

Abstract
Barrett’s esophagus (BE) confers a significant increased risk for development of esophageal adenocarcinoma (EAC), with the pathogenesis appearing to progress through a “metaplasia-dysplasia-carcinoma” (MDC) sequence. Many of the genetic insults driving this MDC sequence have recently been characterized, providing targets for candidate biomarkers with potential clinical utility to stratify risk in individual patients. Many clinical risk factors have been investigated, and associations with a variety of genetic, specific gastrointestinal and other modifiable factors have been proposed in the literature. This review summarizes the current understanding of the mechanisms involved in neoplastic progression of BE to EAC and critically appraises the relative roles and contributions of these putative risk factors from the published evidence currently available.
Keywords: Barrett’s esophagus, Esophageal adenocarcinoma, Metaplasia-dysplasia-carcinoma, Neoplastic progression, Risk factors
INTRODUCTION
Barrett’s esophagus (BE) describes a condition where native esophageal stratified squamous epithelium is replaced by metaplastic columnar epithelium, with cephalad displacement of the squamocolumnar junction. BE represents the only identified precursor lesion and most important risk factor for esophageal adenocarcinoma (EAC)[1]. Patients with BE have an estimated 30- to 125-fold greater risk of developing EAC than the general population[2]. A systematic review of 27 studies suggested annual progression rates of 0.5%[3], whereas a review of 8 UK studies by Jankowski et al[4] showed cancer risk of 1.0% per year.
BE PATHOGENESIS AND MECHANISMS OF NEOPLASTIC PROGRESSION
BE is an acquired condition where healing from esophageal mucosal injury [typically triggered by gastro-esophageal reflux disease (GERD)] is metaplastic, with replacement of damaged squamous cells by columnar epithelium. Ordinarily, esophageal healing involves regeneration of squamous cells; it remains unclear why the response is metaplastic in some individuals, since only a minority of patients with GERD develop BE. Progression of BE to EAC occurs by a metaplasia-dysplasia-carcinoma (MDC) sequence. Metaplastic columnar epithelial cells are predisposed to genetic damage with potential for developing dysplasia[5]. Dysplasia represents a histological spectrum from low- to high-grade, defined by degree of cytological and architectural disruption present, with genetic instability resulting in progressive acquisition of genetic abnormalities towards a frankly neoplastic phenotype. These can be considered within the framework of Hanahan and Weinberg’s[6] model of “cancer hallmarks” necessary for carcinogenesis, whereby cancer cells must acquire growth self-sufficiency, insensitivity to anti-growth signals, avoidance of apoptosis, limitless replicative potential, sustained angiogenesis, and invasive and metastatic potential[7].
Many genetic insults conferring these advantages in the BE MDC sequence have been characterized. Initiating events probably involve genes regulating cell cycle progression, notably p16. Mutations, loss of heterozygosity (LOH) or promoter hypermethylation (i.e. silencing) of p16 have been identified in 80% of BE, whilst p16 hypermethylation correlated with the degree of dysplasia in some studies[8]. Additional changes identified include upregulation of cyclins D1 and E, transforming growth factor-α and epidermal growth factor (EGF), each contributing towards growth autonomy[9,10]. These mutations should trigger apoptosis via p53-dependent pathways. However, subsequent accrual of p53 lesions confers resistance to apoptosis, and has been identified in 52%-93% of EACs (compared with 1%-5% non-malignant BE cell lines)[11]. Inactivation of p53 increases clonal genomic instability, predisposing to widespread DNA changes and evolution of ploidy lesions, late events in cancer progression. Many other genetic and molecular alterations have been described[8,9,12-64] (Table 1).
Table 1.
Published evidence from selected studies investigating genetic and epigenetic changes implicated in the metaplasia-dysplasia-carcinoma sequence of Barrett’s esophagus
| Factor | Summary of major findings/conclusions | Ref. |
| Growth self-sufficiency | ||
| Cyclin D1 | ↑ nuclear cyclin D1 immunostaining in 46% BE specimens: -?cyclin D1 overexpression early event in MDC sequence | Arber et al[9] |
| ↑ nuclear cyclin D1 immunostaining in 64% EAC specimens | Arber et al[13] | |
| Cyclin D1 expression correlates with degree of dysplasia in BE | Coppola et al[14] | |
| Cyclin D1 expression 43% BE mucosa (vs 0% normal mucosa) | Umansky et al[15] | |
| Polyphenon E inhibits growth of BE and EAC cells via downregulation of cyclin D1 expression | Song et al[16] | |
| Cyclin E | ↑ cyclin E expression in neoplastic cells in BE | Coppola et al[14] |
| Cyclin E expression 37% BE mucosa (vs 0% normal mucosa) | Umansky et al[15] | |
| p27Kip-1 | 83% EAC specimens displayed low p27 protein levels (despite high p27 mRNA): -p27 inactivated in most BE-associated EAC (post-transcriptional modification)→loss of cell cycle inhibition | Singh et al[17] |
| Experimentally-induced BE and EAC development in mouse model significantly enhanced by p27 gene knockout | Ellis et al[18] | |
| EGF (and EGF-R) | ↑ EGF in cytoplasm of BE epithelial cells (vs gastric mucosa) | Jankowski et al[19] |
| EGF-R expression area in inflamed mucosa (43.1%) significantly > normal mucosa (29.5%); all BE showed positive EGF-R staining | Jankowski et al[20] | |
| EGF/EGF-R expression significantly ↑ in BE and EAC mucosa (vs normal mucosa) by flow cytometry (P < 0.01) | Jankowski et al[21] | |
| EGF-R expression positive in 64% of BE-associated EAC; ↑ staining associated with poorer survival (P = 0.004) | Yacoub et al[22] | |
| EGF A61G G/G genotype associated with >double EAC risk in BE pts (vs A/A or A/G) (OR 2.2) | Lanuti et al[23] | |
| TGF-α | ↑ TGF-α expression in cells from BE and EAC mucosa (vs normal gastric mucosa) by flow cytometry (P < 0.01) | Jankowski et al[21] |
| TGF-α expression positive in 100% of BE-associated EAC | Yacoub et al[22] | |
| HGF (and HGF-R) | HGF expression significantly ↑ in BE specimens (vs normal esophageal mucosa) | Konturek et al[24] |
| Intense HGF-R immunostaining in 100% EAC and dysplastic BE specimens (vs minimal staining in non-dysplastic BE or normal mucosa); HGF-R mRNA and protein levels ↑ in EAC cell lines | Herrera et al[25] | |
| Erb family tyrosine kinases | Membranous c-erbB2 overexpressed in 26% EAC (vs 0% BE with dysplasia): -?later event in MDC sequence | Hardwick et al[26] |
| c-erbB-2 gene amplification in 14% EAC vs 11% HG-dysplasia vs 0% metaplasia/LG-dysplasia specimens | Geddert et al[27] | |
| FGF | Immunostaining intensity for FGF sequentially ↑ from metaplasia/LG-dysplasia (negligible)→HG-dysplasia (weak/moderate)→EAC (moderate/strong) | Soslow et al[28] |
| FGF-1 mRNA and protein expression sequentially ↑ in HG-dysplasia/EAC (vs metaplasia/LG-dysplasia/controls) | Soslow et al[29] | |
| Src family tyrosine kinases | Src-specific activity 3-4-fold ↑ in BE and 6-fold ↑ in EAC (vs controls): -?Src activation early event in MDC sequence | Kumble et al[30] |
| Strong Src expression in 85% EAC vs 93% BE HG-dysplasia vs 72% BE LG-dysplasia vs 27% BE specimens | Iravani et al[31] | |
| Insensitivity to anti-growth signals | ||
| p16 | 9p21 (p16) LOH observed in 89% EAC specimens (vs 0% non-dysplastic BE); homozygous p16 deletion in only 25% | González et al[32] |
| p16 promoter hypermethylation (inactivation) in 75% BE with HG-dysplasia vs 56% LG-dysplasia (vs 3% non-dysplastic BE) | Klump et al[8] | |
| APC | 5q (APC) LOH seen in 80% EAC specimens (and surrounding mucosa) | Barrett et al[33] |
| APC gene LOH observed in 60% EAC specimens (vs 0% non-dysplastic BE) | González et al[32] | |
| APC promoter hypermethylation in 92% EAC vs 40% BE (vs 0% normal esophageal tissues) | Kawakami et al[34] | |
| Avoidance of apoptosis | ||
| p53 | Positive p53 immunostaining in 87% EAC vs 55% BE with HG-dysplasia vs 9% LG-dysplasia vs 0% non-dysplastic BE | Younes et al[35] |
| 17p (p53) LOH found in 91% BE pts who developed aneuploid cell populations: -17p allelic losses precede aneuploidy | Blount et al[36] | |
| p53 overexpression in 64% EAC vs 31% dysplastic BE vs 0% non-dysplastic BE; trend of ↑ p53 expression with ↑tumour grade: -?p53 mutation early event in malignant progression | Symmans et al[37] | |
| p53 immunoreactivity only in EAC/BE with HG-dysplasia (not in BE with LG-/no dysplasia); mutated p53 in 69%: -?late event in MDC sequence (during transition to HG-dysplasia) | Rice et al[38] | |
| p53 protein expression in 85% EAC specimens vs 60% BE with HG-dysplasia vs 7% LG-dysplasia (P < 0.001) | Rioux-Leclercq et al[39] | |
| p53 mutations identified in 75% EAC specimens; p53 overexpression in 58% EAC vs 60% BE with HG-dysplasia vs 12% LG-dysplasia vs 0% non-dysplastic BE | Chung et al[40] | |
| Fas (CD95) | ↓ surface expression of Fas observed in EAC specimens; impaired translocation of Fas to membrane wild-type Fas protein retained in cytoplasm in EAC cell line: -?potential mechanism by which EAC cells evade Fas-mediated apoptosis | Hughes et al[41] |
| ↓ surface expression of Fas and resistance to Fas-mediated apoptosis observed in EAC cell lines | Mahidhara et al[42] | |
| Bcl-xl/Bax/Bcl-2 | Bcl-xl positive in all dysplasia and EAC cells, but negative in 47% non-dysplastic BE: -?switch to anti-apoptotic phenotype in transformation from metaplasia to EAC | van der Woude et al[43] |
| Bcl-2 expression in 84% LG-dysplasia vs 0% HG-dysplasia or EAC | Rioux-Leclercq et al[39] | |
| Cytoplasmic Bcl-xl immunostaining in 59% EAC vs 71% BE/HG-dysplasia vs 60% LG-dysplasia vs 27% non-dysplastic | Iravani et al[31] | |
| COX-2 | ↑ COX-2 mRNA levels in 80% BE and 100% EAC specimens (vs normal gastric controls) (P < 0.001); COX-2 immunostaining strongly positive in 100% BE samples (> gastric controls) | Wilson et al[44] |
| COX-2 immunopositivity in 91% non-dysplastic BE vs 94% dysplastic vs 97% EAC | Lagorce et al[45] | |
| Natural/synthetic COX-2 inhibitors suppressed proliferation, induced apoptosis and blocked cell cycle in EAC cell lines | Cheong et al[46] | |
| Cox-2 mRNA strongly upregulated in experimentally-induced BE epithelium in rat model (vs absent in control animals); COX-2 overexpression observed in human BE patients with dysplasia | Majka et al[47] | |
| Limitless replicative potential | ||
| Telomerase | Telomerase RNA positive in 100% EAC/BE with HG-dysplasia vs 90% LG-dysplasia vs 70% non-dysplastic BE: marked ↑ telomerase RNA accompanies transition along MDC sequence | Morales et al[48] |
| human telomerase reverse transcriptase (catalytic subunit of telomerase) expression ↑ at all stages of BE vs normal controls, and in EAC (P = 0.003) and dysplastic BE (P = 0.056) vs non-dysplastic BE | Lord et al[49] | |
| Telomerase activity (by telomeric repeat amplification protocol assay) ↑ in EAC samples vs adjacent mucosa (P = 0.0002) and in EAC vs BE (P = 0.001); no difference BE vs adjacent mucosa | Barclay et al[50] | |
| Telomerase inhibition (by small interference RNAs) induced senescence in 40% and apoptosis in 86% in BE cell lines | Shammas et al[51] | |
| Sustained angiogenesis | ||
| VEGF (and VEGF-R) | VEGF expression correlated with higher vascularisation in BE and EAC specimens | Couvelard et al[52] |
| VEGF-A expressed in BE epithelium; VEGFR-2 strongly expressed in immature endothelial cells feeding BE epithelium; ↑ VEGF-C expression in BE (vs absent in normal epithelium); ↑ VEGFR-3 in EAC: ?aberrant neovasculature early in MDC sequence | Auvinen et al[53] | |
| VEGF expressed in 64% EAC specimens; significantly correlated with angiolymphatic invasion/survival | Saad et al[54] | |
| VEGF expression significantly ↑ in EAC (> dysplastic BE > BE > normal epithelium) | Griffiths et al[55] | |
| Invasive/metastatic potential | ||
| CAMs | ↓ expression in EAC specimens of E-cadherin (in 74%), α-catenin (60%) and β-catenin (72%) | Krishnadath et al[56] |
| Abnormal expression of β-catenin (P = 0.022), α-catenin (P < 0.01) and E-cadherin (P = 0.049) significantly associated with higher degrees of BE-related dysplasia | Washington et al[57] | |
| ↓ expression of E-cadherin with progression along MDC sequence (P < 0.01); in contrast P-cadherin absent from BE (± dysplasia) but expressed in 67% EAC specimens | Bailey et al[58] | |
| Slug (E-cadherin repressor) immunostaining and mRNA levels overexpressed in EAC vs BE metaplasia specimens: -?Slug upregulation represents mechanism of E-cadherin silencing | Jethwa et al[59] | |
| Cathepsins | Detected amplicon at chromosome 8p22-23 resulting in cathepsin B overexpression (observed in 73% EAC samples) | Hughes et al[60] |
| ↑ cathepsin C expression in EAC (vs BE vs normal) in rat model | Cheng et al[61] | |
| CD44 | Stepwise ↑ cathepsin D mRNA levels in GERD→BE→EAC tissue | Breton et al[62] |
| CD44-H and -V6 variant frequently expressed in BE; differing expression patterns along spectrum normal→dysplastic BE→EAC: -?CD44H and V6 involved in carcinogenesis of BE mucosa | Lagorce-Pages et al[63] | |
| ↓ CD44 expression in EAC/HG-dysplasia (vs BE/LG-dysplasia) | Darlavoix et al[64] | |
BE: Barrett’s esophagus; MDC: Metaplasia-dysplasia-carcinoma; EAC: Esophageal adenocarcinoma; EGF: Epidermal growth factor; EGF-R: EGF receptor; pts: Patients; OR: Odds ratio; TGF: Transforming growth factor; HGF: Hepatocyte growth factor; HGF-R: HGF receptor; mRNA: Messenger RNA; FGF: Fibroblast growth factor; HG: High grade; LG: Low grade; LOH: Loss of heterozygosity; APC: Adenomatous polyposis coli; COX-2: Cyclooxygenase-2; VEGF: Vascular endothelial growth factor; VEGF-R: VEGF receptor; CAM: Cell adhesion molecule; GERD: Gastro-esophageal reflux disease.
The concept of a linear, stepwise evolution of tumor suppressor gene mutations in which clonal expansion of a solitary mutated clone expands to fill the entire Barrett’s
segment has been termed the “selective sweep to fixation” model. However, an alternative model has been proposed by Leedham et al[65], who performed genetic analysis of individual crypts rather than a flow purified whole biopsy specimen. This technique permitted identification of certain mutations masked by whole biopsy segment analysis (attributed to dilution effect of the normal stroma on whole biopsy analysis), whilst also revealing a greater degree of genotypical and phenotypical heterogeneity within the same biopsy sample than previously appreciated. The demonstrated lack of a single founder mutation present in every crypt suggested that the clonal expansion arose from multiple independent clones rather than a single common founder mutation[65,66] (Figure 1).
Figure 1.
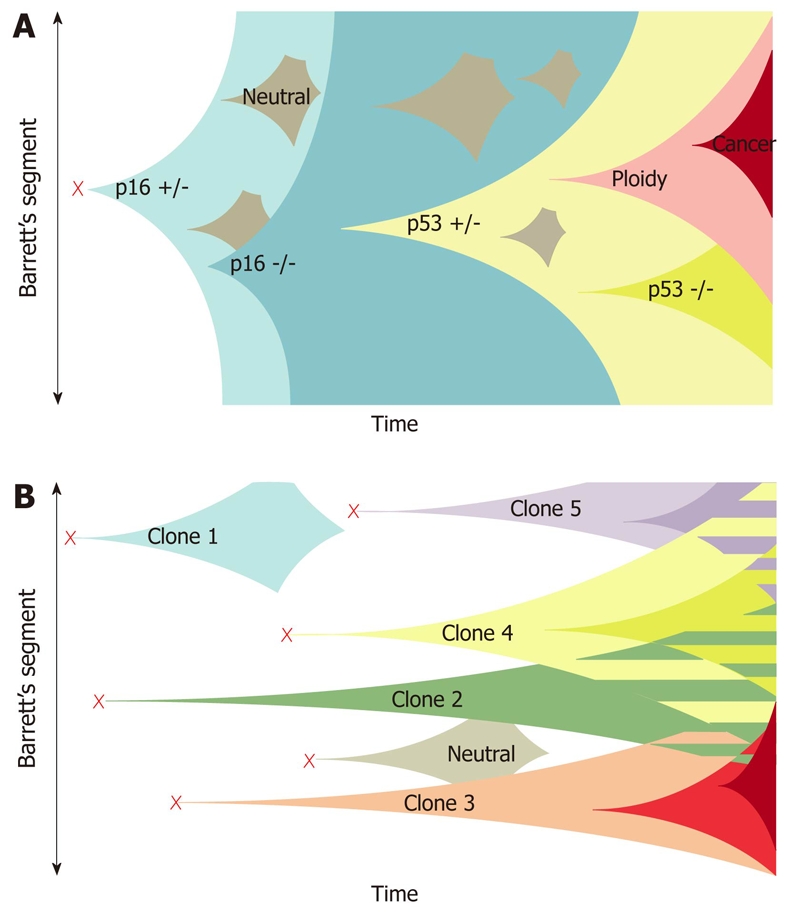
Clonal evolution models in Barrett’s esophagus. A: The current model of clonal evolution adapted from Maley et al[66]. Founder mutation (red cross) occurs in a single progenitor and provides a growth advantage that predisposes to a selective sweep. Successive selective sweeps result in progression along the metaplasia dysplasia pathway. Clone bifurcation is responsible for the genetic heterogeneity in this model; B: The newly proposed model of evolution based on the mutation of multiple progenitor cells situated in esophageal gland squamous ducts located throughout the length of the esophagus (red crosses). Multiple independent clones then arise and evolve separately. The presence of multiple different clones gives rise to a mosaic interdigitating clonal pattern of the Barrett’s segment represented as the striped areas[65].
This enhanced understanding prompted research into > 200 candidate novel biomarkers of disease progression in BE/EAC. Several, including 17p LOH, cyclin D1, tetraploidy and aneuploidy, have undergone phase 3/4 validation and in future might have clinical/prognostic utility as intermediate markers of progression[67]. However, Leedham’s recent findings call into question the reliability of “surveillance” biomarker identification via genetic analysis of whole biopsy specimens, since minority clones within the sample (harboring neoplastic potential) might not be detected[65].
Currently, dysplasia remains the only validated marker for identifying BE patients at risk, and forms the basis of EAC surveillance. However, this is imperfect. The tempo of progression towards EAC is highly variable and it remains unclear whether relentless progression through the MDC sequence is inevitable; some evidence suggests that high-grade dysplasia may remain stable for years or even regress[68]. Patients with BE may develop EAC during surveillance without detection of earlier MDC stages. This might relate to pace of progression, sampling error or lesions skipping directly from non-dysplastic disease to cancer. Other limitations of dysplasia as a prognostic marker include inter-observer variability in histological interpretation, and that inflammation may mimic dysplastic changes[69].
RISK FACTORS FOR NEOPLASTIC PROGRESSION
Until molecular biomarkers enter clinical practice it remains important to identify other clinical risk factors for malignant progression to effectively allocate resources and individualize surveillance programs, targeting those at highest risk. Identifying modifiable risk factors will also inform disease prevention strategies. Epidemiological studies of EAC have described a “birth cohort effect”, with higher incidence rates observed in recent cohorts suggesting exposure to an exogenous risk factor in early life contributing increased risk in all ages of the cohort[70] (Figure 2). Multiple risk factors for neoplastic progression of BE have been investigated (Table 2).
Figure 2.
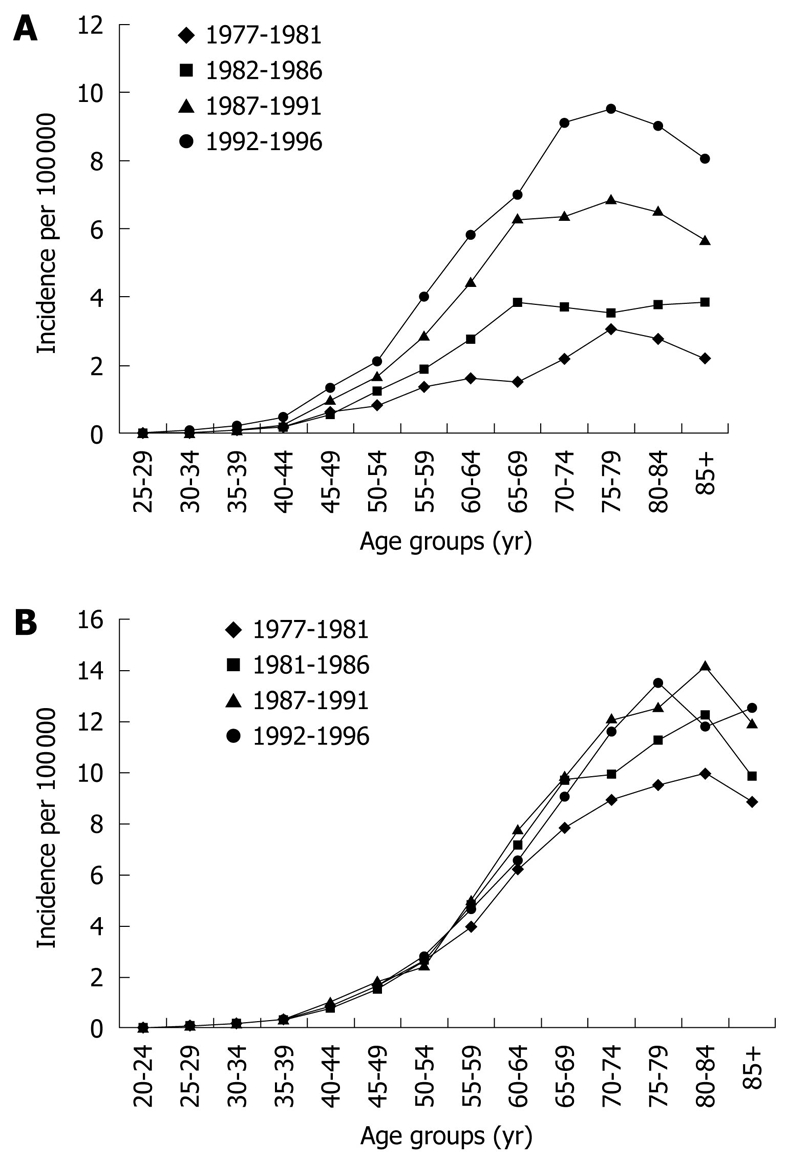
Age distribution of cases diagnosed with oesophageal adenocarcinoma (A) and gastric cardia adenocarcinoma (B) in the USA between 1977-1996, displaying the "birth cohort effect". Each individual curve represents the age-specific incidence rates in a five year period (from El-Serag et al[70]).
Table 2.
Clinical and demographic risk factors for neoplastic progression of Barrett’s esophagus
| Innate factors | Gastrointestinal factors | Other modifiable factors |
| Age | Bile and acid reflux | Obesity |
| Gender | Anti-reflux surgery | Diet |
| Ethnicity | Proton pump inhibition | Alcohol |
| Pharmacological lower esophageal sphincter relaxation | Smoking | |
| Salivary nitrates | Socioeconomic status | |
| Barrett’s segment length | Pharmacological COX-2 inhibition |
COX-2: Cyclooxygenase-2.
INNATE HOST FACTORS
Age is a well-recognized risk for both BE and EAC. Corley et al[71] reported an incidence of BE of 2/100 000 for 21-30-year-old and 31/100 000 for 61-70-year-old, whilst El-Serag et al[70] calculated the risk of EAC to increase by 6.6% for each 5-year age increase. Evidence specifically linking age to risk of neoplastic progression within BE is lacking, but it seems intuitive to propose advancing age as an independent risk factor.
BE displays a male preponderance of approximately 2:1, rising to 4:1 for BE-associated EAC, suggesting an independent influence of gender on risk of neoplastic progression[71,72]. Why male gender should confer additional risk is unknown; some have speculated that male propensity toward visceral pattern of obesity might be relevant[73].
A higher prevalence of BE in Caucasians has long been recognized[74]; again, this association strengthens with development of BE-associated EAC[75]. Analysis of the US Surveillance, Epidemiology and End Results registry found that the annual incidence of EAC for Caucasian males was double that for Hispanic males and four times higher than Black, Asian, Pacific Island and Native American males[76]. Although selection bias and differing endoscopy uptakes between ethnic groups might partially explain this, other factors seem to be involved. Whilst environmental influences are probably important, hitherto-unidentified genetic variations influencing protection against reflux-induced mucosal damage seem likely. A US study found similar GERD prevalence in Caucasian and Black Americans from the same geographical population, yet the latter displayed significantly less esophagitis and almost no BE[77].
GASTROINTESTINAL FACTORS
Bile/acid reflux
The relationship between GERD and BE is well established, and whilst reflux of gastric acid is known to induce chronic mucosal esophageal injury the contribution of bile salts and acids (from duodenal refluxate) is increasingly recognized. Vaezi and Richter demonstrated patients with complicated BE (dysplasia/stricture/ulceration) reflux significantly greater amounts of both gastric and bile acids than those with uncomplicated BE, and postulated that complications might result from synergism between the two[78]. Bile salts induce esophageal injury over a wide pH range, and patients with BE display significantly more bile salts in aspiration studies than patients with mild reflux only[79]. Menges et al[80] observed a strong correlation between duration of esophageal exposure to acid and bile with severity of pathological change in BE. Furthermore, proton pump inhibitor (PPI) therapy predisposes to upper gastrointestinal bacterial colonization and consequent bile salt-deconjugation, which, in this high pH environment, has been linked to chronic inflammation[81].
Refluxate-mediated inflammation might promote carcinogenesis via both the arachidonic acid (AA) pathway and induction of oxidative stress. Low pH and bile salts promote expression of cyclooxygenase-2 (COX-2), catalyzing conversion of AA into various prostaglandins, including PGE2. PGE2 increases proliferation of BE epithelial cells and inhibits tumor surveillance through suppressing natural killer cell function. Consequently, abnormal cells displaying genomic instability may accumulate. COX-2 expression has been shown to increase with neoplastic progression of BE, supporting a role for the AA pathway in EAC carcinogenesis[44]. Chronic mucosal injury also induces production of reactive oxygen species (ROS), depletes antioxidants and increases expression of oxidative stress-related genes. High levels of oxygen radicals and lipid peroxidation products have been demonstrated in BE epithelial cells, with reduced levels of vitamin C and glutathione, indicating compromised oxidant defences[82]. ROS have well-established mutagenic capacity, whilst subsequent apoptosis of mutated cells is additionally suppressed by capacity of bile salts to induce proteasomal degradation of p53[83].
The Factors Influencing the Barrett’s Adenocarcinoma Relationship (FINBAR) study suggested GERD symptom chronicity and frequency appeared better predictors for neoplastic progression than severity[84]. However, a significant proportion of EAC patients (40%-50%) do not recall ever having prior reflux[85]. Furthermore, reflux of gastroduodenal contents correlates poorly with heartburn symptoms, BE is frequently asymptomatic and development of less sensitive Barrett’s epithelium may ameliorate symptoms. Thus, symptom-based risk scores for assessing progression risk have so far not proved useful in clinical practice.
PPIs
PPIs increase pH of gastric refluxate, attenuating acid-induced damage. Ouatu-Lascar et al[86] showed “normalization” of intraesophageal pH with acid suppression favors differentiation and reduces cellular proliferation in BE biopsy specimens. However, PPIs have not prevented recent increases in EAC, and the observation of EAC with PPI administration in animal models raises concern they might actually favor progression of BE[87]. This might be mediated via interaction of gastrin with its cholecystokinin receptor, CCK2R. PPIs elevate serum gastrin levels, which on binding to CCK2R, stimulate expression of EGF and trefoil peptide, inducing COX-2 expression. Gastrin exposure increases proliferation in esophageal cell culture, and BE mucosa expresses more CCK2R than normal squamous mucosa. CCK2R stimulation also inactivates pro-apoptotic factors[88].
Despite this, the clinical relevance in humans remains unproven. Three large studies have examined PPI usage and EAC risk in BE patients, each reporting a strong inverse correlation. Two observed a decreased risk with longer duration of PPI, and one showed an increased risk with delayed PPI use[89]. Obszynska et al[90] investigated effects of hypergastrinemia induced by different PPI doses in cell models and BE patients. Despite increased cell proliferation in vitro, COX2 induction and enhanced epithelial restitution, they found no evidence of longer-term harm using surrogate biomarkers of proliferation or apoptosis in vivo. The Aspirin Esomeprazole Chemoprevention Trial (AspECT) is currently investigating effects of different PPI doses in combination with aspirin on EAC risk.
Anti-reflux surgery
Theoretically, anti-reflux surgery should prevent reflux of duodenal contents, against which PPIs have no effect, potentially mitigating against progression of BE. Unfortunately this is not supported by the available evidence. Two large cohort studies failed to show cancer protection in GERD patients[91,92], whilst a meta-analysis by Corey et al[93] concluded no reduction in progression risk for BE. However, different surgical procedures were employed and effectiveness of reflux control was not always assessed.
Lower esophageal sphincter-relaxing drugs
Pharmacological lower esophageal sphincter (LES) relaxation might promote development/progression of BE by increasing reflux, suggested by the observation that drugs with these effects (e.g. tricyclic antidepressants) have increased in use alongside the rise in EAC. A Swedish population-based study by Lagergren et al[94] reported a positive association between EAC and long-term use of LES-relaxing drugs, with the strongest association for anticholinergics; this association disappeared after adjustment for reflux symptoms.
Helicobacter pylori infection
An increase in BE-associated EAC alongside falling rates of Helicobacter pylori (H. pylori) infection has led some to propose a protective effect of H. pylori, mediated by its influence in reducing gastric acidity. The virulent cagA strain is particularly associated with high-grade gastric inflammation and atrophy[95]. A meta-analysis by Rokkas et al[96] reported statistically significant inverse relationships between H. pylori infection and both EAC and BE [odds ratio (OR), 0.52% and 0.64%, respectively]. Furthermore, a large prospective study of BE patients and GERD controls found less H. pylori infection with increasing “severity” of disease: 44% in GERD; 35% in uncomplicated BE; 14%-15% in BE with high-grade dysplasia/EAC[97].
However, another study, controlling for demographic and lifestyle factors, failed to demonstrate reduced EAC with cagA+ infection[98]. A confounding factor might be the degree of bile acid reflux, since excessive bile reflux may prevent H. pylori colonization and contribute to chronic mucosal injury[88]. The protective role for H. pylori is debatable and since H. pylori is a World Health Orgaisation class 1 mutagen for gastric adenocarcinoma it is difficult to argue against its eradication whenever it is detected.
Salivary nitrates
Dietary nitrate, concentrated in saliva and reduced to nitrites by oral flora, produces intraesophageal nitric oxide (NO) during reflux. Achlorhydria induced by PPI or atrophic gastritis may cause overgrowth of nitrate-reducing bacteria in the upper gut, providing another source of nitrite[88]. Clemons demonstrated the capacity of NO to induce double-strand DNA breaks in esophageal BE cells in vitro, which could promote neoplastic progression[99]. Increasing agricultural nitrate use in the latter 20th century caused significant increases in nitrate content of leafy vegetables and drinking water[100] and could have partially contributed to the increase in EAC incidence.
Barrett’s segment length
Although EAC can develop in BE segments of any length, several observational studies support the intuitive notion that longer segments confer greater risk[101]. However, a meta-analysis by Thomas et al[102] showed only a non-significant trend towards reduced progression with shorter BE segments, and evidence remains insufficient to advocate surveillance strategies based on segment length alone.
OTHER MODIFIABLE RISK FACTORS
Obesity
Increasing obesity has also paralleled increased rates of BE and EAC. Strong links between obesity and both GERD and erosive esophagitis have been established[103]. It is logical that this might predispose to BE, but a meta-analysis specifically comparing body mass index (BMI) in BE cases with population controls showed only a modest risk increase[104]. However, elevated BMI is a strong risk factor for EAC (OR, 1.8 and 2.4 for BMI > 25 and BMI > 30, respectively)[105]. Increased risk may relate more to distribution of body fat than BMI alone, with visceral (abdominal) obesity conferring greater risk[106]. Other studies noted an association between obesity in early life and EAC risk, suggesting adiposity may act early in the disease process[84,107].
Although a small prospective study by Oberg and colleagues failed to identify any association between BMI and progression from BE to low- or high-grade dysplasia[108], it had limited power, and a larger study from the Seattle Barrett’s Esophagus Program revealed strong correlations between waist-to-hip ratio and intermediate biomarkers of progression[109]; again, associations were less apparent for elevated BMI per se.
Obesity causes GERD through several mechanical and physiological mechanisms. However, part of the association between obesity and EAC is independent of GERD, suggesting a role for reflux-independent mechanisms, probably linked to important endocrine actions of adipose tissue. Many recent studies have linked several adipokines (metabolically active factors) to plausible actions in the MDC process[110-117] (Table 3).
Table 3.
Selected published evidence linking adipokines (and ghrelin) with Barrett’s esophagus and progression to esophageal adenocarcinoma
| Adipokine |
Evidence in BE and EAC |
|
| Relevant study findings | Ref. | |
| Adiponectin (↓ in obesity) | ↓ adiponectin receptors in Barrett’s mucosa compared with normal mucosa from controls | Konturek et al[110] |
| ↑ Bax (pro-apoptotic), ↓ Bcl-2 (anti-apoptotic) and ↑ apoptosis of EAC cell lines on incubation with adiponectin | Konturek et al[110] | |
| Plasma adiponectin levels inversely associated with BE risk in 50 matched cases (OR 4.7 for each 10 μg/mL ↓ in level) (independent of BMI) | Rubenstein et al[111] | |
| No difference in adiponectin levels between 51 BE patients and 67 controls | Kendall et al[112] | |
| Leptin (↑ in obesity) | Leptin receptors expressed in esophagus | Francois et al[113] |
| ↑ proliferation and ↓ apoptosis (via various signalling pathways) in EAC cell lines | Ogunwobi et al[114] | |
| Leptin levels strongly associated with ↑ risk of BE in males (no association in females) | Kendall et al[112] | |
| Gastric (fundic) leptin levels positively associated with BE (no association with serum leptin) | Francois et al[113] | |
| Ghrelin (↓ in obesity) | ↑ gastric emptying (so may ↓ gastric reflux) | Dornonville et al[115] |
| ↓ TNF-α-induced COX-2 and interleukin-1-β expression in BE cell line | Konturek et al[110] | |
| Ghrelin expression negligible in archived EAC cell specimens (vs rich expression in normal mucosa) | Mottershead et al[116] | |
| ↑ serum ghrelin associated with ↓ EAC risk (in overweight subjects) | de Martel et al[117] | |
BE: Barrett’s esophagus; EAC: Esophageal adenocarcinoma; OR: Odds ratio; BMI: Body mass index; COX-2: Cyclooxygenase-2; TNF: Tumor necrosis factor.
Kristal et al[118] investigated whether weight loss (alongside other dietary measures) impacted upon measured biomarkers of cellular proliferation in BE. Despite weight loss (mean 3.6 kg) at 18 mo no differences in biomarkers were observed. This study was relatively small, and the lack of response might relate to the relatively modest weight loss, and/or choice of proliferation markers employed.
Diet
Several studies have shown an association between a diet high in fruit and vegetables and reduced EAC. A large population-based Swedish study found individuals in the highest exposure quartile of fruit and vegetable intake to have approximately 50% less EAC compared to the lowest quartile[119]. However, Kristal et al’s study observed no effect on biomarkers of BE cell proliferation despite a net increase in fruit and vegetable consumption[118], whilst the FINBAR study observed a reduction in EAC with increased fruit, but not vegetable, consumption[84]. A protective effect for the natural anti-oxidants in fruit was proposed. A well-controlled, prospective study by Dong et al[120] showed patients who took multivitamin pills had significantly decreased risk of tetraploidy [hazard ratio (HR), 0.19] and frank EAC (HR, 0.38). Significant inverse associations with EAC were also observed for supplemental vitamins C (HR, 0.25) and E (HR, 0.25), both well-recognized antioxidants.
Chen et al[121] observed a significant inverse association between zinc intake and EAC risk compared with controls (OR, 0.5); inverse associations were also noted for vitamin A, β-cryptoxanthin, riboflavin, folate, fiber, protein and carbohydrate, whilst saturated fat intake was positively associated with EAC. Rudolph et al[122] investigated selenium levels in 396 BE patients: those with levels in the upper three quartiles were less likely to display high-grade dysplasia (OR, 0.5), aneuploidy (OR, 0.4) or 17p LOH (OR, 0.5) than the lowest quartile. No association was observed with p16 LOH (an early event in the MDC sequence), indicating selenium’s protective effects might occur late in progression to EAC.
Alcohol
Data supporting links between alcohol and BE/EAC are sparse. The UK BE registry found no association between alcohol consumption in patients with BE compared with reflux esophagitis[123]. Although at least eleven studies have investigated the relationship between alcohol and EAC only six have shown a positive association, and in most it was weak[124-134]. One study even seemed to suggest wine to be protective[133].
Smoking
Studies of smoking and BE/EAC are contradictory. An Australian population-based case-control study found smoking was associated with 2- to 3-fold increased risk of BE and BE with dysplasia[135]. However, there was no dose-response effect. Other small studies found no clear association[131]. Whilst smoking is a strong risk factor for esophageal squamous cell carcinoma, studies of EAC have been inconsistent, yielding conclusions ranging from complete absence of association[132-134] to a significant OR of 3.4 for current smokers[128]. Problems with study methodology occur and certainly smoking has rarely been a primary endpoint for studies of BE/EAC.
Socioeconomic status
There are no clear associations between socioeconomic status and neoplastic progression of BE. Some studies suggest increased EAC risk in higher socioeconomic groups, others the reverse[72].
COX-2 inhibition
Given the role of the AA pathway in neoplastic progression, pharmacological inhibition of COX-2 might modify the natural history of BE. Various studies have investigated whether aspirin and non-steroidal anti-inflammatory drugs (NSAIDs) might confer protection against EAC. A meta-analysis by Corley et al[136] including 1813 EAC patients suggested a protective association (OR, 0.67). Both intermittent and frequent use appeared advantageous, with evidence of a dose-effect, whilst aspirin conferred greater protection than NSAIDs.
However the Chemoprevention for Barrett’s Oesophagus Trial randomized 100 BE patients with dysplasia to either celecoxib 200 mg twice daily or placebo, with negative results[137]. A retrospective analysis of the UK BE registry with a total follow-up of 3683 patient-years also failed to demonstrate a protective effect of aspirin[138]. AspECT should provide further useful information.
CONCLUSION
The etiology of progression of BE is probably multi-factorial, with contributions from environmental risk factors interacting with genetically-determined characteristics. Obesity and ongoing bile and acid reflux are emerging as potentially modifiable risk factors, though designing practical interventions has so far proved difficult. Developments in understanding the MDC process in BE may provide future testable therapeutic targets.
ACKNOWLEDGMENTS
The authors would like to thank British Medical Journal Group publishing for permission to use the illustrations and graph. The clinical review was initially conducted by the first author (Wiseman EF) as part of a MSc degree at the University of Salford, Salford, UK. The manuscript has since been modified and updated by Ang YS with the latest developments in the field of BE.
Footnotes
Peer reviewer: Evangelos Tsiambas, MD, PhD, Lecturer in Molecular Cytopathology, Department of Pathology, Medical School, University of Athens, Ag Paraskevi Attiki 15341, Greece
S- Editor Sun H L- Editor Cant MR E- Editor Zheng XM
References
- 1.Reid BJ. Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterol Clin North Am. 1991;20:817–834. [PubMed] [Google Scholar]
- 2.Hage M, Siersema PD, van Dekken H, Steyerberg EW, Dees J, Kuipers EJ. Oesophageal cancer incidence and mortality in patients with long-segment Barrett’s oesophagus after a mean follow-up of 12.7 years. Scand J Gastroenterol. 2004;39:1175–1179. doi: 10.1080/00365520410003524. [DOI] [PubMed] [Google Scholar]
- 3.Shaheen NJ, Crosby MA, Bozymski EM, Sandler RS. Is there publication bias in the reporting of cancer risk in Barrett’s esophagus? Gastroenterology. 2000;119:333–338. doi: 10.1053/gast.2000.9302. [DOI] [PubMed] [Google Scholar]
- 4.Jankowski JA, Provenzale D, Moayyedi P. Esophageal adenocarcinoma arising from Barrett’s metaplasia has regional variations in the west. Gastroenterology. 2002;122:588–590. doi: 10.1053/gast.2002.31599. [DOI] [PubMed] [Google Scholar]
- 5.Boulton RA, Usselmann B, Mohammed I, Jankowski J. Barrett’s esophagus: environmental influences in the progression of dysplasia. World J Surg. 2003;27:1014–1017. doi: 10.1007/s00268-003-7054-0. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 7.Morales CP, Souza RF, Spechler SJ. Hallmarks of cancer progression in Barrett’s oesophagus. Lancet. 2002;360:1587–1589. doi: 10.1016/S0140-6736(02)11569-8. [DOI] [PubMed] [Google Scholar]
- 8.Klump B, Hsieh CJ, Holzmann K, Gregor M, Porschen R. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett’s esophagus. Gastroenterology. 1998;115:1381–1386. doi: 10.1016/s0016-5085(98)70016-2. [DOI] [PubMed] [Google Scholar]
- 9.Arber N, Lightdale C, Rotterdam H, Han KH, Sgambato A, Yap E, Ahsan H, Finegold J, Stevens PD, Green PH, et al. Increased expression of the cyclin D1 gene in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 1996;5:457–459. [PubMed] [Google Scholar]
- 10.Jankowski J, Hopwood D, Wormsley KG. Expression of epidermal growth factor, transforming growth factor alpha and their receptor in gastro-oesophageal diseases. Dig Dis. 1993;11:1–11. doi: 10.1159/000171396. [DOI] [PubMed] [Google Scholar]
- 11.Ireland AP, Clark GW, DeMeester TR. Barrett’s esophagus. The significance of p53 in clinical practice. Ann Surg. 1997;225:17–30. doi: 10.1097/00000658-199701000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wijnhoven BP, Tilanus HW, Dinjens WN. Molecular biology of Barrett’s adenocarcinoma. Ann Surg. 2001;233:322–337. doi: 10.1097/00000658-200103000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arber N, Gammon MD, Hibshoosh H, Britton JA, Zhang Y, Schonberg JB, Roterdam H, Fabian I, Holt PR, Weinstein IB. Overexpression of cyclin D1 occurs in both squamous carcinomas and adenocarcinomas of the esophagus and in adenocarcinomas of the stomach. Hum Pathol. 1999;30:1087–1092. doi: 10.1016/s0046-8177(99)90227-7. [DOI] [PubMed] [Google Scholar]
- 14.Coppola D, Falcone R, Livingston S, Karl R, Nicosia S, Cacho CM. Cyclin D1 expression correlates with degrees of dysplasia in Barrett's esophagus. Lab Invest. 1997;76:298–302. [Google Scholar]
- 15.Umansky M, Yasui W, Hallak A, Brill S, Shapira I, Halpern Z, Hibshoosh H, Rattan J, Meltzer S, Tahara E, et al. Proton pump inhibitors reduce cell cycle abnormalities in Barrett’s esophagus. Oncogene. 2001;20:7987–7991. doi: 10.1038/sj.onc.1204947. [DOI] [PubMed] [Google Scholar]
- 16.Song S, Krishnan K, Liu K, Bresalier RS. Polyphenon E inhibits the growth of human Barrett’s and aerodigestive adenocarcinoma cells by suppressing cyclin D1 expression. Clin Cancer Res. 2009;15:622–631. doi: 10.1158/1078-0432.CCR-08-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh SP, Lipman J, Goldman H, Ellis FH, Aizenman L, Cangi MG, Signoretti S, Chiaur DS, Pagano M, Loda M. Loss or altered subcellular localization of p27 in Barrett’s associated adenocarcinoma. Cancer Res. 1998;58:1730–1735. [PubMed] [Google Scholar]
- 18.Ellis FH, Xu X, Kulke MH, LoCicero J, Loda M. Malignant transformation of the esophageal mucosa is enhanced in p27 knockout mice. J Thorac Cardiovasc Surg. 2001;122:809–814. doi: 10.1067/mtc.2001.116471. [DOI] [PubMed] [Google Scholar]
- 19.Jankowski J, Coghill G, Tregaskis B, Hopwood D, Wormsley KG. Epidermal growth factor in the oesophagus. Gut. 1992;33:1448–1453. doi: 10.1136/gut.33.11.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jankowski J, Murphy S, Coghill G, Grant A, Wormsley KG, Sanders DS, Kerr M, Hopwood D. Epidermal growth factor receptors in the oesophagus. Gut. 1992;33:439–443. doi: 10.1136/gut.33.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jankowski J, Hopwood D, Wormsley KG. Flow-cytometric analysis of growth-regulatory peptides and their receptors in Barrett’s oesophagus and oesophageal adenocarcinoma. Scand J Gastroenterol. 1992;27:147–154. doi: 10.3109/00365529209165436. [DOI] [PubMed] [Google Scholar]
- 22.Yacoub L, Goldman H, Odze RD. Transforming growth factor-alpha, epidermal growth factor receptor, and MiB-1 expression in Barrett’s-associated neoplasia: correlation with prognosis. Mod Pathol. 1997;10:105–112. [PubMed] [Google Scholar]
- 23.Lanuti M, Liu G, Goodwin JM, Zhai R, Fuchs BC, Asomaning K, Su L, Nishioka NS, Tanabe KK, Christiani DC. A functional epidermal growth factor (EGF) polymorphism, EGF serum levels, and esophageal adenocarcinoma risk and outcome. Clin Cancer Res. 2008;14:3216–3222. doi: 10.1158/1078-0432.CCR-07-4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konturek PC, Nikiforuk A, Kania J, Raithel M, Hahn EG, Mühldorfer S. Activation of NFkappaB represents the central event in the neoplastic progression associated with Barrett’s esophagus: a possible link to the inflammation and overexpression of COX-2, PPARgamma and growth factors. Dig Dis Sci. 2004;49:1075–1083. doi: 10.1023/b:ddas.0000037790.11724.70. [DOI] [PubMed] [Google Scholar]
- 25.Herrera LJ, El-Hefnawy T, Queiroz de Oliveira PE, Raja S, Finkelstein S, Gooding W, Luketich JD, Godfrey TE, Hughes SJ. The HGF receptor c-Met is overexpressed in esophageal adenocarcinoma. Neoplasia. 2005;7:75–84. doi: 10.1593/neo.04367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardwick RH, Shepherd NA, Moorghen M, Newcomb PV, Alderson D. c-erbB-2 overexpression in the dysplasia/carcinoma sequence of Barrett’s oesophagus. J Clin Pathol. 1995;48:129–132. doi: 10.1136/jcp.48.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geddert H, Zeriouh M, Wolter M, Heise JW, Gabbert HE, Sarbia M. Gene amplification and protein overexpression of c-erb-b2 in Barrett carcinoma and its precursor lesions. Am J Clin Pathol. 2002;118:60–66. doi: 10.1309/KG1Y-XNGD-54NK-PP66. [DOI] [PubMed] [Google Scholar]
- 28.Soslow RA, Ying L, Altorki NK. Expression of acidic fibroblast growth factor in Barrett’s esophagus and associated esophageal adenocarcinoma. J Thorac Cardiovasc Surg. 1997;114:838–843. doi: 10.1016/S0022-5223(97)70089-8. [DOI] [PubMed] [Google Scholar]
- 29.Soslow RA, Nabeya Y, Ying L, Blundell M, Altorki NK. Acidic fibroblast growth factor is progressively increased in the development of oesophageal glandular dysplasia and adenocarcinoma. Histopathology. 1999;35:31–37. doi: 10.1046/j.1365-2559.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 30.Kumble S, Omary MB, Cartwright CA, Triadafilopoulos G. Src activation in malignant and premalignant epithelia of Barrett’s esophagus. Gastroenterology. 1997;112:348–356. doi: 10.1053/gast.1997.v112.pm9024288. [DOI] [PubMed] [Google Scholar]
- 31.Iravani S, Zhang HQ, Yuan ZQ, Cheng JQ, Karl RC, Jove R, Coppola D. Modification of insulin-like growth factor 1 receptor, c-Src, and Bcl-XL protein expression during the progression of Barrett’s neoplasia. Hum Pathol. 2003;34:975–982. doi: 10.1053/s0046-8177(03)00354-x. [DOI] [PubMed] [Google Scholar]
- 32.González MV, Artímez ML, Rodrigo L, López-Larrea C, Menéndez MJ, Alvarez V, Pérez R, Fresno MF, Pérez MJ, Sampedro A, et al. Mutation analysis of the p53, APC, and p16 genes in the Barrett’s oesophagus, dysplasia, and adenocarcinoma. J Clin Pathol. 1997;50:212–217. doi: 10.1136/jcp.50.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrett MT, Galipeau PC, Sanchez CA, Emond MJ, Reid BJ. Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene. 1996;12:1873–1878. [PubMed] [Google Scholar]
- 34.Kawakami K, Brabender J, Lord RV, Groshen S, Greenwald BD, Krasna MJ, Yin J, Fleisher AS, Abraham JM, Beer DG, et al. Hypermethylated APC DNA in plasma and prognosis of patients with esophageal adenocarcinoma. J Natl Cancer Inst. 2000;92:1805–1811. doi: 10.1093/jnci/92.22.1805. [DOI] [PubMed] [Google Scholar]
- 35.Younes M, Lebovitz RM, Lechago LV, Lechago J. p53 protein accumulation in Barrett’s metaplasia, dysplasia, and carcinoma: a follow-up study. Gastroenterology. 1993;105:1637–1642. doi: 10.1016/0016-5085(93)91058-p. [DOI] [PubMed] [Google Scholar]
- 36.Blount PL, Galipeau PC, Sanchez CA, Neshat K, Levine DS, Yin J, Suzuki H, Abraham JM, Meltzer SJ, Reid BJ. 17p allelic losses in diploid cells of patients with Barrett’s esophagus who develop aneuploidy. Cancer Res. 1994;54:2292–2295. [PubMed] [Google Scholar]
- 37.Symmans PJ, Linehan JM, Brito MJ, Filipe MI. p53 expression in Barrett’s oesophagus, dysplasia, and adenocarcinoma using antibody DO-7. J Pathol. 1994;173:221–226. doi: 10.1002/path.1711730304. [DOI] [PubMed] [Google Scholar]
- 38.Rice TW, Goldblum JR, Falk GW, Tubbs RR, Kirby TJ, Casey G. p53 immunoreactivity in Barrett’s metaplasia, dysplasia, and carcinoma. J Thorac Cardiovasc Surg. 1994;108:1132–1137. [PubMed] [Google Scholar]
- 39.Rioux-Leclercq N, Turlin B, Sutherland F, Heresbach N, Launois B, Campion JP, Ramee MP. Analysis of Ki-67, p53 and Bcl-2 expression in the dysplasia-carcinoma sequence of Barrett’s esophagus. Oncol Rep. 1999;6:877–882. doi: 10.3892/or.6.4.877. [DOI] [PubMed] [Google Scholar]
- 40.Chung SM, Kao J, Hyjek E, Chen YT. p53 in esophageal adenocarcinoma: a critical reassessment of mutation frequency and identification of 72Arg as the dominant allele. Int J Oncol. 2007;31:1351–1355. [PubMed] [Google Scholar]
- 41.Hughes SJ, Nambu Y, Soldes OS, Hamstra D, Rehemtulla A, Iannettoni MD, Orringer MB, Beer DG. Fas/APO-1 (CD95) is not translocated to the cell membrane in esophageal adenocarcinoma. Cancer Res. 1997;57:5571–5578. [PubMed] [Google Scholar]
- 42.Mahidhara RS, Queiroz De Oliveira PE, Kohout J, Beer DG, Lin J, Watkins SC, Robbins PD, Hughes SJ. Altered trafficking of Fas and subsequent resistance to Fas-mediated apoptosis occurs by a wild-type p53 independent mechanism in esophageal adenocarcinoma. J Surg Res. 2005;123:302–311. doi: 10.1016/j.jss.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 43.van der Woude CJ, Jansen PL, Tiebosch AT, Beuving A, Homan M, Kleibeuker JH, Moshage H. Expression of apoptosis-related proteins in Barrett’s metaplasia-dysplasia-carcinoma sequence: a switch to a more resistant phenotype. Hum Pathol. 2002;33:686–692. doi: 10.1053/hupa.2002.124908. [DOI] [PubMed] [Google Scholar]
- 44.Wilson KT, Fu S, Ramanujam KS, Meltzer SJ. Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in Barrett’s esophagus and associated adenocarcinomas. Cancer Res. 1998;58:2929–2934. [PubMed] [Google Scholar]
- 45.Lagorce C, Paraf F, Vidaud D, Couvelard A, Wendum D, Martin A, Fléjou JF. Cyclooxygenase-2 is expressed frequently and early in Barrett’s oesophagus and associated adenocarcinoma. Histopathology. 2003;42:457–465. doi: 10.1046/j.1365-2559.2003.01627.x. [DOI] [PubMed] [Google Scholar]
- 46.Cheong E, Ivory K, Doleman J, Parker ML, Rhodes M, Johnson IT. Synthetic and naturally occurring COX-2 inhibitors suppress proliferation in a human oesophageal adenocarcinoma cell line (OE33) by inducing apoptosis and cell cycle arrest. Carcinogenesis. 2004;25:1945–1952. doi: 10.1093/carcin/bgh184. [DOI] [PubMed] [Google Scholar]
- 47.Majka J, Rembiasz K, Migaczewski M, Budzynski A, Ptak-Belowska A, Pabianczyk R, Urbanczyk K, Zub-Pokrowiecka A, Matlok M, Brzozowski T. Cyclooxygenase-2 (COX-2) is the key event in pathophysiology of Barrett’s esophagus. Lesson from experimental animal model and human subjects. J Physiol Pharmacol. 2010;61:409–418. [PubMed] [Google Scholar]
- 48.Morales CP, Lee EL, Shay JW. In situ hybridization for the detection of telomerase RNA in the progression from Barrett’s esophagus to esophageal adenocarcinoma. Cancer. 1998;83:652–659. [PubMed] [Google Scholar]
- 49.Lord RV, Salonga D, Danenberg KD, Peters JH, DeMeester TR, Park JM, Johansson J, Skinner KA, Chandrasoma P, DeMeester SR, et al. Telomerase reverse transcriptase expression is increased early in the Barrett’s metaplasia, dysplasia, adenocarcinoma sequence. J Gastrointest Surg. 2000;4:135–142. doi: 10.1016/s1091-255x(00)80049-9. [DOI] [PubMed] [Google Scholar]
- 50.Barclay JY, Morris A, Nwokolo CU. Telomerase, hTERT and splice variants in Barrett’s oesophagus and oesophageal adenocarcinoma. Eur J Gastroenterol Hepatol. 2005;17:221–227. doi: 10.1097/00042737-200502000-00014. [DOI] [PubMed] [Google Scholar]
- 51.Shammas MA, Koley H, Batchu RB, Bertheau RC, Protopopov A, Munshi NC, Goyal RK. Telomerase inhibition by siRNA causes senescence and apoptosis in Barrett’s adenocarcinoma cells: mechanism and therapeutic potential. Mol Cancer. 2005;4:24. doi: 10.1186/1476-4598-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Couvelard A, Paraf F, Gratio V, Scoazec JY, Hénin D, Degott C, Fléjou JF. Angiogenesis in the neoplastic sequence of Barrett’s oesophagus. Correlation with VEGF expression. J Pathol. 2000;192:14–18. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH709>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 53.Auvinen MI, Sihvo EI, Ruohtula T, Salminen JT, Koivistoinen A, Siivola P, Rönnholm R, Rämö JO, Bergman M, Salo JA. Incipient angiogenesis in Barrett’s epithelium and lymphangiogenesis in Barrett’s adenocarcinoma. J Clin Oncol. 2002;20:2971–2979. doi: 10.1200/JCO.2002.09.011. [DOI] [PubMed] [Google Scholar]
- 54.Saad RS, El-Gohary Y, Memari E, Liu YL, Silverman JF. Endoglin (CD105) and vascular endothelial growth factor as prognostic markers in esophageal adenocarcinoma. Hum Pathol. 2005;36:955–961. doi: 10.1016/j.humpath.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 55.Griffiths EA, Pritchard SA, McGrath SM, Valentine HR, Price PM, Welch IM, West CM. Increasing expression of hypoxia-inducible proteins in the Barrett’s metaplasia-dysplasia-adenocarcinoma sequence. Br J Cancer. 2007;96:1377–1383. doi: 10.1038/sj.bjc.6603744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krishnadath KK, Tilanus HW, van Blankenstein M, Hop WC, Kremers ED, Dinjens WN, Bosman FT. Reduced expression of the cadherin-catenin complex in oesophageal adenocarcinoma correlates with poor prognosis. J Pathol. 1997;182:331–338. doi: 10.1002/(SICI)1096-9896(199707)182:3<331::AID-PATH860>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 57.Washington K, Chiappori A, Hamilton K, Shyr Y, Blanke C, Johnson D, Sawyers J, Beauchamp D. Expression of beta-catenin, alpha-catenin, and E-cadherin in Barrett’s esophagus and esophageal adenocarcinomas. Mod Pathol. 1998;11:805–813. [PubMed] [Google Scholar]
- 58.Bailey T, Biddlestone L, Shepherd N, Barr H, Warner P, Jankowski J. Altered cadherin and catenin complexes in the Barrett’s esophagus-dysplasia-adenocarcinoma sequence: correlation with disease progression and dedifferentiation. Am J Pathol. 1998;152:135–144. [PMC free article] [PubMed] [Google Scholar]
- 59.Jethwa P, Naqvi M, Hardy RG, Hotchin NA, Roberts S, Spychal R, Tselepis C. Overexpression of Slug is associated with malignant progression of esophageal adenocarcinoma. World J Gastroenterol. 2008;14:1044–1052. doi: 10.3748/wjg.14.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hughes SJ, Glover TW, Zhu XX, Kuick R, Thoraval D, Orringer MB, Beer DG, Hanash S. A novel amplicon at 8p22-23 results in overexpression of cathepsin B in esophageal adenocarcinoma. Proc Natl Acad Sci USA. 1998;95:12410–12415. doi: 10.1073/pnas.95.21.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng P, Gong J, Wang T, Chen J, Liu GS, Zhang R. Gene expression in rats with Barrett’s esophagus and esophageal adenocarcinoma induced by gastroduodenoesophageal reflux. World J Gastroenterol. 2005;11:5117–5122. doi: 10.3748/wjg.v11.i33.5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Breton J, Gage MC, Hay AW, Keen JN, Wild CP, Donnellan C, Findlay JB, Hardie LJ. Proteomic screening of a cell line model of esophageal carcinogenesis identifies cathepsin D and aldo-keto reductase 1C2 and 1B10 dysregulation in Barrett’s esophagus and esophageal adenocarcinoma. J Proteome Res. 2008;7:1953–1962. doi: 10.1021/pr7007835. [DOI] [PubMed] [Google Scholar]
- 63.Lagorce-Pages C, Paraf F, Dubois S, Belghiti J, Fléjou JF. Expression of CD44 in premalignant and malignant Barrett’s oesophagus. Histopathology. 1998;32:7–14. doi: 10.1046/j.1365-2559.1998.00316.x. [DOI] [PubMed] [Google Scholar]
- 64.Darlavoix T, Seelentag W, Yan P, Bachmann A, Bosman FT. Altered expression of CD44 and DKK1 in the progression of Barrett’s esophagus to esophageal adenocarcinoma. Virchows Arch. 2009;454:629–637. doi: 10.1007/s00428-009-0769-z. [DOI] [PubMed] [Google Scholar]
- 65.Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, Harrison R, Novelli MR, Jankowski JA, Wright NA. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut. 2008;57:1041–1048. doi: 10.1136/gut.2007.143339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 2004;64:3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
- 67.Paulson TG, Reid BJ. Focus on Barrett’s esophagus and esophageal adenocarcinoma. Cancer Cell. 2004;6:11–16. doi: 10.1016/j.ccr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 68.Ramel S. Barrett’s esophagus: model of neoplastic progression. World J Surg. 2003;27:1009–1013. doi: 10.1007/s00268-003-7053-1. [DOI] [PubMed] [Google Scholar]
- 69.Falk GW, Goldblum JR. Extent of low-grade dysplasia in Barrett’s esophagus: is it useful for risk stratification? Am J Gastroenterol. 2007;102:494–496. doi: 10.1111/j.1572-0241.2007.01067.x. [DOI] [PubMed] [Google Scholar]
- 70.El-Serag HB, Mason AC, Petersen N, Key CR. Epidemiological differences between adenocarcinoma of the oesophagus and adenocarcinoma of the gastric cardia in the USA. Gut. 2002;50:368–372. doi: 10.1136/gut.50.3.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corley DA, Kubo A, Levin TR, Block G, Habel L, Rumore G, Quesenberry C, Buffler P. Race, ethnicity, sex and temporal differences in Barrett’s oesophagus diagnosis: a large community-based study, 1994-2006. Gut. 2009;58:182–188. doi: 10.1136/gut.2008.163360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wong A, Fitzgerald RC. Epidemiologic risk factors for Barrett’s esophagus and associated adenocarcinoma. Clin Gastroenterol Hepatol. 2005;3:1–10. doi: 10.1016/s1542-3565(04)00602-0. [DOI] [PubMed] [Google Scholar]
- 73.von Rahden BH, Stein HJ, Siewert JR. Barrett’s esophagus and Barrett’s carcinoma. Curr Oncol Rep. 2003;5:203–209. doi: 10.1007/s11912-003-0111-x. [DOI] [PubMed] [Google Scholar]
- 74.Rogers EL, Goldkind SF, Iseri OA, Bustin M, Goldkind L, Hamilton SR, Smith RL. Adenocarcinoma of the lower esophagus. A disease primarily of white men with Barrett’s esophagus. J Clin Gastroenterol. 1986;8:613–618. doi: 10.1097/00004836-198612000-00004. [DOI] [PubMed] [Google Scholar]
- 75.Pondugula K, Wani S, Sharma P. Barrett’s esophagus and esophageal adenocarcinoma in adults: long-term GERD or something else? Curr Gastroenterol Rep. 2007;9:468–474. doi: 10.1007/s11894-007-0061-9. [DOI] [PubMed] [Google Scholar]
- 76.Kubo A, Corley DA. Marked multi-ethnic variation of esophageal and gastric cardia carcinomas within the United States. Am J Gastroenterol. 2004;99:582–588. doi: 10.1111/j.1572-0241.2004.04131.x. [DOI] [PubMed] [Google Scholar]
- 77.El-Serag HB, Petersen NJ, Carter J, Graham DY, Richardson P, Genta RM, Rabeneck L. Gastroesophageal reflux among different racial groups in the United States. Gastroenterology. 2004;126:1692–1699. doi: 10.1053/j.gastro.2004.03.077. [DOI] [PubMed] [Google Scholar]
- 78.Vaezi MF, Richter JE. Synergism of acid and duodenogastroesophageal reflux in complicated Barrett’s esophagus. Surgery. 1995;117:699–704. doi: 10.1016/s0039-6060(95)80015-8. [DOI] [PubMed] [Google Scholar]
- 79.Nehra D, Howell P, Williams CP, Pye JK, Beynon J. Toxic bile acids in gastro-oesophageal reflux disease: influence of gastric acidity. Gut. 1999;44:598–602. doi: 10.1136/gut.44.5.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Menges M, Müller M, Zeitz M. Increased acid and bile reflux in Barrett’s esophagus compared to reflux esophagitis, and effect of proton pump inhibitor therapy. Am J Gastroenterol. 2001;96:331–337. doi: 10.1111/j.1572-0241.2001.03515.x. [DOI] [PubMed] [Google Scholar]
- 81.Theisen J, Nehra D, Citron D, Johansson J, Hagen JA, Crookes PF, DeMeester SR, Bremner CG, DeMeester TR, Peters JH. Suppression of gastric acid secretion in patients with gastroesophageal reflux disease results in gastric bacterial overgrowth and deconjugation of bile acids. J Gastrointest Surg. 2000;4:50–54. doi: 10.1016/s1091-255x(00)80032-3. [DOI] [PubMed] [Google Scholar]
- 82.Wild CP, Hardie LJ. Reflux, Barrett’s oesophagus and adenocarcinoma: burning questions. Nat Rev Cancer. 2003;3:676–684. doi: 10.1038/nrc1166. [DOI] [PubMed] [Google Scholar]
- 83.Qiao D, Gaitonde SV, Qi W, Martinez JD. Deoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradation. Carcinogenesis. 2001;22:957–964. doi: 10.1093/carcin/22.6.957. [DOI] [PubMed] [Google Scholar]
- 84.Anderson LA, Watson RG, Murphy SJ, Johnston BT, Comber H, Mc Guigan J, Reynolds JV, Murray LJ. Risk factors for Barrett’s oesophagus and oesophageal adenocarcinoma: results from the FINBAR study. World J Gastroenterol. 2007;13:1585–1594. doi: 10.3748/wjg.v13.i10.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chak A, Faulx A, Eng C, Grady W, Kinnard M, Ochs-Balcom H, Falk G. Gastroesophageal reflux symptoms in patients with adenocarcinoma of the esophagus or cardia. Cancer. 2006;107:2160–2166. doi: 10.1002/cncr.22245. [DOI] [PubMed] [Google Scholar]
- 86.Ouatu-Lascar R, Fitzgerald RC, Triadafilopoulos G. Differentiation and proliferation in Barrett’s esophagus and the effects of acid suppression. Gastroenterology. 1999;117:327–335. doi: 10.1053/gast.1999.0029900327. [DOI] [PubMed] [Google Scholar]
- 87.Attwood SE, Harrison LA, Preston SL, Jankowski JA. Esophageal adenocarcinoma in & quot; mice and men& quot; : back to basics! Am J Gastroenterol. 2008;103:2367–2372. doi: 10.1111/j.1572-0241.2008.02004.x. [DOI] [PubMed] [Google Scholar]
- 88.Buttar NS, Wang KK. Mechanisms of disease: Carcinogenesis in Barrett’s esophagus. Nat Clin Pract Gastroenterol Hepatol. 2004;1:106–112. doi: 10.1038/ncpgasthep0057. [DOI] [PubMed] [Google Scholar]
- 89.Islami F, Kamangar F, Boffetta P. Use of proton pump inhibitors and risk of progression of Barrett’s esophagus to neoplastic lesions. Am J Gastroenterol. 2009;104:2646–2648. doi: 10.1038/ajg.2009.369. [DOI] [PubMed] [Google Scholar]
- 90.Obszynska JA, Atherfold PA, Nanji M, Glancy D, Santander S, Graham TA, Otto WR, West K, Harrison RF, Jankowski JA. Long-term proton pump induced hypergastrinaemia does induce lineage-specific restitution but not clonal expansion in benign Barrett’s oesophagus in vivo. Gut. 2010;59:156–163. doi: 10.1136/gut.2009.186775. [DOI] [PubMed] [Google Scholar]
- 91.Ye W, Chow WH, Lagergren J, Yin L, Nyrén O. Risk of adenocarcinomas of the esophagus and gastric cardia in patients with gastroesophageal reflux diseases and after antireflux surgery. Gastroenterology. 2001;121:1286–1293. doi: 10.1053/gast.2001.29569. [DOI] [PubMed] [Google Scholar]
- 92.Tran T, Spechler SJ, Richardson P, El-Serag HB. Fundoplication and the risk of esophageal cancer in gastroesophageal reflux disease: a Veterans Affairs cohort study. Am J Gastroenterol. 2005;100:1002–1008. doi: 10.1111/j.1572-0241.2005.41007.x. [DOI] [PubMed] [Google Scholar]
- 93.Corey KE, Schmitz SM, Shaheen NJ. Does a surgical antireflux procedure decrease the incidence of esophageal adenocarcinoma in Barrett’s esophagus? A meta-analysis. Am J Gastroenterol. 2003;98:2390–2394. doi: 10.1111/j.1572-0241.2003.08702.x. [DOI] [PubMed] [Google Scholar]
- 94.Lagergren J, Bergström R, Adami HO, Nyrén O. Association between medications that relax the lower esophageal sphincter and risk for esophageal adenocarcinoma. Ann Intern Med. 2000;133:165–175. doi: 10.7326/0003-4819-133-3-200008010-00007. [DOI] [PubMed] [Google Scholar]
- 95.Maaroos HI, Vorobjova T, Sipponen P, Tammur R, Uibo R, Wadström T, Keevallik R, Villako K. An 18-year follow-up study of chronic gastritis and Helicobacter pylori association of CagA positivity with development of atrophy and activity of gastritis. Scand J Gastroenterol. 1999;34:864–869. doi: 10.1080/003655299750025318. [DOI] [PubMed] [Google Scholar]
- 96.Rokkas T, Pistiolas D, Sechopoulos P, Robotis I, Margantinis G. Relationship between Helicobacter pylori infection and esophageal neoplasia: a meta-analysis. Clin Gastroenterol Hepatol. 2007;5:1413–1417, 1417.e1-1417.e2. doi: 10.1016/j.cgh.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 97.Weston AP, Badr AS, Topalovski M, Cherian R, Dixon A, Hassanein RS. Prospective evaluation of the prevalence of gastric Helicobacter pylori infection in patients with GERD, Barrett’s esophagus, Barrett’s dysplasia, and Barrett’s adenocarcinoma. Am J Gastroenterol. 2000;95:387–394. doi: 10.1111/j.1572-0241.2000.01758.x. [DOI] [PubMed] [Google Scholar]
- 98.Wu AH, Crabtree JE, Bernstein L, Hawtin P, Cockburn M, Tseng CC, Forman D. Role of Helicobacter pylori CagA+ strains and risk of adenocarcinoma of the stomach and esophagus. Int J Cancer. 2003;103:815–821. doi: 10.1002/ijc.10887. [DOI] [PubMed] [Google Scholar]
- 99.Clemons NJ, McColl KE, Fitzgerald RC. Nitric oxide and acid induce double-strand DNA breaks in Barrett’s esophagus carcinogenesis via distinct mechanisms. Gastroenterology. 2007;133:1198–1209. doi: 10.1053/j.gastro.2007.06.061. [DOI] [PubMed] [Google Scholar]
- 100.Forman D, Al-Dabbagh S, Doll R. Nitrates, nitrites and gastric cancer in Great Britain. Nature. 1985;313:620–625. doi: 10.1038/313620a0. [DOI] [PubMed] [Google Scholar]
- 101.Falk GW. Risk factors for esophageal cancer development. Surg Oncol Clin N Am. 2009;18:469–485. doi: 10.1016/j.soc.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 102.Thomas T, Abrams KR, De Caestecker JS, Robinson RJ. Meta analysis: Cancer risk in Barrett’s oesophagus. Aliment Pharmacol Ther. 2007;26:1465–1477. doi: 10.1111/j.1365-2036.2007.03528.x. [DOI] [PubMed] [Google Scholar]
- 103.Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143:199–211. doi: 10.7326/0003-4819-143-3-200508020-00006. [DOI] [PubMed] [Google Scholar]
- 104.Cook MB, Greenwood DC, Hardie LJ, Wild CP, Forman D. A systematic review and meta-analysis of the risk of increasing adiposity on Barrett’s esophagus. Am J Gastroenterol. 2008;103:292–300. doi: 10.1111/j.1572-0241.2007.01621.x. [DOI] [PubMed] [Google Scholar]
- 105.Kubo A, Corley DA. Body mass index and adenocarcinomas of the esophagus or gastric cardia: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:872–878. doi: 10.1158/1055-9965.EPI-05-0860. [DOI] [PubMed] [Google Scholar]
- 106.Murray L, Romero Y. Role of obesity in Barrett’s esophagus and cancer. Surg Oncol Clin N Am. 2009;18:439–452. doi: 10.1016/j.soc.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 107.de Jonge PJ, Steyerberg EW, Kuipers EJ, Honkoop P, Wolters LM, Kerkhof M, van Dekken H, Siersema PD. Risk factors for the development of esophageal adenocarcinoma in Barrett’s esophagus. Am J Gastroenterol. 2006;101:1421–1429. doi: 10.1111/j.1572-0241.2006.00626.x. [DOI] [PubMed] [Google Scholar]
- 108.Oberg S, Wenner J, Johansson J, Walther B, Willén R. Barrett esophagus: risk factors for progression to dysplasia and adenocarcinoma. Ann Surg. 2005;242:49–54. doi: 10.1097/01.sla.0000167864.46462.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vaughan TL, Kristal AR, Blount PL, Levine DS, Galipeau PC, Prevo LJ, Sanchez CA, Rabinovitch PS, Reid BJ. Nonsteroidal anti-inflammatory drug use, body mass index, and anthropometry in relation to genetic and flow cytometric abnormalities in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2002;11:745–752. [PubMed] [Google Scholar]
- 110.Konturek PC, Burnat G, Rau T, Hahn EG, Konturek S. Effect of adiponectin and ghrelin on apoptosis of Barrett adenocarcinoma cell line. Dig Dis Sci. 2008;53:597–605. doi: 10.1007/s10620-007-9922-1. [DOI] [PubMed] [Google Scholar]
- 111.Rubenstein JH, Dahlkemper A, Kao JY, Zhang M, Morgenstern H, McMahon L, Inadomi JM. A pilot study of the association of low plasma adiponectin and Barrett’s esophagus. Am J Gastroenterol. 2008;103:1358–1364. doi: 10.1111/j.1572-0241.2008.01823.x. [DOI] [PubMed] [Google Scholar]
- 112.Kendall BJ, Macdonald GA, Hayward NK, Prins JB, Brown I, Walker N, Pandeya N, Green AC, Webb PM, Whiteman DC. Leptin and the risk of Barrett’s oesophagus. Gut. 2008;57:448–454. doi: 10.1136/gut.2007.131243. [DOI] [PubMed] [Google Scholar]
- 113.Francois F, Roper J, Goodman AJ, Pei Z, Ghumman M, Mourad M, de Perez AZ, Perez-Perez GI, Tseng CH, Blaser MJ. The association of gastric leptin with oesophageal inflammation and metaplasia. Gut. 2008;57:16–24. doi: 10.1136/gut.2007.131672. [DOI] [PubMed] [Google Scholar]
- 114.Ogunwobi O, Mutungi G, Beales IL. Leptin stimulates proliferation and inhibits apoptosis in Barrett’s esophageal adenocarcinoma cells by cyclooxygenase-2-dependent, prostaglandin-E2-mediated transactivation of the epidermal growth factor receptor and c-Jun NH2-terminal kinase activation. Endocrinology. 2006;147:4505–4516. doi: 10.1210/en.2006-0224. [DOI] [PubMed] [Google Scholar]
- 115.Dornonville de la Cour C, Lindström E, Norlén P, Håkanson R. Ghrelin stimulates gastric emptying but is without effect on acid secretion and gastric endocrine cells. Regul Pept. 2004;120:23–32. doi: 10.1016/j.regpep.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 116.Mottershead M, Karteris E, Barclay JY, Suortamo S, Newbold M, Randeva H, Nwokolo CU. Immunohistochemical and quantitative mRNA assessment of ghrelin expression in gastric and oesophageal adenocarcinoma. J Clin Pathol. 2007;60:405–409. doi: 10.1136/jcp.2006.038356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.de Martel C, Haggerty TD, Corley DA, Vogelman JH, Orentreich N, Parsonnet J. Serum ghrelin levels and risk of subsequent adenocarcinoma of the esophagus. Am J Gastroenterol. 2007;102:1166–1172. doi: 10.1111/j.1572-0241.2007.01116.x. [DOI] [PubMed] [Google Scholar]
- 118.Kristal AR, Blount PL, Schenk JM, Sanchez CA, Rabinovitch PS, Odze RD, Standley J, Vaughan TL, Reid BJ. Low-fat, high fruit and vegetable diets and weight loss do not affect biomarkers of cellular proliferation in Barrett esophagus. Cancer Epidemiol Biomarkers Prev. 2005;14:2377–2383. doi: 10.1158/1055-9965.EPI-05-0158. [DOI] [PubMed] [Google Scholar]
- 119.Terry P, Lagergren J, Hansen H, Wolk A, Nyrén O. Fruit and vegetable consumption in the prevention of oesophageal and cardia cancers. Eur J Cancer Prev. 2001;10:365–369. doi: 10.1097/00008469-200108000-00010. [DOI] [PubMed] [Google Scholar]
- 120.Dong LM, Kristal AR, Peters U, Schenk JM, Sanchez CA, Rabinovitch PS, Blount PL, Odze RD, Ayub K, Reid BJ, et al. Dietary supplement use and risk of neoplastic progression in esophageal adenocarcinoma: a prospective study. Nutr Cancer. 2008;60:39–48. doi: 10.1080/01635580701586762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen H, Tucker KL, Graubard BI, Heineman EF, Markin RS, Potischman NA, Russell RM, Weisenburger DD, Ward MH. Nutrient intakes and adenocarcinoma of the esophagus and distal stomach. Nutr Cancer. 2002;42:33–40. doi: 10.1207/S15327914NC421_5. [DOI] [PubMed] [Google Scholar]
- 122.Rudolph RE, Vaughan TL, Kristal AR, Blount PL, Levine DS, Galipeau PC, Prevo LJ, Sanchez CA, Rabinovitch PS, Reid BJ. Serum selenium levels in relation to markers of neoplastic progression among persons with Barrett’s esophagus. J Natl Cancer Inst. 2003;95:750–757. doi: 10.1093/jnci/95.10.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Caygill CP, Johnston DA, Lopez M, Johnston BJ, Watson A, Reed PI, Hill MJ. Lifestyle factors and Barrett’s esophagus. Am J Gastroenterol. 2002;97:1328–1331. doi: 10.1111/j.1572-0241.2002.05768.x. [DOI] [PubMed] [Google Scholar]
- 124.Gammon MD, Schoenberg JB, Ahsan H, Risch HA, Vaughan TL, Chow WH, Rotterdam H, West AB, Dubrow R, Stanford JL, et al. Tobacco, alcohol, and socioeconomic status and adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst. 1997;89:1277–1284. doi: 10.1093/jnci/89.17.1277. [DOI] [PubMed] [Google Scholar]
- 125.Kabat GC, Ng SK, Wynder EL. Tobacco, alcohol intake, and diet in relation to adenocarcinoma of the esophagus and gastric cardia. Cancer Causes Control. 1993;4:123–132. doi: 10.1007/BF00053153. [DOI] [PubMed] [Google Scholar]
- 126.Zhang ZF, Kurtz RC, Sun M, Karpeh M, Yu GP, Gargon N, Fein JS, Georgopoulos SK, Harlap S. Adenocarcinomas of the esophagus and gastric cardia: medical conditions, tobacco, alcohol, and socioeconomic factors. Cancer Epidemiol Biomarkers Prev. 1996;5:761–768. [PubMed] [Google Scholar]
- 127.Brown LM, Silverman DT, Pottern LM, Schoenberg JB, Greenberg RS, Swanson GM, Liff JM, Schwartz AG, Hayes RB, Blot WJ. Adenocarcinoma of the esophagus and esophagogastric junction in white men in the United States: alcohol, tobacco, and socioeconomic factors. Cancer Causes Control. 1994;5:333–340. doi: 10.1007/BF01804984. [DOI] [PubMed] [Google Scholar]
- 128.Vaughan TL, Davis S, Kristal A, Thomas DB. Obesity, alcohol, and tobacco as risk factors for cancers of the esophagus and gastric cardia: adenocarcinoma versus squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 1995;4:85–92. [PubMed] [Google Scholar]
- 129.Menke-Pluymers MB, Hop WC, Dees J, van Blankenstein M, Tilanus HW. Risk factors for the development of an adenocarcinoma in columnar-lined (Barrett) esophagus. The Rotterdam Esophageal Tumor Study Group. Cancer. 1993;72:1155–1158. doi: 10.1002/1097-0142(19930815)72:4<1155::aid-cncr2820720404>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 130.Achkar JP, Post AB, Achkar E, Carey WD. Risk of extraesophageal malignancy in patients with adenocarcinoma arising in Barrett’s esophagus. Am J Gastroenterol. 1995;90:39–43. [PubMed] [Google Scholar]
- 131.Gray MR, Donnelly RJ, Kingsnorth AN. The role of smoking and alcohol in metaplasia and cancer risk in Barrett’s columnar lined oesophagus. Gut. 1993;34:727–731. doi: 10.1136/gut.34.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Levi F, Ollyo JB, La Vecchia C, Boyle P, Monnier P, Savary M. The consumption of tobacco, alcohol and the risk of adenocarcinoma in Barrett’s oesophagus. Int J Cancer. 1990;45:852–854. doi: 10.1002/ijc.2910450511. [DOI] [PubMed] [Google Scholar]
- 133.Lagergren J, Bergström R, Lindgren A, Nyrén O. The role of tobacco, snuff and alcohol use in the aetiology of cancer of the oesophagus and gastric cardia. Int J Cancer. 2000;85:340–346. [PubMed] [Google Scholar]
- 134.Avidan B, Sonnenberg A, Schnell TG, Chejfec G, Metz A, Sontag SJ. Hiatal hernia size, Barrett’s length, and severity of acid reflux are all risk factors for esophageal adenocarcinoma. Am J Gastroenterol. 2002;97:1930–1936. doi: 10.1111/j.1572-0241.2002.05902.x. [DOI] [PubMed] [Google Scholar]
- 135.Smith KJ, O’Brien SM, Smithers BM, Gotley DC, Webb PM, Green AC, Whiteman DC. Interactions among smoking, obesity, and symptoms of acid reflux in Barrett’s esophagus. Cancer Epidemiol Biomarkers Prev. 2005;14:2481–2486. doi: 10.1158/1055-9965.EPI-05-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology. 2003;124:47–56. doi: 10.1053/gast.2003.50008. [DOI] [PubMed] [Google Scholar]
- 137.Heath EI, Canto MI, Piantadosi S, Montgomery E, Weinstein WM, Herman JG, Dannenberg AJ, Yang VW, Shar AO, Hawk E, et al. Secondary chemoprevention of Barrett’s esophagus with celecoxib: results of a randomized trial. J Natl Cancer Inst. 2007;99:545–557. doi: 10.1093/jnci/djk112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gatenby PA, Ramus JR, Caygill CP, Winslet MC, Watson A. Aspirin is not chemoprotective for Barrett’s adenocarcinoma of the oesophagus in multicentre cohort. Eur J Cancer Prev. 2009;18:381–384. doi: 10.1097/CEJ.0b013e32832e0955. [DOI] [PubMed] [Google Scholar]