MYC is known to regulate C13orf25; surprisingly, mutation of a putative promoter MYC-binding site enhanced promoter activity. We found that the inhibitory MYC family member MXI1 bound to this region. The chromatin structure of a >22.5-kb region encompassing the gene contains peaks of activating histone marks, suggesting the presence of enhancers, and we confirmed that at least two regions have enhancer activity. Because the miR-17-92 cluster acts as an important oncogene in several cancers and targets genes important in regulating cell proliferation and survival, further studies of its transcriptional control are warranted.
MicroRNAs (miRNAs) are single-stranded RNAs of ∼22 nucleotides that negatively regulate expression of their target genes posttranscriptionally.1–4 Various miRNAs are overexpressed or underexpressed in different types of cancer in humans and may function as either tumor suppressors or oncogenes.5–13 A cluster of six miRNAs (miR-17-92, comprising miR-17, miR-18a, miR-20a, miR-19a, miR-19b-1, and miR-92a-1) is processed from the transcript of C13orf25 (also known as MIR17HG or MIRHG1), a gene amplified in some lymphomas and solid tumors5,14–17 and overexpressed in a large fraction of lymphomas.5 Overexpression of miR-17-92 accelerates lymphomagenesis in mouse models.5,8 The miRNAs in the cluster can target transcripts of genes, such as E2F1, PTEN, and BIM, which are important in cell proliferation and apoptosis.18–22 The transcriptional regulation of the miR-17-92 cluster, however, is poorly understood.
C13orf25 is activated by MYC and E2F transcription factors, and MYC was first shown to bind to a region containing a conserved CATGTG sequence in the first intron of the gene locus.8 By chromatin immunoprecipitation (ChIP) in HeLa cells, endogenous E2F1, E2F2, and E2F3 were found to directly bind the promoter of the gene and regulate its transcription.21 The primary transcript initiates from a consensus initiator sequence downstream of a nonconsensus TATA box.23 This TATA box is flanked by a TP53 binding site that mediates repression under hypoxic conditions.24 A recent study showed a direct link between the action of IL-6 and the expression of C13orf25 mediated by STAT3.25
To explore transcriptional regulation of the mir17-92 cluster, we have begun to analyze the C13orf25 promoter and the first intron of the gene in several cell culture model systems. Because it is now clear that epigenetic mechanisms regulate miRNA expression,26–30 we also investigated histone posttranslational modifications (PTMs) in C13orf25 in normal B cells and in B-cell lines. Further studies of the transcriptional control of C13orf25 are warranted because the miR-17-92 cluster targets genes important in regulating cell proliferation and survival and acts as an important oncogene in several cancers, including lymphoma.
Materials and Methods
Cell Lines and Normal B Cells
Mantle cell lymphoma–derived cell lines (JeKo-1 and JVM2), Burkitt's lymphoma cell lines (CL-01, Daudi, Raji, and P3HR-1), a diffuse large B-cell lymphoma (DLBCL) cell line of germinal center B-cell type [SU-DHL16 (hereafter, DHL16)], a mediastinal large B-cell lymphoma cell line (Karpas 1106), a Hodgkin's lymphoma cell line (L428), and a myeloma cell line (U266) were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen). A DLBCL line of germinal center B-cell type (DHL6) and two DLBCL lines of activated B-cell type (OCI-Ly3 and OCI-Ly10) were maintained in Iscove's modified Dulbecco's medium (Invitrogen) supplemented with 20% human serum, 50 μmol/L 2-mercaptoethanol, and 1% penicillin/streptomycin. HEK293T and NIH3T3 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. HEK293T, NIH3T3, JeKo-1, JVM2, and U266 were obtained from American Type Culture Collection (Manassas, VA) and Karpas 1106 from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). P3HR-1 and CL-01 were gifts from Dr. Luwen Zhang (University of Nebraska, Lincoln, NE) and Dr. Paolo Casali (University of California, Irvine, CA), respectively. All the other lines were a gift from Dr. Louis Staudt (National Cancer Institute, Bethesda, MD).
Primary cells isolated for miRNA studies, including naive B cells (IgD+) and centroblasts (CD77+), were isolated from tonsils using magnetic microbeads (Miltenyi Biotec Inc., Auburn, CA), as described.31
miRNA Expression Levels
miRNA expression profiling was conducted using the TaqMan human miRNA array set v2.0 (Applied Biosystems, Foster City, CA) with 300 ng of total RNA from cell lines or normal cells. All the RNA samples were reverse transcribed using Megaplex RT primers (Invitrogen) for 40 cycles at 16°C for 2 minutes, 42°C for 1 minute, and 50°C for 1 second; were held at 85°C for 5 minutes to inactivate the reverse transcriptase; and were preamplified for 12 cycles using Megaplex PreAmp primers (Invitrogen). The diluted preamplified DNA was loaded onto the 384-well plate per the manufacturer's instructions, and real-time PCR was run using a 7900HT fast real-time PCR system (Applied Biosystems) in 384-well plates at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. The CT is defined as the fractional cycle number at which the fluorescence exceeds the fixed threshold of 0.1 using RQ Manager 2.1 software (Applied Biosystems). Data from each 384-well plate were normalized with the average expression of U6 small nuclear RNA, the least variable control RNA analyzed on the plate. RNA was isolated using the RNeasy mini kit (Qiagen Inc., Valencia, CA), and miR-17-92 pri-miRNA and RPL13A mRNA, for normalization, were quantified by SYBR Green PCR (New England BioLabs, Inc., Ipswich, MA) using the DNA Engine Opticon 2 real-time PCR detection system (MJ Research, Waltham, MA) according to the manufacturer's instructions; the primers used are listed in Table 1. To determine the expression levels of mature miRNAs, TaqMan miRNA assays (Applied Biosystems) were performed using an Applied Biosystems 7900HT fast real-time PCR system and were normalized to U6 expression.
Table 1.
Oligonucleotides Used in This Study
| Primer name | Sequence | Purpose |
|---|---|---|
| miR-17∼92-pri-F | 5′-CAGTAAAGGTAAGGAGAGCTCAATCTG-3′ | RT-PCR |
| miR-17∼92-pri-R | 5′-CATACAACCACTAAGCTAAAGAATAATCTGA-3′ | RT-PCR |
| RPL13A-F | 5′-ACCGTCTCAAGGTGTTTGACG-3′ | RT-PCR (normalization) |
| RPL13A-R | 5′-GTACTTCCAGCCAACCTCGTG-3′ | RT-PCR (normalization) |
| ODC1-F | 5′-TGGGCGCTCTGAGGTGAGGG-3′ | ChIP of ODC1 (positive control) |
| ODC1-R | 5′-ATCCAGCCGCGGGAGAACCC-3′ | ChIP of ODC1 (positive control) |
| C13orf25-1-F | 5′-AAACGTTCTGAATGTTCTGGATTGT-3′ | ChIP of site 1 |
| C13orf25-1-R | 5′-CACAGCCTTCTCAAGTCAGCTAAA-3′ | ChIP of site 1 |
| C13orf25-2-F | 5′-AAGGAAGTATGAGCGAAACCCT-3′ | ChIP of site 2 |
| C13orf25-2-R | 5′-CAGGTTTCTTAGCAGGAGTTATTCA-3′ | ChIP of site 2 |
| C13orf25-3-F | 5′-GACTTGTACAATCACGAACCAGTG-3′ | ChIP of site 3 |
| C13orf25-3-R | 5′-TCTGGGTTCTCCAGTAGAAATAGC-3′ | ChIP of site 3 |
| C13orf25-4-F | 5′-TTTATGCTAATGAGGGAGTGG-3′ | ChIP of site 4 |
| C13orf25-4-R | 5′-GCTTTACTACGACCGGAGG-3′ | ChIP of site 4 |
| C13orf25-5-F | 5′-GACGGCGAACACAATGGC-3′ | ChIP of site 5 |
| C13orf25-5-R | 5′-GCGTACAAAGTTTGGGGAACC-3′ | ChIP of site 5 |
| C13orf25-6-F | 5′-AAGAATAGTCTGTGGGCTGCT-3′ | ChIP of site 6 |
| C13orf25-6-R | 5′-GCTATTTTCCAACCTCCTAACAGC-3′ | ChIP of site 6 |
| Cloning and deletions⁎ | ||
| LucSeqR2 | 5′-CCAGGAACCAGGGCGTATC-3′ | Preparing promoter deletions and mutations |
| P482-F | 5′-atttggtaccGCTCCCGGCTCCCGCTCTC-3′ | Preparing P482 deletion |
| P435-F | 5′-atttggtacCGGGGCCTGCGGTGATTGG-3′ | Preparing P435 deletion |
| P512-F | 5′-ataggtaccgagGGGGAAGGCTG-3′ | Preparing 3′ promoter deletions |
| P339-R | 5′-gaaactcgagCCCGCGAGGAGAGCTTCG-3′ | Preparing P339 deletion |
| 301-R | 5′-caaactcgaGCGGCGCTGCGGTAGTCGTC-3′ | Preparing P301 deletion |
| 217-R | 5′-caaactcgagGGCACCTCGAAGGACCATG-3′ | Preparing P217 deletion |
| IntronA-F | 5′-gaaaggatccCGGACGGCGAACACAATG-3′ | Cloning “intron A” from BAC |
| IntronA-R | 5′-cattgtcgaCCTCCTTTGTGTCCCCCAG-3′ | Cloning “intron A” from BAC |
| IntronB-F | 5′-caaaggatccCTGGGGGACACAAAGGAG-3′ | Cloning “intron B” from BAC |
| IntronB-R | 5′-catagtcgacCTTTTACTCTCTAAGAAACCAATCC-3′ | Cloning “intron B” from BAC |
| Mutations by overlap PCR† | ||
| P512-F2 | 5′-gtttttcgtacgaattcCCGGGGAAGGCTGC-3′ | Preparing promoter mutations |
| NFYmut-F | 5′-GGCCTGCGGTGGCTAGCGGGCGGGCG-3′ | Preparing promoter NFY mutation |
| NFYmut-R | 5′-CGCCCGCCCGCTAGCCACCGCAGGCC-3′ | Preparing promoter NFY mutation |
| SP1mut1-F | 5′-CGAGCCGCCGTTAACCGGGCCTGCGGTG-3′ | Preparing promoter SP1 mutation 1 |
| SP1mut1-R | 5′-CGAGCCGCCGTTAACCGGGCCTGCGGTG-3′ | Preparing promoter SP1 mutation 1 |
| SP1mut2-F | 5′-GGTGATTGGCGAGAATTCGGGGAGGTCGG-3′ | Preparing promoter SP1 mutation 2 |
| SP1mut2-R | 5′-CCGACCTCCCCGAATTCTCGCCAATCACC-3′ | Preparing promoter SP1 mutation 2 |
| ETSmut-F | 5′-GCGGGGAGGTCCGCGGTACTTTGTTTTTTATG-3′ | Preparing promoter ETS mutation |
| ETSmut-R | 5′-CATAAAAAACAAAGTACCGCGGACCTCCCCGC-3′ | Preparing promoter ETS mutation |
| OCTmut-F | 5′-CTTTGTTTTTTATCGATTTGAGGGAGTGGG p-3′ | Reparing promoter OCT mutation |
| OCTmut-R | 5′-CCCACTCCCTCAAATCGATAAAAAACAAAG-3′ | Preparing promoter OCT mutation |
| Ebox1mut-F | 5′-CCTTCATTCACCCGATCGGTCCTTCGAG-3′ | Preparing promoter E-box mutation |
| Ebox1mut-R | 5′-CTCGAAGGACCGATCGGGTGAATGAAGG-3′ | Preparing promoter E-box mutation |
| E2Fmut1-F | 5′-CTGCGCCTTGGGCCCACTTCGCGCCCTCGGGCG-3′ | Preparing promoter E2F mutation 1 |
| E2Fmut1-R | 5′-CGCCCGAGGGCGCGAAGTGGGCCCAAGGCGCAG-3′ | Preparing promoter E2F mutation 1 |
| E2Fmut2-F | 5′-CGCCACTTCGGTACCTCGGGCGAG-3′ | Preparing promoter E2F mutation 2 |
| E2Fmut2-R | 5′-CTCGCCCGAGGTACCGAAGTGGCG-3′ | Preparing promoter E2F mutation 2 |
| E2Fmut1,2-F | 5′-CCTTGGGCCCACTTCGGTACCTCGGGCGAGGCC-3′ | Preparing combined E2F mutations 1 & 2 |
| E2Fmut1,2-R | 5′-CGCCCGAGGTACCGAAGTGGGCCCAAGGCGCAG-3′ | Preparing combined E2F mutations 1 & 2 |
| Ebox2mut-F | 5′-GCGGCGGCGGCGATCGGGGCAGGCCG-3′ | Preparing intron E-box mutation 2 |
| Ebox2mut-R | 5′-CGGCCTGCCCCGATCGCCGCCGCCGC-3′ | Preparing intron E-box mutation 2 |
| Ebox3mut-F | 5′-CCTTGTGCGACGATCGCTGCCGGCCC-3′ | Preparing intron E-box mutation 3 |
| Ebox3mut-R | 5′-GGGCCGGCAGCGATCGTCGCACAAGG-3′ | Preparing intron E-box mutation 3 |
| Ebox4mut-F | 5′-CCAGTCATACGATCGGACCTAACTGC-3′ | Preparing intron E-box mutation 4 |
| Ebox4mut-R | 5′-GCAGTTAGGTCCGATCGTATGACTGG-3′ | Preparing intron E-box mutation 4 |
In the “Cloning and deletions” section, added nucleotides are in lowercase and restriction sites are underlined.
In the “Mutations by overlap PCR” section, altered nucleotides are in italics and new restriction sites are underlined.
Plasmids
The firefly luciferase reporter vectors pGL3 basic, pGL3 promoter, and Renilla luciferase vectors pRL-SV40 are from Promega (Madison, WI). From BAC RP11-97P7 (BACPAC Resources Center, Children's Hospital Oakland Research Institute, Oakland, CA), a fragment containing the human C13orf25 promoter (−659 to 323 bp) was PCR amplified with the addition of an XhoI site at the 3′ end and was cloned into the pBLCAT7 plasmid. Four fragments of the promoter region were cut out from the human C13orf25-pBLCAT7 plasmid by digestion with XhoI and either PvuII (P982; names correspond to the insert size in bp), HpaI (P606), SmaI (P512), or ScaI (P395) and were ligated into the Ecl136II and XhoI sites of pGL3 basic vector. Additional 5′ deletions (P482 and P435) and 3′ deletions (P339, P301, and P217) of P512 were generated by PCR. Downstream regions A, B, and AB were PCR amplified from BAC DNA and then inserted into BamHI-SalI sites located downstream of the luciferase gene in the pGL3 C13orf25-P301 promoter or pGL3 promoter reporter vector. All the mutations were generated by the overlap PCR method from P512. Primers used in preparing these mutations and other constructs are listed in Table 1. The sequences of all the constructs prepared using PCR were confirmed by complete sequencing.
Luciferase Reporter Assays
HEK293T or NIH3T3 cells were plated at 5 × 104 cells per well in a 24-well plate 24 hours before transfection. The plasmids were cotransfected with pRL-SV40 using TurboFect transfection reagent (Fermentas, Hanover, MD) according to the manufacturer's instructions. DHL16 cells were transiently transfected using an Amaxa Nucleofector (Lonza, Basel, Switzerland) with cell line solution V combined with program L-29. Luciferase assays were performed 24 hours after transfection using the Dual-Luciferase reporter assay system (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity for each reaction. Triplicate transfected wells were analyzed for each group.
ChIP
ChIP assay was performed in P3HR-1 and DHL16 cells according to the protocol of the ChampionChIP one-day kit (SABiosciences Corp., Frederick, MD). The antibodies used in this study were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA): MYC (N-262, sc-764), MNT (M-132, sc-769), and MXI1 (G-16, sc-1042). Real-time PCR amplifications were performed using DyNAmo Capillary SYBR Green PCR master mix (New England BioLabs, Inc.) and the DNA Engine Opticon 2 real-time PCR detection system according to the manufacturer's instruction. Primer sequences are listed in Table 1.
Labeling and Hybridization of ChIP Samples to a Custom Oligonucleotide Chip
ChIP of modified histones and RNA polymerase II (polII), amplification of ChIP DNA, and hybridization to a custom chip were performed as described previously.31 For analysis of histone modifications, we performed native ChIP on 20 × 106 cells without formaldehyde cross-linking. Nuclei were digested by micrococcal nuclease enzyme followed by mild sonication to obtain mononucleosomes. For analysis of polII binding, we performed ChIP after sonication after formaldehyde cross-linking to obtain DNA fragments ranging from 200 to 400 bp. Chromatin was then dialyzed into radioimmunoprecipitation assay buffer at 4°C. We conjugated 4 μg of H3-K9, K14-diacetyl–specific, and lysine-27 trimethylation–specific antibodies from Upstate Biotechnology (Millipore, Billerica, MA) (06–559 and 07–449) and lysine-4 monomethylation–specific, histone 3 lysine-4 trimethylation (H3K4me3)–specific, and polII-specific antibodies from Abcam Inc. (Cambridge, MA) (ab8895, ab8580, and ab5408) to Dynabeads protein A (Dynal Biotech, Oslo, Norway). We performed ChIP by overnight incubation of dialyzed chromatin with the antibody-loaded beads. We thoroughly washed the beads containing antibody and chromatin complexes, and the DNA was finally eluted from the beads. The eluted DNA was tested for enrichment by quantitative PCR. Input and enriched ChIP DNA were amplified by ligation-mediated PCR. Ligation-mediated PCR–amplified input DNA and ChIP-enriched DNA were labeled with Cy3 and Cy5, respectively, and were hybridized to a custom 385K array (Roche NimbleGen, Inc., Madison, WI) by the NimbleGen service laboratory in Reykjavik, Iceland.
Analysis of Publicly Available ChIP-Seq Data
Coordinates for individual reads from genome-wide ChIP-Seq experiments in K562 and HeLa-S3 cells and DNase sensitivity analysis in K562 were downloaded (http://genome.ucsc.edu, last accessed July 27, 2010) and sorted; for each experimental replicate, the average fragment size was estimated using an Excel spreadsheet (Microsoft Corp., Redmond, WA) by determining the coordinate displacement that resulted in maximal overlap of forward and reverse reads in a continuous block of ≥200,000 reads. Coordinates of forward and reverse reads in the C13orf25 region were adjusted based on these values, and the numbers of reads over all replicates and both orientations were combined and plotted in 20-bp increments using a 100-bp window. Evolutionary conservation in the C13orf25 promoter and close matches conserved consensus binding sites for transcription factors were determined using the MatInspector program (Genomatix Software GmbH, Munich, Germany).
Results
Expression of the miR-17-92 Cluster in B Cells
miRNA expression profiling was initially conducted using the TaqMan human miRNA array. Because the miR-17-92 cluster is expressed in proliferating cells, we normalized the values to those of normal centroblasts, which proliferate rapidly. We observed decreased miR-17-92 expression in normal naive B cells compared with that in centroblasts for each of the six miRNAs in the cluster but to varying extents, ranging from 1.28-fold in miR-92a to 7.8-fold in miR-18a (Figure 1A). Various tumor cell lines showed higher expression of the miRNA cluster than in normal centroblasts, with a ratio varying from 1.8 to 12.7, averaging over the six miRNAs (Figure 1A). In general, there are strong correlations between the levels of the different miRNAs in the cluster in the 11 cell lines analyzed, representing different stages of B-cell differentiation. The miRNA whose level correlated least well with the others was miR-18a (Figure 1A and inset). Processing of this miRNA is known to be regulated by an RNA-binding protein32; variation in efficiency of miR-18a processing among different cell lines is likely to be responsible for the relatively low correlation between miR-18a levels and those of the other members of the cluster.
Figure 1.
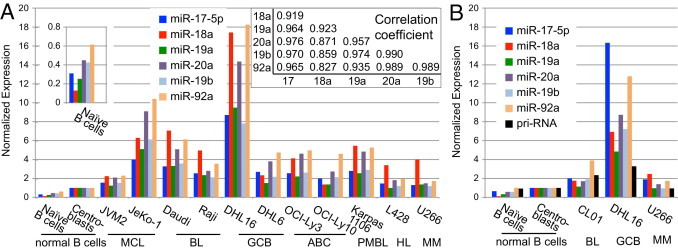
miRNA expression. A: Expression (linear scale) of the six miRNAs of the miR-17-92 cluster in normal naive human B cells and centroblasts and in 11 cell lines representative of various stages of mature B-cell differentiation analyzed in a PCR plate format and normalized to U6 RNA. Values are given as 2−ΔCT. Values of the centroblast samples were set to 1. The inset shows the correlation coefficients among the samples for each miRNA. B: Expression (linear scale) of the miR-17-92 cluster and of the primary RNA (pri-RNA), analyzed in individual PCR reactions in naive B cells and centroblasts, two of the cell lines analyzed in A, and a third cell line, CL01. Values of the centroblast samples were set to 1. ABC, activated B cell; BL, Burkitt's lymphoma; GCB, germinal center B cell; HL, Hodgkin's lymphoma; MCL, mantle cell lymphoma; MM, multiple myeloma; PMBL, primary mediastinal B cell lymphoma.
Of the 11 cell lines, the greatest amounts of miR-17-92 expression were seen in JeKo-1 (a mantle cell lymphoma line) and in DHL16 (a DLBCL line of the germinal center B-cell subtype). Because JeKo-1 cells are known to have amplification of the C13orf25 locus, we wished to determine whether gene amplification was also responsible for the high-level miR-17-92 expression in DHL16 cells. This was done using two approaches. First, we performed quantitative PCR of genomic DNA with primers in C13orf25 and the control locus RPL13 in DHL16 and seven other lines. Four of the lines (DHL6, OCI-Ly1, OCI-Ly3, and JVM2) had virtually identical ratios of C13orf25 to RPL13, and it was assumed that they contain two copies of C13orf25. Relative to the average normalized quantity of C13orf25 DNA in these four lines, DHL16 showed a mean ± SD ratio of 1.33 ± 0.60 and JeKo-1 a ratio of 5.8 ± 1.6 (see Supplemental Figure S1A at http://ajp.amjpathol.org). These data suggest that DHL16 has either two or three copies of C13orf25, whereas JeKo-1 has approximately 12. As a second approach to test for possible C13orf25 amplification in DHL16, we used the ChIP with hybridization to a custom oligonucleotide chip (ChIP-chip) data (described later herein) to estimate the relative copy numbers of the locus in DHL16 cells compared with those in normal centroblasts or naive B cells. We determined the ratio of the hybridization of input nucleosomal DNA to probes within C13orf25 to the hybridization to all the >100 genes on the custom array for the four histone modifications examined. The mean ± SD ratio of DHL16 to normal centroblasts was 0.883 ± 0.218 and of DHL16 to naive B cells was 0.784 ± 0.177 (see Supplemental Figure S1B at http://ajp.amjpathol.org). Thus, gene amplification does not seem to be responsible for the increased expression of the miR-17-92 cluster in DHL16 cells.
Results of miRNA profiling were also confirmed by real-time RT-PCR amplification in at least triplicate for each of the miRNAs of the cluster in normal naive B cells, centroblasts, and three cell lines (Figure 1B). Because the levels of the miRNAs in the cluster did not vary completely in parallel, we also analyzed a dilution series to determine whether differences in PCR efficiency among the primer sets might be responsible for this variation; however, all the primer sets showed similar amplification efficiencies (data not shown).
Elements Required for Full Promoter Activity Extend Approximately from −159 to 112 bp Relative to the Transcriptional Start Site
miRNAs are first transcribed as primary miRNAs by polII.33 As a preliminary indication of sequence elements that may be important in regulating the C13orf25 gene, we analyzed the degree of evolutionary conservation in the promoter region (Figure 2) and identified close matches to well-defined consensus binding sites for transcription factors. The region of highest conservation extends from approximately −118 to 70 bp relative to the transcription start site (TSS). Within this region are numerous close matches to consensus transcription factor binding sites, most of which are conserved even in zebra fish. The promoter lies within a 1.9-kb CpG island [as defined at the UCSC Genome Bioinformatics website (http://genome.ucsc.edu, last accessed July 27, 2010)], which, overall, has 63.5% guanine and cytosine content. As commonly found in promoters associated with CpG islands, several consensus sites for binding to the SP1 family are present. In addition to those noted in Figure 2, several close matches to SP1 consensus are found more proximal to the TSS. Despite being embedded in a CpG island, the promoter contains several AT-rich elements, including an octamer (OCT element), a possible LEF/SOX site, an atypical TATA box, and an initiator element. Other potentially important sites are described later herein. Additional highly conserved sequences likely represent binding sites for unidentified transcription factors.
Figure 2.
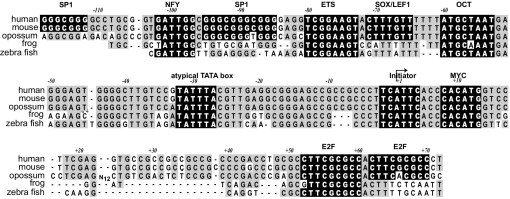
Promoter conservation. Alignment of sequences from human, mouse, opossum, frog, and zebra fish C13orf25 orthologs using ClustalW, with further manual modifications to compact nonconserved regions. Strong matches to consensus transcription factor binding motifs are shown based on analysis using the MatInspector program.
We used transient transfection of promoter luciferase reporter constructs to identify the DNA elements important in regulating transcription. Although we are especially interested in transcription of the gene in B cells, miRNAs seem to be expressed in all proliferating cells; for example, miR-17-92 miRNAs are expressed in a variety of mouse embryonic tissues,34 and unlike most miRNAs tested, in situ hybridization showed them to be ubiquitously expressed in zebra fish embryos.35 The promoter contains consensus binding sites for many widely expressed transcription factors, suggesting that much of the regulation of the gene may be cell-type nonspecific. Thus, because B cells are notoriously difficult to transfect, detailed promoter analysis was performed in HEK293T cells.
To determine elements important for promoter activity, a nested 5′ deletion series of six DNA fragments from the promoter region were cloned into the pGL3 basic luciferase reporter vector. Of these, initially four were prepared using convenient restriction enzyme sites (P982, P606, P512, and P395; names correspond to the length of promoter DNA included in the construct). In initial results (not shown), we determined that the P512 fragment, from −189 to 323 bp, had full activity in transient transfection experiments, whereas P395 had reduced activity. Based on these results, two additional constructs of size intermediate between P512 and P395 were generated by PCR cloning, and a small 5′ deletion of P512, removing 30 additional 5′ bp (to −159 bp, P482) was found to result in little change in activity (Figure 3). The similarity in activity of P982, P606, P512, and P482 shows that deleting less conserved regions from −659 to −159 bp does not reduce promoter activity, whereas a further 47-bp deletion (to −112 bp, P435) reduced activity ∼37% compared with P435. Correlating these results with the affected putative transcription factor binding sites, the deletion in P435 resulted in loss of the most 5′ putative SP1 site, and deletion of the 5′ cluster of SP1 sites and the NFY and ETS sites in P395 reduced activity by ∼66% compared with P435.
Figure 3.
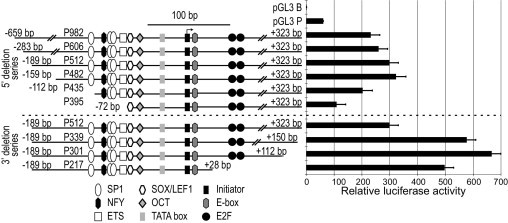
Firefly luciferase reporter assays in HEK293T cells of nested deletions of the C13orf25 promoter region normalized to cotransfected Renilla luciferase. Expression is relative to the promoterless pGL3 basic vector (pGL3 B), which was set to 1. Results with the SV40 promoter (pGL3 P) are also shown. Diagrams at the left show the location of the motifs shown in Figure 2. Note that the results with P512 are shown twice for ease of comparison. Data are given as mean ± SD.
A series of 3′ deletions was also analyzed (Figure 3). The P339 construct showed almost double the activity of the P512 construct. The P339 3′ deletion removed poorly conserved regions that contain a potential initiation codon in a good Kozak context that is out of frame of the downstream luciferase gene. Thus, although we have not ruled out the possibility that the deleted region binds factors with repressor activity, the most likely reason for increased activity of these 3′ deletions is increased productive translation of the message. A further deletion (to 28 bp) that removed two consensus E2F sites (comparing P217 with P301) modestly decreased activity. These results imply that positive regulatory elements in the promoter extend approximately from −159 to 112 bp from the transcriptional start site, encompassing the region of highest evolutionary conservation.
The Promoter Is Regulated by the Collaborative Activity of Several Transcription Factors, Most of Which Individually Have Only a Moderate Effect
To assess the importance of specific sequence elements in the region important for promoter activity, we prepared site-directed mutations. Mutations affecting all the SP1 sites reduced activity to 30%, similar to deletion of the corresponding region. Mutations of individual SP1 sites and the NFY and OCT sites reduced activity by ∼40% to 50% each, whereas mutation of the putative ETS site did not affect the activity (Figure 4A).
Figure 4.
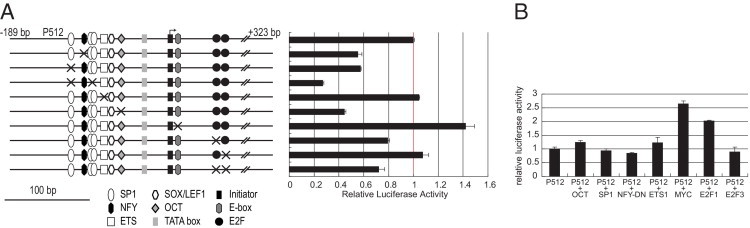
Multiple transcription factors control the C13orf25 promoter. A: Luciferase reporter assays in HEK293T cells of various C13orf25 promoter-reporter constructs derived from plasmid P512 with mutations in transcription factor binding motifs, as shown in the diagrams at the left. B: Results in HEK293T cells of cotransfection of P512 with expression constructs for transcription factors as indicated. Data are given as mean ± SD.
Mutation of the first E2F site or of both E2F sites reduced activity by ∼25%, similar to the effect of deleting the sites (comparing P217 with P301; Figure 3); mutation of the second E2F site alone had minimal effects. The promoter E-box is only one of several in the C13orf25 locus, and we refer to it as E1 in further analysis described later herein. Mutation of the E1 E-box increased activity by up to ∼40% (Figure 4A). Because C13orf25 is known to be regulated positively by MYC, this result was initially surprising. However, a variety of transcriptional repressors closely related to MYC, such as MNT and MXI1, are able to compete with it for heterodimerization with its partner MAX and to compete for binding to DNA. The effect of mutating the E-box suggests that C13orf25 is not only activated by MYC but also is inhibited by repressors of the MYC family. This repressive effect of the site predominates in HEK293T cells. ChIP data described later herein support this possibility.
These results of mutational analysis indicate that the promoter is regulated by the collaborative activity of several transcription factors, most of which individually have only a moderate effect. We further investigated the function of these sites by cotransfection of the P512 reporter construct with constructs expressing various transcription factors, namely, OCT2, SP1, ETS1, MYC, E2F1, and E2F3, and a dominant negative NFY vector (pNFYA29).36 The data indicated that only MYC and E2F1 expression vectors enhanced the activity (Figure 4B).
Histone Modifications in the C13orf25 Locus
As part of a larger analysis of ∼100 genes important in B cells, we analyzed histone PTMs in a 22.65-kb region using ChIP-chip.31 We analyzed the three B-cell lines for which miRNA abundances are shown in Figure 1B and normal naive B cells and centroblasts. Four PTMs were quantified: H3K4me3 and lysine-9 and -14 diacetylation (H3K9,14ac), associated with promoters and CpG islands; lysine-4 monomethylation, associated with enhancers or adjacent to regions of H3K4me3; and lysine-27 trimethylation, associated with repression by polycomb complexes. In addition, for the three cell lines, we also analyzed binding of polII. To facilitate comparison among samples, data were globally normalized based on hybridization to the entire chip of ∼388,000 oligonucleotides.
The histone PTMs in the locus varied considerably among the samples (Figure 5). In the proximal portion of the gene, H3K4me3 varied the least, with peaks immediately 5′ and 3′ of the CpG island, a small peak near the TSS, and a peak ∼0.7 kb downstream of the TSS. In the three cell lines, H3K4me3 extends more 3′ and is found over the miRNA sequences. The location of H3K9,14ac is similar but varies quantitatively, with very low values in naive B cells and centroblasts but higher values in the cell lines. This analysis of the other genes in this array showed that the transcript level for a given gene correlated with the quantity of H3K9,14ac surrounding the promoter.31
Figure 5.
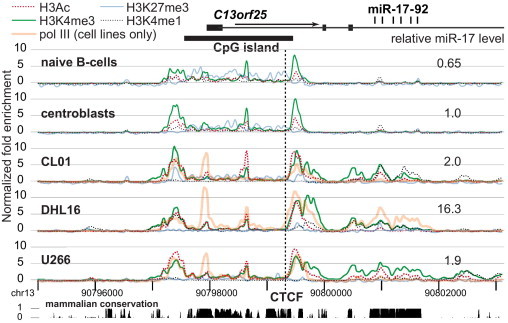
Chromatin structure. ChIP-chip results using a custom NimbleGen oligonucleotide array with the five samples analyzed in Figure 1B, namely, normal naive B cells and centroblasts and cell lines DHL16, CL01, and U266. The results from each hybridization experiment were globally normalized over the array, setting the enrichment of the top 0.1% of probes to 10. At the top are shown the structure of the gene, the sequences encoding the mature miRNAs, and the extent of the CpG island encompassing the promoter. Expression of miR-17-5p (Figure 1B) is also shown. Sequence positions on chromosome 13 (chr13) are in hg18 coordinates. Conservation with 17 other placental mammals (from http://genome.ucsc.edu) is shown at the bottom of the figure; height indicates the relative Multiz37 conservation score (maximum is 1). Also shown is the position of a strong consensus site (5′-CGCCGCCCCCTGGCGGCTAC-3′) for CCCTC-binding factor (CTCF), close to the peak maximum by ChIP-Seq in K562 (see Figure 7) and a variety of other cell lines.
A strong peak of polII binding was found near the TSS; polII was also found, to a variable extent, at the other sites of H3K4me3. Among the three cell line samples for which polII data were obtained, the most striking difference was the presence of polII over the miRNA sequences only in DHL16, which showed, by far, the highest levels of the mature miRNAs.
Enhancers for the C13orf25 Locus
Recently, specific histone PTMs have been identified as markers for distal enhancers.38 The presence of activating histone modifications in the C13orf25 gene body at considerable distances from the TSS raised the possibility that these are associated with regulatory regions, in particular, enhancers. In an initial step to test this possibility, we analyzed two neighboring regions for enhancer activity: region A (586 to 1,265 bp) in intron 1, which encompasses a small peak of active histone modifications in the ChIP-chip data, and region B (1,248 to 2,183 bp), extending into intron 2, containing the largest peak of active histone modifications. The luciferase reporter pGL3 basic containing the C13orf25 promoter and region A displayed at least 2.3-fold greater luciferase activity than the control that contained only the promoter (Figure 6A). The plasmid C13orf25 promoter/region B showed modestly greater activity than the parental C13orf25 promoter construct in HEK293T, NIH3T3, and DHL16 cells but considerably less activity than the construct containing region A. There was no evidence of synergy when a construct containing both regions A and B was analyzed.
Figure 6.
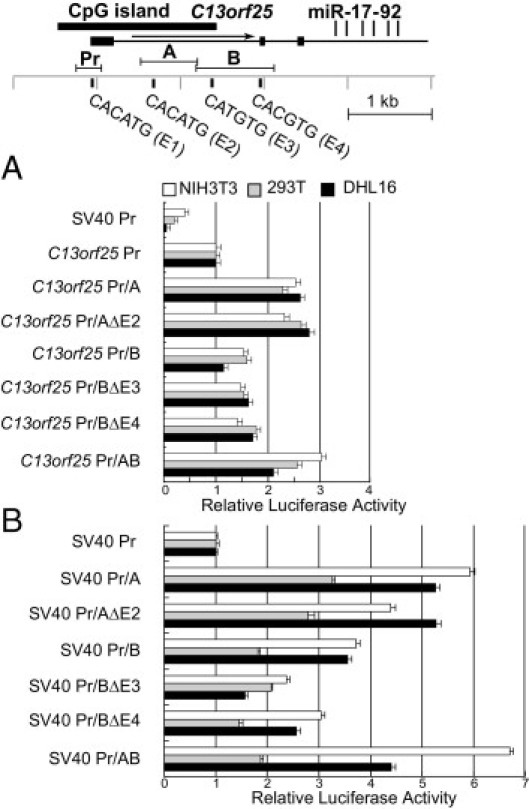
Potential C13orf25 enhancers. Luciferase activity in three cell lines (HEK293T, NIH3T3, and DHL16) of various constructs, some of which contain two regions, A and B, being analyzed for enhancer activity. Where indicated, constructs containing point mutations were created in one or more putative E-boxes. The positions of the C13orf25 promoter region (Pr) being analyzed, of regions A and B, and of the four putative E-boxes are shown at the top. A: Results using constructs containing the C13orf25 promoter. B: Results using constructs containing the SV40 promoter. Data are given as mean ± SD.
To confirm the enhancer activity of the intronic regions, we cloned them into the pGL3 promoter luciferase vector, in which luciferase expression is driven by the SV40 promoter. The pGL3 P/A luciferase construct showed more than fivefold luciferase activity compared with control vector in DHL16 and NIH3T3 cells (Figure 6B). The pGL3 P/B vector showing 3.5-fold activity compared with control vector in the same two cell lines. Compared with pGL3 P/A and pGL3 P/B, pGL3 P/AB possessed the highest activity in NIH3T3 cells, whereas pGL3 P/A yielded the strongest luciferase activity in DHL16 and HEK293T cells.
E-Box Motif Function in the C13orf25 Locus
Because of the reported role of MYC in regulating C13orf25, we were especially interested in potential MYC binding sites. MYC and its family members normally bind DNA as heterodimers with a common partner, MAX. The highest affinity site for MAX homodimers and its heterodimers is CACGTG; only one such conserved site, in exon 2 (E4, region B), is found in or neighboring the region of active histone PTMs. Slightly lower affinity sites have the sequence CATGTG; three such sites are found in the region of active histone modification, including one site each in regions A and B (E2 and E3) and a site in the promoter region (E1), discussed previously herein, whose mutation led to an increase in promoter activity.
Mutations of E2, E3, and E4 were prepared in the C13orf25 promoter/A and C13orf25 promoter/B constructs and showed little effect (Figure 6A). Some of the same mutations in the SV40 promoter background (pGL3 P/A and pGL3 P/B) reduced, but did not eliminate, enhancer activity (Figure 6B), with the effects varying among the cell lines tested. For DHL16, no effect was seen with the E2 mutation in region A, but mutation of E4 reduced enhancer activity in the B region, whereas mutation of E3 strongly reduced enhancer activity.
MXI1 Binds to the Promoter
A previous study showed that the miR-17-92 cluster is directly transactivated by MYC, which directly binds to the first intron of C13orf 25.8 To identify the binding sites for MYC family members to the endogenous C13orf25 locus, we made use of publicly available genome-wide ChIP sequencing data (http://genome.ucsc.edu).39 Among the many cell lines analyzed, data for MAX and MYC binding are particularly extensive for HeLa-S3 and for the erythroleukemia cell line K562. For both lines and both transcription factors, by far the highest binding was at E3, followed by the promoter E1 (Figure 7, A and B). Binding at E2 was low, and at E4, it was almost undetectable. Two additional sites upstream of the promoter were found in both cell lines, centered approximately 610 and 300 bp from the TSS. Within 50 bp of each peak maximum was found the sequence CACGCG, with a single mismatch from the highest affinity site; a detailed analysis of MAX binding revealed that MAX binds detectably to such sites.40 Both of these sites are present in the construct P982 (Figure 3) but not in any of the smaller constructs; thus, these sites do not contribute obviously to reporter construct expression.
Figure 7.
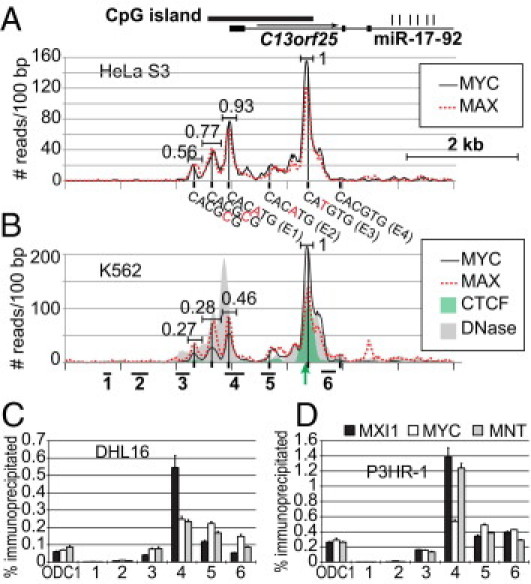
Binding of MYC family transcription factors. A: Genome-wide ChIP-Seq data in HeLa-S3 cells using antibodies that recognize MYC and MAX are shown. The positions of four putative E-boxes and of two E-box variants are also shown. The relative binding of MYC and MAX at various peaks was quantified, being normalized by setting the MYC/MAX ratio to 1 for E-box E3. B: Data for K562, as in A. Also shown are ChIP-Seq with antibodies to CCCTC-bindng factor (CTCF) and sites of DNase I cleavage (“digital” DNase hypersensitivity analysis; sequencing reads prepared by the Stamatoyannopoulos Laboratory at the University of Washington were downloaded from http://genome.ucsc.edu). The arrow shows the position of a close match to the consensus CTCF DNA-binding sequence. The positions of the PCR products analyzed by conventional ChIP in DHL16 (C) and P3HR-1 (D) are shown at the bottom. C: Results of ChIP and quantitative real-time PCR with probes across the C13orf25 locus in DHL16 cells using antibodies that recognize MYC, MXI1, and MNT. D: As in C, analyzing ChIP in P2HR-1 cells. Note that E1 and E2 lie within the amplicons of primer sets 4 and 5, respectively, whereas the primer set 6 amplicon lies between E3 and E4.
A possible explanation for the minimal MYC and MAX binding at E2 and E4 in K562 cells is chromatin inaccessibility. DNase I sensitivity is frequently used to identify accessible DNA, as found at enhancers, promoters, and other regulatory regions. Publicly available genome-wide DNase sensitivity for the K562 cell line (http://genome.ucsc.edu, last accessed July 27, 2010) revealed that the sequences surrounding E2 and E4 were insensitive to DNase digestion (Figure 7B), consistent with the hypothesis that a closed chromatin conformation blocks MYC and MAX binding to these sites.
Although in HeLa the relative binding of MYC and MAX was similar at E1 and E3, in K562, considerably lower relative binding of MYC was found at E1 than at E3, suggesting that at least half of the MAX bound to E1 was in heterodimers with proteins other than MYC and that different E-boxes preferentially bind different MYC family members. If so, this would provide a potential explanation for the paradoxical increase in luciferase activity on mutating E1 and the different effects of mutating the other E-boxes (Figures 4 and 6).
The results in K562 suggested that family members in addition to MYC may bind to E1 at the promoter. To test this possibility, we performed ChIP with antibodies to MYC and to two MYC family members, MNT and MXI1, which bind to E-boxes as heterodimers with MAX to function as dedicated transcriptional repressors.41 Isolated DNA was analyzed by quantitative real-time PCR using several probes across the C13orf25 locus and in the promoter of the known MYC target ODC1. Two cell lines were used for ChIP: DHL16 and, for comparison, another B-cell line, the Burkitt's lymphoma cell line P3HR-1, which expresses high levels of MYC. The immunoprecipitated DNA was enriched at the promoter and intronic regions for all three antibodies, with highest enrichment at the promoter (Figure 7C). The relative enrichment of MYC, MXI1, and MNT, however, varied at the various sites, with greater enrichment of MXI1 and, in P3HR-1, of MNT at the promoter than at intronic sites. This is consistent with the ChIP-seq data available for K562 and for the increased activity of the C13orf25 promoter when the E1 E-box is disrupted (Figure 4A).
Discussion
C13orf25, containing the miR-17-92 cluster, is amplified in some lymphomas, but the miRNAs themselves are much more frequently overexpressed in lymphoma.5 In the cell lines we analyzed, overexpression was particularly striking in the case of DHL16, in which the locus is not amplified. These data suggest that abundance of the miRNAs in the miR-17-92 cluster is regulated at several levels. Although this analysis was centered on transcriptional regulation, the results suggest that differential processing or stability of the miRNAs also influences their abundance. The abundance of the six miRNAs correlate relatively well except for miR-18a, whose processing has been shown to be regulated by binding of hnRNPA1 to its conserved terminal loop.32,42 In particular, the abundance of miR-18a is strikingly low in naive B cells. A study of miR-17-92 during mouse development also showed that expression of the individual miRNAs of the cluster did not vary entirely in parallel,34 with miR-18a showing a disproportionate decrease in several postnatal tissues compared with the embryo. In addition to the variation among the miRNAs in the cluster, the overall degree of processing is likely to vary because the levels of the primary RNA do not correlate well with the levels of the mature miRNAs. This is especially striking in the case of DHL16, in which the primary RNA levels are 3.3-fold those of normal centroblasts, whereas the ratios for the mature miRNAs vary from 4.8 to 12.8.
These results suggest that the primary RNA is processed more efficiently in DHL16 than in the other cell lines and normal samples. Because processing requires cleavage of the primary RNA, more efficient processing would be expected to result in lower steady-state levels of the primary RNA. Thus, the modestly enhanced levels of primary RNA in DHL16 cells may not fully reflect the rate of transcription in this cell line. The most striking difference in ChIP-chip results between DHL16 and the other two lines that were analyzed is the presence of polII over the miRNA sequences in DHL16 only. An intriguing possibility is that the presence of polII over the miRNA-encoding sequences is due to slowing of transcriptional elongation through the region by unknown mechanisms, allowing for proper folding of the RNA hairpins and subsequent cleavage by the Drosha complex.
The present analysis of vertebrate sequence conservation revealed that the promoter region comprises several putative SP1 sites; sites for NFY, ETS, OCT, and MYC; and two tandem E2F sites. This region is highly conserved in vertebrates, and most of the putative transcription factor binding sites are conserved even in fish. Additional highly conserved sequences likely represent binding sites for unidentified transcription factors.
Several early studies demonstrated that the E2F family of transcription factors can directly bind C13orf25 and stimulate its transcription.21,23 The present studies indicated that a 3′ deletion, eliminating the second E2F site, reduced activity by ∼30%, whereas mutation of both sites reduced activity to a similar extent. The mutation of putative SP1 sites reduced activity of promoter-reporter constructs by 70%, suggesting that the SP/KLF family of transcription factors may be important in regulating this gene. To investigate the role of SP1 in the transcription of this gene, SP1 expression vectors were cotransfected with the promoter reporter. The failure to affect transcription is likely because SP1 is abundantly and ubiquitously expressed and cotransfection with its expression vector did not sufficiently perturb the overall level of the protein to affect transcription.
From ChIP-chip analysis of SP1 binding to chromosomes 21 and 22, a minimal estimate of the number of SP1-binding sites in the human genome was 12,000.43 Initially, SP1 was thought to have no role other than to constitutively activate housekeeping genes, but evidence has mounted for important roles in cell proliferation and tumorigenesis.44 Additional specificity is likely provided by its direct interaction with numerous other transcription factors, including several that may affect C13orf25 expression, including E2F1, OCT1, NFYA, and NFYC. Interactions with E2F2 and E2F3, ETS1, and MYC have also been identified, but these may be indirect.44 Thus, the SP1 sites may be important in promoting cooperative recruitment of SP1 along with other transcription factors that bind the promoter.
Histone modifications are now known to play a central role in the regulation of gene transcription.45–47 We performed ChIP-chip to identify the histone modifications associated with the C13orf25 locus in normal B cells and in various B-cell lines. The various samples showed considerable variation in the pattern of histone PTMs. One of the most striking differences was the very low level of H3K9,14ac in naive B cells and normal centroblasts compared with in the three cell lines. H3K9,14ac has been shown to be important in recruiting the general transcription factor complex TFIID to promoters.48 In addition, the observation downstream of the promoter of histone PTMs associated with active transcription suggested the presence of intronic enhancers, which we confirmed using reporter assays.
Previous studies in a Burkitt's cell line with regulated MYC expression demonstrated that the C13orf 25 locus is directly transactivated by MYC and that MYC directly binds to the first intron.8 Resting naive B cells have among the highest levels of MYC mRNA among undividing cells (http://biogps.gnf.org, last accessed July 27, 2010). Surprisingly, in contrast to other dividing cells, normal rapidly dividing centroblasts have less MYC mRNA than resting B cells, and MYC protein is undetectable by immunohistochemical analysis in normal centroblasts.49 In contrast, MYC mRNA is up-regulated in dividing B-lineage cells at other differentiation stages and in B cells induced to divide by mitogens in vitro.50 This unusual regulation of MYC in centroblasts seems to be abrogated in most DLBCLs.51 In normal centroblasts, the low level of MYC protein may contribute to the low level of the activating histone PTM H3K9,14ac found in the C13orf25 locus (Figure 5) and the minimal up-regulation of miR-17-92 expression (Figure 1).
After identifying several conserved E-boxes in the C13orf25 locus that function in transient transfection experiments, we examined which of the putative E-boxes can bind MYC family members in their normal chromosomal context. Publicly available ChIP-seq data allow the sites of binding to be defined more precisely (Figure 7). It is likely that the considerable differences in HeLa and K562 cells in MYC and MAX binding among the four conserved E-boxes result, in part, from differences in chromatin accessibility and structure. E2 and E4 are in regions of low DNase sensitivity in K562, suggesting that these regions are poorly accessible to transcription factor binding. In contrast, E1 and E3 lie in regions of DNase hypersensitivity in K562 (Figure 7B), the first corresponding to the promoter and the second lying only 36 bp from a binding site for CCCTC-binding factor, which is efficient in excluding nucleosomes and positioning neighboring nucleosomes52 and, thus, would be expected to contribute to binding at the neighboring E3 site. CCCTC-binding factor has complex effects on transcription but functions in part by promoting chromatin looping53; thus, it may contribute to interaction of the neighboring intronic enhancers with the promoter. MYC binding is also associated with areas of H3K4me3 and H3 acetylation.54 The ChIP-chip experiments (Figure 5) revealed prominent peaks of H3K4me3 and H3K9,14ac in the three cell lines analyzed immediately neighboring E1 and E3. Peaks of these histone PTMs, albeit smaller, were also detected adjacent to E2. The active histone PTMs are consistent with the ChIP data with MYC family members (Figure 7C), which showed relatively high enrichment with primer pair 5, whose amplicon includes E2. Although mutation of E2 did not affect luciferase activity in DHL16 in transient transfection experiments (Figure 6), these data raise the possibility that E2 is active in B cells in its normal chromosomal context, in contrast to K562 and HeLa S3.
Analysis of high-quality ChIP-seq data on MYC and MAX binding in K562 and HeLa S3 cells showed that binding to the two factors did not occur entirely in parallel across the C13orf25 locus (Figure 7, A and B); in particular, compared with binding at E3, proportionally less MYC binding was found at the promoter (E1) in K562, but little difference was found in HeLa S3 cells. The differential binding at E1 and E3 in K562, despite their identical core sequences, may be due to differences in flanking sequences, to differences in histone modifications, or to binding by cooperating transcription factors. We also found preferential binding of inhibitory MYC family members to the promoter (E1) in B-cell lines (Figure 7C). Preferential binding of inhibitory MYC family members to E1 may explain why mutation of E1 increased promoter-reporter activity (Figure 4A).
Binding to two noncanonical E-boxes (CACGCG) upstream of the promoter showed a large relative excess of MAX binding over MYC binding (Figure 7). MAX has been shown to bind to this sequence40 with an affinity that is appreciable although lower than to canonical sites, and MYC can activate transcription of TAF4B through such a site.55 MNT, however, has been reported to bind with higher affinity to this noncanonical sequence than to canonical E-boxes.56 Thus, it is likely that in some cell types these upstream noncanonical sites are preferentially bound by MNT and perhaps other repressive MYC family members whose sequence specificity has not been analyzed in detail. Although deleting these sequences had little effect in the promoter-reporter assays (Figure 4A), we cannot exclude the possibility that they are functional in their normal chromosomal context.
In summary, we provide evidence that the abundance of C13orf25 miRNAs is affected not only by transcriptional mechanisms but likely also by varying processing efficiency. We mapped the promoter and identified intronic regions with enhancer activity. Finally, these data suggest that C13orf25 is bound not only by MYC but also by inhibitory members of the MYC family, allowing for a complex mode of regulation by MYC family proteins.
Acknowledgments
We thank Drs. Javeed Iqbal for gene expression analysis, Zhong Feng Liu for technical assistance with gene expression analysis, and Brian Boer for initial subcloning of the C13orf25 promoter region.
Footnotes
Supported by University of Nebraska Medical Center Eppley Cancer Center pilot grants (T.W.M. and K.F.), a clinical investigator career development award from Lymphoma Research Foundation/Millennium Pharmaceuticals Inc. (K.F.), a National Cancer Institute grant (U01-CA114778-03 to W.C.C.), and a National Institute of General Medical Sciences grant (GM-080751 to A.R.).
M.J. and E.R. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.06.008.
Contributor Information
Kai Fu, Email: kfu@unmc.edu.
Timothy W. McKeithan, Email: tmckeith@unmc.edu.
Supplementary data
Quantification of copy number of C13orf25 in various B-cell lines. A: Quantification by real-time PCR and normalization to the RPL13 locus. Quantitative real-time PCR was performed as described in the methods for ChIP. The values for cell lines DHL6, OCI-Ly1, OCI-Ly3, and JVM2 were similar, and their average was set at 1. As discussed in the text, JeKo-1 shows marked amplification, and Granta and Z138C, which are also mantle cell lymphoma cell lines from American Type Culture Collection, likely show a small copy number gain. DHL16 shows either a normal 2N copy number or a gain of 1. B: Quantification by comparing hybridization of input DNA used for ChIP-chip. The fraction of hybridization signal of the input DNA (before IP) from nucleosomal DNA to the C13orf25 locus (see Figure 5) was compared with the hybridization signal to the entire custom array interrogating ≥100 genes. The ratio of this value to that obtained for normal naive B cells and for normal centroblasts was calculated for IP experiments in each of four histone modifications. The mean values are shown. Error bars are standard deviations.
References
- 1.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 2.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.He L., Hannon G.J. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 4.Zamore P.D., Haley B. Ribo-gnome: the big world of small RNAs. Science. 2005;309:1519–1524. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
- 5.He L., Thomson J.M., Hemann M.T., Hernando-Monge E., Mu D., Goodson S., Powers S., Cordon-Cardo C., Lowe S.W., Hannon G.J., Hammond S.M. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim H., Huang W., Jiang X., Pennicooke B., Park P.J., Johnson M.D. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc Natl Acad Sci U S A. 2010;107:2183–2188. doi: 10.1073/pnas.0909896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson S.M., Grosshans H., Shingara J., Byrom M., Jarvis R., Cheng A., Labourier E., Reinert K.L., Brown D., Slack F.J. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 8.O'Donnell K.A., Wentzel E.A., Zeller K.I., Dang C.V., Mendell J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 9.Costinean S., Zanesi N., Pekarsky Y., Tili E., Volinia S., Heerema N., Croce C.M. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–7029. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voorhoeve P.M., le Sage C., Schrier M., Gillis A.J., Stoop H., Nagel R., Liu Y.P., van Duijse J., Drost J., Griekspoor A., Zlotorynski E., Yabuta N., De Vita G., Nojima H., Looijenga L.H., Agami R. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 11.He L., He X., Lim L.P., de Stanchina E., Xuan Z., Liang Y., Xue W., Zender L., Magnus J., Ridzon D., Jackson A.L., Linsley P.S., Chen C., Lowe S.W., Cleary M.A., Hannon G.J. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mestdagh P., Fredlund E., Pattyn F., Schulte J.H., Muth D., Vermeulen J., Kumps C., Schlierf S., De Preter K., Van Roy N., Noguera R., Laureys G., Schramm A., Eggert A., Westermann F., Speleman F., Vandesompele J. MYCN/c-MYC-induced microRNAs repress coding gene networks associated with poor outcome in MYCN/c-MYC-activated tumors. Oncogene. 2010;29:1394–1404. doi: 10.1038/onc.2009.429. [DOI] [PubMed] [Google Scholar]
- 13.Trang P., Medina P.P., Wiggins J.F., Ruffino L., Kelnar K., Omotola M., Homer R., Brown D., Bader A.G., Weidhaas J.B., Slack F.J. Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2010;29:1580–1587. doi: 10.1038/onc.2009.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ota A., Tagawa H., Karnan S., Tsuzuki S., Karpas A., Kira S., Yoshida Y., Seto M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 15.Hayashita Y., Osada H., Tatematsu Y., Yamada H., Yanagisawa K., Tomida S., Yatabe Y., Kawahara K., Sekido Y., Takahashi T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65:9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- 16.Tagawa H., Seto M. A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia. 2005;19:2013–2016. doi: 10.1038/sj.leu.2403942. [DOI] [PubMed] [Google Scholar]
- 17.Rinaldi A., Poretti G., Kwee I., Zucca E., Catapano C.V., Tibiletti M.G., Bertoni F. Concomitant MYC and microRNA cluster miR-17-92 (C13orf25) amplification in human mantle cell lymphoma. Leuk Lymphoma. 2007;48:410–412. doi: 10.1080/10428190601059738. [DOI] [PubMed] [Google Scholar]
- 18.Koralov S.B., Muljo S.A., Galler G.R., Krek A., Chakraborty T., Kanellopoulou C., Jensen K., Cobb B.S., Merkenschlager M., Rajewsky N., Rajewsky K. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell. 2008;132:860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Ventura A., Young A.G., Winslow M.M., Lintault L., Meissner A., Erkeland S.J., Newman J., Bronson R.T., Crowley D., Stone J.R., Jaenisch R., Sharp P.A., Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17∼92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao C., Srinivasan L., Calado D.P., Patterson H.C., Zhang B., Wang J., Henderson J.M., Kutok J.L., Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sylvestre Y., De Guire V., Querido E., Mukhopadhyay U.K., Bourdeau V., Major F., Ferbeyre G., Chartrand P. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem. 2007;282:2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 22.Pickering M.T., Stadler B.M., Kowalik T.F. miR-17 and miR-20a temper an E2F1-induced G1 checkpoint to regulate cell cycle progression. Oncogene. 2009;28:140–145. doi: 10.1038/onc.2008.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods K., Thomson J.M., Hammond S.M. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007;282:2130–2134. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- 24.Yan H.L., Xue G., Mei Q., Wang Y.Z., Ding F.X., Liu M.F., Lu M.H., Tang Y., Yu H.Y., Sun S.H. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009;28:2719–2732. doi: 10.1038/emboj.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brock M., Trenkmann M., Gay R.E., Michel B.A., Gay S., Fischler M., Ulrich S., Speich R., Huber L.C. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009;104:1184–1191. doi: 10.1161/CIRCRESAHA.109.197491. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y.W., Liu J.C., Deatherage D.E., Luo J., Mutch D.G., Goodfellow P.J., Miller D.S., Huang T.H. Epigenetic repression of microRNA-129-2 leads to overexpression of SOX4 oncogene in endometrial cancer. Cancer Res. 2009;69:9038–9046. doi: 10.1158/0008-5472.CAN-09-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roman-Gomez J., Agirre X., Jimenez-Velasco A., Arqueros V., Vilas-Zornoza A., Rodriguez-Otero P., Martin-Subero I., Garate L., Cordeu L., San Jose-Eneriz E., Martin V., Castillejo J.A., Bandres E., Calasanz M.J., Siebert R., Heiniger A., Torres A., Prosper F. Epigenetic regulation of microRNAs in acute lymphoblastic leukemia. J Clin Oncol. 2009;27:1316–1322. doi: 10.1200/JCO.2008.19.3441. [DOI] [PubMed] [Google Scholar]
- 28.Tsai K.W., Kao H.W., Chen H.C., Chen S.J., Lin W.C. Epigenetic control of the expression of a primate-specific microRNA cluster in human cancer cells. Epigenetics. 2009;4:587–592. doi: 10.4161/epi.4.8.10230. [DOI] [PubMed] [Google Scholar]
- 29.Saetrom P., Snove O., Jr, Rossi J.J. Epigenetics and microRNAs. Pediatr Res. 2007;61:17R–23R. doi: 10.1203/pdr.0b013e318045760e. [DOI] [PubMed] [Google Scholar]
- 30.Saito Y., Jones P.A. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle. 2006;5:2220–2222. doi: 10.4161/cc.5.19.3340. [DOI] [PubMed] [Google Scholar]
- 31.Ramachandrareddy H., Bouska A., Shen Y., Ji M., Rizzino A., Chan W.C., McKeithan T.W. BCL6 promoter interacts with far upstream sequences with greatly enhanced activating histone modifications in germinal center B cells. Proc Natl Acad Sci U S A. 2010;107:11930–11935. doi: 10.1073/pnas.1004962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michlewski G., Guil S., Semple C.A., Caceres J.F. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol Cell. 2008;32:383–393. doi: 10.1016/j.molcel.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai X., Hagedorn C.H., Cullen B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jevnaker A.M., Khuu C., Kjole E., Bryne M., Osmundsen H. Expression of members of the miRNA17-92 cluster during development and in carcinogenesis. J Cell Physiol. 2011;226:2257–2266. doi: 10.1002/jcp.22562. [DOI] [PubMed] [Google Scholar]
- 35.Wienholds E., Kloosterman W.P., Miska E., Alvarez-Saavedra E., Berezikov E., de Bruijn E., Horvitz H.R., Kauppinen S., Plasterk R.H. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani R., Li X.Y., Pessara U., Hooft van Huisjduijnen R., Benoist C., Mathis D. Dominant negative analogs of NF-YA. J Biol Chem. 1994;269:20340–20346. [PubMed] [Google Scholar]
- 37.Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D., Haussler D., Miller W. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z., Zang C., Rosenfeld J.A., Schones D.E., Barski A., Cuddapah S., Cui K., Roh T.Y., Peng W., Zhang M.Q., Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Euskirchen G.M., Rozowsky J.S., Wei C.L., Lee W.H., Zhang Z.D., Hartman S., Emanuelsson O., Stolc V., Weissman S., Gerstein M.B., Ruan Y., Snyder M. Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 2007;17:898–909. doi: 10.1101/gr.5583007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maerkl S.J., Quake S.R. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 2007;315:233–237. doi: 10.1126/science.1131007. [DOI] [PubMed] [Google Scholar]
- 41.Hooker C.W., Hurlin P.J. Of Myc and Mnt. J Cell Sci. 2006;119:208–216. doi: 10.1242/jcs.02815. [DOI] [PubMed] [Google Scholar]
- 42.Guil S., Caceres J.F. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol. 2007;14:591–596. doi: 10.1038/nsmb1250. [DOI] [PubMed] [Google Scholar]
- 43.Cawley S., Bekiranov S., Ng H.H., Kapranov P., Sekinger E.A., Kampa D., Piccolboni A., Sementchenko V., Cheng J., Williams A.J., Wheeler R., Wong B., Drenkow J., Yamanaka M., Patel S., Brubaker S., Tammana H., Helt G., Struhl K., Gingeras T.R. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 44.Wierstra I. Sp1: emerging roles–beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun. 2008;372:1–13. doi: 10.1016/j.bbrc.2008.03.074. [DOI] [PubMed] [Google Scholar]
- 45.Ting A.H., McGarvey K.M., Baylin S.B. The cancer epigenome–components and functional correlates. Genes Dev. 2006;20:3215–3231. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- 46.Lennartsson A., Ekwall K. Histone modification patterns and epigenetic codes. Biochim Biophys Acta. 2009;1790:863–868. doi: 10.1016/j.bbagen.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 47.Jones P.A., Baylin S.B. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agalioti T., Chen G., Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–392. doi: 10.1016/s0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- 49.Klein U., Tu Y., Stolovitzky G.A., Keller J.L., Haddad J., Jr, Miljkovic V., Cattoretti G., Califano A., Dalla-Favera R. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci USA. 2003;100:2639–2644. doi: 10.1073/pnas.0437996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shaffer A.L., Rosenwald A., Hurt E.M., Giltnane J.M., Lam L.T., Pickeral O.K., Staudt L.M. Signatures of the immune response. Immunity. 2001;15:375–385. doi: 10.1016/s1074-7613(01)00194-7. [DOI] [PubMed] [Google Scholar]
- 51.Ci W., Polo J.M., Cerchietti L., Shaknovich R., Wang L., Yang S.N., Ye K., Farinha P., Horsman D.E., Gascoyne R.D., Elemento O., Melnick A. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood. 2009;113:5536–5548. doi: 10.1182/blood-2008-12-193037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fu Y., Sinha M., Peterson C.L., Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 2008;4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phillips J.E., Corces V.G. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guccione E., Martinato F., Finocchiaro G., Luzi L., Tizzoni L., Dall' Olio V., Zardo G., Nervi C., Bernard L., Amati B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764–770. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 55.Teye K., Okamoto K., Tanaka Y., Umata T., Ohnuma M., Moroi M., Kimura H., Tsuneoka M. Expression of the TAF4b gene is induced by MYC through a non-canonical, but not canonical, E-box which contributes to its specific response to MYC. Int J Oncol. 2008;33:1271–1280. [PubMed] [Google Scholar]
- 56.Meroni G., Reymond A., Alcalay M., Borsani G., Tanigami A., Tonlorenzi R., Lo Nigro C., Messali S., Zollo M., Ledbetter D.H., Brent R., Ballabio A., Carrozzo R. Rox, a novel bHLHZip protein expressed in quiescent cells that heterodimerizes with Max, binds a non-canonical E box and acts as a transcriptional repressor. EMBO J. 1997;16:2892–2906. doi: 10.1093/emboj/16.10.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantification of copy number of C13orf25 in various B-cell lines. A: Quantification by real-time PCR and normalization to the RPL13 locus. Quantitative real-time PCR was performed as described in the methods for ChIP. The values for cell lines DHL6, OCI-Ly1, OCI-Ly3, and JVM2 were similar, and their average was set at 1. As discussed in the text, JeKo-1 shows marked amplification, and Granta and Z138C, which are also mantle cell lymphoma cell lines from American Type Culture Collection, likely show a small copy number gain. DHL16 shows either a normal 2N copy number or a gain of 1. B: Quantification by comparing hybridization of input DNA used for ChIP-chip. The fraction of hybridization signal of the input DNA (before IP) from nucleosomal DNA to the C13orf25 locus (see Figure 5) was compared with the hybridization signal to the entire custom array interrogating ≥100 genes. The ratio of this value to that obtained for normal naive B cells and for normal centroblasts was calculated for IP experiments in each of four histone modifications. The mean values are shown. Error bars are standard deviations.