

Abstract
BACKGROUND
Intraperitoneal tumors expressing high amounts of mesothelin such as malignant mesothelioma and ovarian cancers tend to develop ascites and result in significant morbidity and mortality in the patient. A suitable preclinical intraperitoneal model will assist in the illustration of the mechanisms of molecular oncogenesis and facilitate in addressing issues related to early screening, diagnosis, and therapy for intraperitoneal tumors.
METHODS
In the current study, an ascitogenic malignant tumor model (WF-3) was created. The mobility and proliferation of WF-3 and its precursor cells, WF-0, were characterized using transwell and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays. In addition, the in vivo tumorgenicity of WF-3 and WF-0 was determined using intraperitoneal injection of the tumor cells. Microarray analysis was performed using WF-3 and WF-0. Northern blot analysis was used to characterize the expression of the mesothelin gene in WF-3 and WF-0. Furthermore, the mesothelin levels in serum and ascites were used to correlate with tumor load of WF-3 in tumor challenged mice.
RESULTS
The WF-3 tumor cells demonstrated relatively high proliferation and migration rates compared with the parental cell line, WF-0. The tumors from the WF-3 but not WF-0 were capable of forming ascites and peritoneal-based tumors after tumor challenge. The WF-3 tumor model was also capable of implanting into multiple organs including the diaphragm, intestines, and peritoneal wall. Furthermore, the WF-3 tumor expressed high levels of mesothelin, which is commonly observed in the majority of ovarian cancers, pancreatic cancer, and malignant mesothelioma. In addition, the authors found that the serum and ascites mesothelin levels correlated with tumor loads in tumor-challenged mice.
CONCLUSIONS
The data indicate that the WF-3 murine tumor model may potentially serve as a good model for understanding the molecular oncogenesis of peritoneal tumors. In addition, the preclinical model may potentially be useful for the development of diagnostic and therapeutic methods against intraperitoneal cancers.
Keywords: tumor model, animal model, microarray, mesothelin, ovarian cancer
Peritoneal dissemination of tumors such as ovarian cancer and mesothelioma is often a preterminal event. Ovarian cancer is the sixth most common malignancy in women and the leading cause of death from all gynecological cancers in the U.S.1 Most women are diagnosed in the later stages of the disease and require invasive surgical and chemotherapeutic strategies. Although significant advancement has been made in both surgical and chemotherapeutic approaches, the 5-year survival rate for ovarian cancer remained unchanged (< 30%).2 Likewise, malignant mesothelioma is an aggressive tumor that does not respond to chemotherapy or radiation therapy.3,4 Because the early stages of ovarian cancer and/or malignant mesothelioma generally go unnoticed, little is known of the establishment and progression of the disease in its early stages. Thus, it is important to have suitable preclinical intra-peritoneal tumor models that allow us to investigate both molecular mechanisms of oncogenesis and possible methods of early screening, diagnosis, and therapy of intraperitoneal tumorigenesis.
Many of the animal models for intraperitoneal tumors use immunocompromised mice such as athymic nude mice or SCID mice.5,6 Such models allow direct transplantation of human tumor cell lines in the mice via subcutaneous and/or intraperitoneal injections and provide useful insights into clinical management.7,8 However, these models cannot provide important information regarding the interaction of the host immune system with tumors.
Several intraperitoneal tumor models capable of growing in immunocompetent mice have been developed. For example, Roby et al.9 developed a murine ovarian tumor, MOSEC, that is capable of growing in syngeneic C57BL/6 mice. MOSEC has been further modified to grow more aggressively in C57BL/6 mice.10,11 Davis et al.12 developed a malignant mesothelioma capable of growing in syngeneic BALB/c mice. Such model systems are potentially very useful for the study of various cellular, molecular, and genetic aspects of the diseases and for the preclinical evaluation of potential therapeutic agents including immunotherapy.
Although the aforementioned preclinical tumor models are useful for the research in intraperitoneal tumors, stepwise precursor tumor cells were not available for most of these tumor models. The comprehensive collection of the precursor tumor cells at different stages of tumor progression will allow us to perform various genetic analyses such as microarray gene chip analysis, SAGE (serial analysis of gene expression), or differential display to determine genes that are important for tumor progression.
Therefore, we have created a peritoneal-based tumor model from C57BL/6 mice by introducing various oncogenes in a stepwise fashion. The continuous isolation and passage of early-stage tumor cells (WF-0) from the ascites fluid of the tumor-challenged C57BL/6 mice resulted in an aggressive tumor cell line named WF-3. We performed microarray analysis using WF-0 and WF-3 cells. One of the genes that are highly expressed in WF-3 but not in WF-0 is mesothelin. Mesothelin is a 40-kilodalton (kD) cell surface glyco-protein that has been found to be attached by phosphatidylinositol. The protein may have a role in cell-adhesion and in cell-to-cell recognition and signaling.13,14 It has been shown that mesothelin is expressed in the majority of ovarian cancers and is absent or expressed at low levels in normal tissues.15 Furthermore, mesothelin-related proteins have been found to be raised in most patients with malignant mesothelioma.14 We also found that the levels of mesothelin in sera and ascites of mice correlated with the tumor burden and the severity of intraperitoneal diseases in WF-3-challenged mice. The potential usage of the preclinical intraperitoneal tumor model is discussed.
MATERIALS AND METHODS
Mice
C57BL/6 female mice (6–8 weeks old) were purchased from the National Cancer Institute (Frederick, Md). All animals were maintained under specific pathogen-free conditions at the Johns Hopkins Hospital (Baltimore, Md). All procedures were performed according to approved protocols and in accordance with recommendations for the proper care of laboratory animals.
Generation of WF-0, WF-1, WF-2, and WF-3 Tumor Cells
C57BL/6 mouse peritoneal cells were collected by peritoneal washing with 1× Hanks buffered salt solution (HBSS). The primary single cell suspensions were cultured in vitro in RPMI-1640, supplemented with 10% fetal calf serum, 50 U/mL of penicillin/streptomycin, 2 mM of L-glutamine, 1 mM of sodium pyruvate, and 2 mM of nonessential amino acids and grown at 37°C with 5% carbon dioxide. Transduction of human papillomavirus type 16 (HPV-16) E6 and E7 genes into primary peritoneal cells was performed with the LXSN16E6E7 retroviral vector, a generous gift from Dr. D.A. Galloway.16 The HPV-16 E6- and E7-expressing LXSN16E6E7 were used to infect CRIP cells to generate recombinant virus with a wide host range. The primary peritoneal cells were immortalized by transduction with LXSN16E6E7 using methods described previously.16 After transduction, the retroviral supernatant fluid was removed and the cells were grown in G418 (0.4 mg/mL) culture medium for an additional 3 days to allow for integration and expression of the recombinant retroviral genes. The immortalized peritoneal cells were further transfected with pVEJB DNA, which encodes activated human c-Ha-ras gene (kindly provided by Chi V. Dang at the Johns Hopkins Hospital, Baltimore, Md), and selected with G418 (0.4 mg/mL) and hygromycin (0.2 mg/mL). The transduced cells, named WF cells, were further injected into athymic nude mice. The injected athymic mice eventually developed peritoneal-based tumors and ascites after intraperitoneal injection. The isolated tumor cells from athymic mice were named WF-0.
The generation of the WF-3 tumor cell line is shown in Figure 1. Six months after WF-0 cells were injected into C57BL/6 mice, < 10% of injected C57BL/6 mice had developed peritoneal-based tumors and ascites. The tumor cells grown from ascites in mice challenged with WF-0 were further isolated and cultured in vitro. These cell lines were named WF-1 cells. Mice were then challenged with WF-1 cells intraperitoneally. The tumor cells grown from the ascites of mice challenged with WF-1 were further isolated, expanded in vitro, and then named WF-2 cells. The tumor cells isolated and expanded from the ascites in mice challenged with WF-2 were named WF-3 cells. All of the C57BL/6 mice challenged with WF-3 tumor cells at a dose of 5 × 105 developed peritoneal-based tumors within 2 months. All the tumor cell lines were grown in RPMI-1640, supplemented with 10% (volume/volume) fetal bovine serum, 50 U/mL of penicillin/streptomycin, 2 mM of L-glutamine, 1 mM of sodium pyruvate, 2 mM of nonessential amino acids, and 0.4 mg/mL of G418 at 37°C with 5% carbon dioxide. On the day of tumor challenge, tumor cells were harvested by trypsinization, washed twice with 1× HBSS, and finally resuspended in 1× HBSS to the designated concentration for injection.
FIGURE 1.
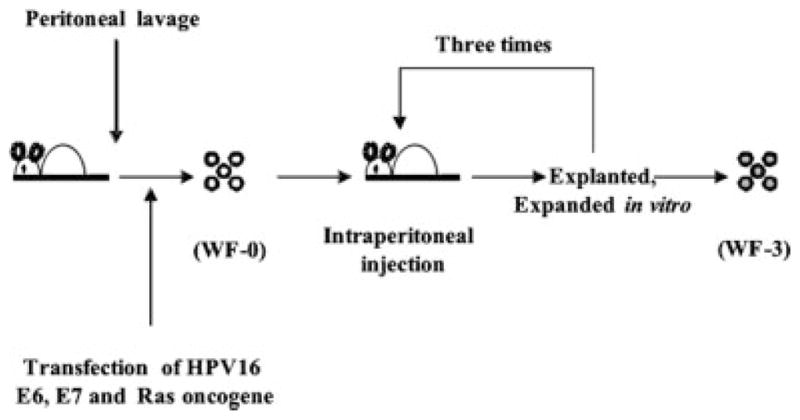
Schematic diagram showing the in vivo selection of WF-0, WF-1, WF-2, and WF-3 cell lines. The peritoneal cells of the C57BL/6 mice were collected, transduced with retrovirus encoding human papillomavirus type 16 (HPV-16) E6 and E7 genes, followed by transfection with DNA encoding human c-Ha-ras gene, and named WF cells. The WF cells were injected intraperitoneally into athymic mice repeatedly. Eventually, athymic mice developed ascites. The tumor cells from ascites in athymic mice previously challenged with WF cells were isolated, expanded in vitro, and named WF-0. When WF-0 cells were injected into C57BL/6 mice, <10% of WF-0 tumor-challenged C57BL/6 mice developed ascites. Tumor cells from the ascites of C57BL/6 mice were isolated and expanded in vitro. These expanded cell lines were called WF-1. The mice were further challenged intraperitoneally with WF-1. The outgrown ascites in mice challenged with WF-1 were further cultured in vitro. These expanded cell lines were called WF-2. The mice were challenged intraperitoneally with WF-2. Cells harvested from the ascites of mice challenged with WF-2 were further cultured in vitro. These expanded cell lines were called WF-3.
In Vitro Cell Migration Assays
The ability of tumor cells to migrate was assessed by counting the number of cells that migrated through transwell inserts with 8-μm pores (Becton Dickinson, Mountain View, Calif), according to the protocol recommended by the manufacturer. Briefly, transwell membranes were coated with 100 μL of Matrigel (Collaborative Research, Boston, Mass) at a final concentration of 0.1 mg/mL and dried. Tumor cells (5 × 104) in 100 μL of RPMI-1640 supplemented with 0.1% FBS medium were added to the upper chamber triplicate wells and allowed to migrate through Matrigel overnight at 37°C in a 5% carbon dioxide atmosphere. The lower compartment of the transwell chamber was filled with complete tumor medium. The cells that migrated through the membranes into the lower wells were assessed by hematoxylin uptake. Then the number of cells was counted within a field at ×200 under a light microscope. For each membrane, a total of 5 fields were selected at random and the numbers were averaged. For another experiment, after removing the cells on the top with a cotton swab, the cells were incubated in MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution (30 μL; 5 mg/mL) for 5 hours. Then, 100 μL of DMSO was added to dissolve formazan crystals under vigorous shaking for 30 minutes and detected using the absorption at OD 570 nanometers (nm) with an enzyme-linked immunoadsorbent assay (ELISA) reader.
In Vitro Measurement of Proliferation of Tumor Cell Lines
The in vitro tumor cell proliferation was performed as done previously by Behrens et al.,17 with some modifications. Briefly, WF-0 and WF-3 tumor cells (2 × 103) were seeded in triplicate in flat-bottom 96-well microtiter plates and incubated for 24, 48, 72, or 96 hours with the culture media to be tested. MTT solution (30 μL; 5 mg/mL) was added and incubated for 4 hours. By the addition of 100 μL of DMSO to dissolve formazan crystals under vigorous shaking for 30 minutes, detection of the absorption at OD 570 nm was performed with an ELISA reader.
In another experiment, WF-0 and WF-3 tumor cells (2 × 103/well) were seeded in triplicate in flat-bottom 6-well plates and incubated with the culture media. The tumor cells were trypsinized and counted with counting chamber under light microscope directly at the indicated time.
In Vivo Tumor Growth Kinetics
C57BL/6 mice (5 per group) were challenged with 1 × 104, 5 × 104, or 1 × 105/mouse of WF-0 or WF-3 tumor cells intraperitoneally. Mice were monitored regularly for evidence of tumor growth by palpation and inspection twice a week for tumor growth and overall survival until they were sacrificed at Day 90.
In a second set of experiments, C57BL/6 mice were challenged with 5 × 104/mouse of WF-0 or WF-3 tumor cells subcutaneously. Mice were monitored regularly for evidence of tumor growth by palpation and inspection twice a week for tumor growth. The tumor size were measured and recorded serially until they were sacrificed at Day 90. The data were used to plot graphs of tumor growth kinetics.
Histologic and Pathologic Features of WF-3
C57BL/6 mice were challenged with 5 × 104/mouse of WF-3 tumor cells intraperitoneally. Five to 6 weeks later, the mice were sacrificed and the invasiveness of the tumor was examined macroscopically. In addition, the removed internal organs were fixed with 4% buffered formaldehyde and histologic sections were performed and stained with routine hematoxylin and eosin stain. The slides were observed under light microscope.
RNA Preparation, Microarray, and Data Analysis
Poly(A)+ RNA from approximately 1 × 107 of WF-0 or WF-3 were isolated according to the manufacturer’s protocol using a FastTrack 2.0 mRNA isolation kit (Invitrogen, Carlsbad, Calif). cDNA generation, hybridization, and data collection were performed by Incyte Genomics (Wilmington, Del). In brief, alterations in gene expression were evaluated by reverse transcription of poly(A)+ RNAs in the presence of Cy3 or Cy5 fluorescent labeling dyes followed by hybridization to a mouse GEM2 (UniGene 1, Fair-field, NJ) microarray chip. Each chip displays a total of 8734 elements, 7854 of which are unique genes/clusters. These unique gene/clusters can be further defined as 3205 annotated and 4649 nonannotated sequences. Subsets of genes were selected for further study based on differential Cy3/Cy5 expression ratios that were ≥ 2.5 in response to antibody cross-linking treatment. Differential expression of representative selected genes was confirmed by reverse-transcriptase polymerase chain reaction (RT-PCR) and/or Northern hybridization.
Northern Blot Analysis of WF-0 and WF-3 Cells
Total cellular RNA was isolated by the Trizol RNA isolation kit (Life Technologies, Gaithersburg, Md) according to the manufacturer. Northern blot analysis was performed according to the method described previously.18 A 20-μg portion of total RNA was resolved on a 1.5% formaldehyde-agarose gel and blotted onto a nylon membrane (Hybond, Amer-sham Biosciences, Arlington Heights, Ill) by the standard procedure. The hybridization was performed with a 32P-labeled mouse mesothelin DNA fragment probe generated by the random-priming method. The mouse mesothelin cDNA probe was prepared from a monolayer of WF-3 by the RT-PCR method using primers: 5′-TTGTGCCCACTTCTTC TCCCTCA-3′ (corresponding to nucleotides 522–544) and 3′-CTCATCCAACACTGCTACCAAGC-3′ (corresponding to nucleotides863–885). The PCR products were confirmed by sequence. After overnight hybridization at 42°C with standard buffer containing 10% dextran sulfate, the blot was washed twice in 2× standard saline citrate (SSC) (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-1% sodium dodecyl sulfate (SDS) and once in 2× SSC-0.1% SDS and subjected to either autoradiography or direct quantification with Fuji film image guide software.
ELISA to Determine Levels of Mesothelin in Sera or Ascites of Mice Challenged With WF-0 or WF-3 Cells
Mice were challenged with WF-0 or WF-3 cells intraperitoneally. The sera and ascites were collected at the indicated schedule and frozen at −20°C until analyzed. A direct ELISA was used, with some modifications.19 Briefly, a 96-microwell plate was coated with 100 μL of anti-mesothelin antibody (1:20,000 dilution; Labvision, Fremont, Calif) and incubated at 4°C overnight. The wells were then blocked with phosphate-buffered saline (PBS) containing 20% fetal bovine serum. Sera or ascites were serially diluted in PBS, added to the ELISA wells, and incubated at 37°C for 2 hours. After washing with PBS containing 0.05% Tween 20, the plate was incubated with a 1:2000 dilution of a peroxidase-conjugated goat anti-mouse IgG antibody (Biosource, Camarillo, Calif) at room temperature for 1 hour. The plate was washed, developed with 1-Step Turbo TMB-ELISA (Clinical Science Lab, Mansfield, Mass), and stopped with 1 MH2SO4. The ELISA plate was read with a standard ELISA reader at 450 nm.
Statistical Analysis
All data are expressed as mean ± the standard deviation (SD). Data shown are representative of at least 2 different experiments. Data for tumor treatment experiments were evaluated by analysis of variance (ANOVA). Comparisons between individual data points were made using a Student t test. In the tumor protection experiment, the principal outcome of interest was time to development of tumor. The survival experiments for different mice were compared by use of the method of Kaplan-Meier and the log-rank statistic. Differences were considered significant if P <.05.
RESULTS
Migration of WF-3 Cells Is Greater Than That of WF-0 Cells
We first characterized the migration of WF-0 and WF-3 cells by counting the number of cells that migrated through the transwell. The representative figures of the migration of WF-0 and WF-3 cells through the Matrigel-coated membrane are shown in Figure 2A. The numbers of WF-3 cells that migrated through the transwell membrane were significantly higher than those of WF-0 cells (94 ± 5.1 vs 2.6 ± 1.2; P <.01, 1-way ANOVA) (Fig. 2B). In addition, MTT assay also showed that the absorbance at 570 nm in WF-3 cells is also significantly higher than that in the WF-0 cells (0.850 ± 0.016 vs 0.004 ± 0.003; P <.01, ANOVA) (Fig. 2C). Taken together, our results revealed that WF-3 cells have a higher migration ability than WF-0 cells.
FIGURE 2.

In vitro tumor cell migration assays. The migration of tumor cells was assessed by counting the number of cells that migrated through transwell. The cells that migrated through the membranes into the lower wells were assessed by hematoxylin uptake and the number of cells was counted, or by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. (A) Representative figures of cell migration in WF-0 and WF-3 cells. (B) Bar graph depicting total number of tumor cells that migrated through transwell. Note that the cell numbers that migrated through the membranes in the WF-3 group were significantly higher than those in the WF-0 group (P < .05, 1-way analysis of variance [ANOVA]). (C) Bar graph depicting absorbance at 570 nanometers (nm) using enzyme-linked immunoadsorbent assay (ELISA) in WF-0 or WF-3 tumor cells treated with MTT solution. Note that the absorbance at 570 nm of the WF-3 group were also significantly higher than those of the WF-0 group (P < .05, 1-way ANOVA).
WF-3 Cells Demonstrate a Higher Proliferation Rate Compared With WF-0 In Vitro and Are More Tumorigenic In Vivo
To evaluate if the proliferation rate of WF-3 and WF-0 tumor cell lines are different in vitro, we performed the MTT assays as well as a cell counting assay using microscopic examination. As shown in Figures 3A and 3B, both MTT assays and cell counting assay demonstrated that WF-3 tumor cells has a significantly higher proliferation rate than WF-0 tumor cells.
FIGURE 3.

In vitro measurement of cell proliferation and in vivo tumor growth experiments. We characterized the proliferative rates of WF-0 and WF-3 cell lines in vitro. The WF-0 and WF-3 cells were seeded with the same number of tumor cells. The tumor cells were collected at different time points and characterized by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays (A) or direct cell counting under the microscope (B). Note that the absorption at OD 570 nanometers (nm) of the WF-3 cells was significantly higher than that of the WF-0 cells (P < .05, 1-way analysis of variance [ANOVA]); the total numbers of the WF-3 cells were significantly higher than those of the WF-0 cells (P < .05, 1-way ANOVA). (C) In vivo tumor growth experiments. C57BL/6 mice were challenged with WF-0 (closed circle) or WF-3 (open circle) subcutaneously. Tumor growth was measured twice a week. Note that no subcutaneous tumor growth was identified in mice challenged with WF-0. In contrast, all of the mice challenged with WF-3 developed tumor growth. SD indicates standard deviation.
We also evaluated the in vivo tumorigenesis of WF-0 and WF-3 cells in C57BL/6 mice. As shown in Figure 3C, whereas mice challenged with WF-0 tumor cells subcutaneously did not develop a tumor 90 days after tumor challenge, all mice challenged with WF-3 tumor cells subcutaneously developed significant tumors over time. The size of WF-3 subcutaneous tumors increased over time. Thus, our data indicated that WF-3 cells grow faster and are more tumorigenic than WF-0 cells.
Mice Challenged With WF-3 Tumor Cells Intraperitoneally Develop Ascites and Tumor Implants
To demonstrate the ascites formation in mice challenged with WF-3 tumor cells, C57BL/6 mice (5 per group) were challenged at a dose of 1 × 106 tumor cells/mouse or not challenged. Six weeks after the WF-3 tumor challenge all of the tumor-challenged mice developed ascites. Figure 4A is a representative photo of mice with (left panel) or without (right panel) WF-3 tumor challenge. Mice challenged with WF-3 tumor cells were found to have a distended abdominal wall (left). In contrast, mice without tumor challenge demonstrated a scaphoid abdomen (right). As mentioned above, < 10% of C57BL/6 mice challenged with WF-0 tumor cells developed tumor implants or ascites. We further observed the abdominal cavities of mice challenged with WF-3 tumor cells. Macroscopically, the WF-3-challenged mice were emaciated and their abdomen was bulging. The peritoneal cavity was filled with a significant amount of bloody ascites. Furthermore, multiple, friable, and grayish-white tumor nodules of various sizes were found in the peritoneal cavity (arrows, Fig. 4B). The tumor nodules involved multiple organs, including liver, pancreas, small intestine, and diaphragm. Thus, the WF-3 tumor is capable of forming ascites and developing tumor implants when challenged intraperitoneally.
FIGURE 4.
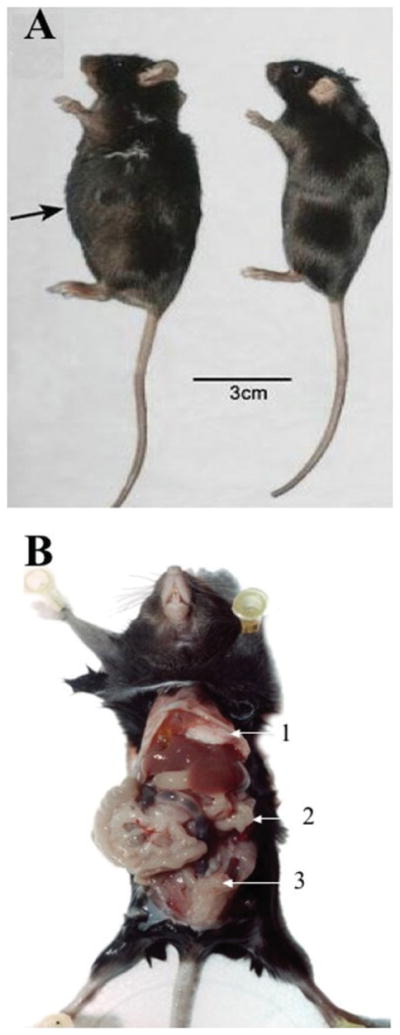
Ascitogenic features of mice challenged with WF-3 cells intraperitoneally. C57BL/6 mice were challenged with 5 × 104/mouse of WF-3 tumor cells intraperitoneally. The tumor-challenged mice were sacrificed 12 weeks after tumor challenge. (A) Representative figures of tumor-challenged mice showing ascites formation. The mice challenged with WF-3 cells were found to have a distended abdomen (left mouse). In contrast, the control mouse without tumor challenge revealed a scaphoid abdomen (right mouse). (B) Representative figures showing the distribution of WF-3 tumors in the peritoneal cavity. Note: multiple, friable, and grayish-white tumor nodules of various sizes were found in the peritoneal cavity. The tumor implants were identified in multiple abdominal organs, including diaphragm (arrow 1), peritoneal wall (arrow 2), and intestine (arrow 3).
WF-3 Tumors Share Malignant Features Commonly Seen in Intraperitoneal Tumors
We also examined the morphologic features of WF-3 tumors derived from tumor-challenged mice. Histologically, the tumors were characterized by bizarre anaplastic tumor cells with marked pleomorphism, high nucleus/cytoplasmic ratio, hyperchromatism, and numerous mitotic figures (arrows, Fig. 5A). These features were consistent with malignant tumors. In addition, the tumor cells showed a carcinomatous component (Fig. 5B) as well as a sarcomatous component (Fig. 5C). The biphasic morphological features resemble malignant mesothelioma. Furthermore, foci of papillary configurations were also appreciated in the intraperitoneal tumor nodules (Fig. 5D). The papillary configurations of the tumor are commonly observed in intraperitoneal tumors such as mesothelioma and ovarian tumors. Thus, WF-3 tumors share morphological features with malignant tumors.
FIGURE 5.

Morphologic features of WF-3 tumor cells. WF-3 tumor cells were challenged intraperitoneally into C57BL/6 mice at a dose of 5 × 104/mouse. The mice were sacrificed 12 weeks after the tumor challenge. Tissue sections derived from the tumors were prepared by hematoxylin and eosin staining. The morphology as well as the invasiveness of the tumor was characterized by histology. (A) The tumors were characterized by sheets of bizarre anaplastic tumor cells with marked pleomorphism, a high nuclear:cytoplasmic ratio, and hyperchromatism with numerous mitotic figures (arrows). (B) Some areas of the WF-3 tumors showed a carcinomatous component. (C) Some areas of the WF-3 tumor showed a sarcomatous component. (D) Focal areas of the WF-3 tumor also demonstrated papillary configuration.
Tumor Growth Kinetics of WF-0 and WF-3 Tumor Cells Challenged Intraperitoneally
To determine the tumor growth kinetics in mice, we challenged C57BL/6 mice with WF-0 or WF-3 tumor cells intraperitoneally at various doses (1 × 104, 5 × 104, 1 × 105, and 1 × 106 tumor cells/mouse). As shown in Figure 6A,B, none of the WF-0 tumor cell-challenged mice developed tumors or ascites or died up to 90 days after tumor challenge. In comparison, all of the C57BL/6 mice challenged with WF-3 tumor cells at doses of 5 × 104 and 1 × 105 tumor cells/mouse intraperitoneally developed tumors and ascites within 21 days after tumor challenge (Fig. 6C). 80% of mice challenged with WF-3 tumor cells at a dose of 1 × 104/mouse also developed tumors and ascites within 50 days after tumor challenge.
FIGURE 6.

In vivo tumor growth kinetics of WF-0 and WF-3 cells. C57BL/6 mice were challenged with different doses of WF-0 or WF-3 tumor cells (1 × 104, 5 × 104, or 1 × 105/mouse) intraperitoneally. Tumor growth was monitored twice a week. In addition, survival of mice was characterized by Kaplan-Meier survival analysis. (A) In vivo tumor growth experiments in mice challenged with WF-0 cells. (B) Survival analysis in mice challenged with WF-0 cells. (C) In vivo tumor growth experiments in mice challenged with WF-3 cells. (D) Survival analysis in mice challenged with WF-3 cells. Note that all of the mice challenged with WF-3 tumor cells developed tumors and ascites within 25 days after challenging with 5 × 104 or 1 × 105/mouse WF-3 tumor cells. 80% of mice challenged with WF-3 at a dose of 1 × 104 developed a tumor 49 days after tumor challenge. In contrast, none of the mice challenged with WF-0 developed tumor growth. Furthermore, all of the mice challenged with 5 × 104 or 1 × 105/mouse of WF-3 tumor cells died within 60 days after tumor challenge. 80% of mice challenged with 1 × 104/mouse of WF-3 tumor cells died within 90 days after tumor challenge. In contrast, none of the mice challenged with WF-0 died 90 days after tumor challenge.
Furthermore, all of the mice challenged with WF-3 tumor cells at doses of 1 × 105/mouse or 5 × 104/mouse died within 60 days after tumor challenge (Fig. 6D). 80% of mice challenged with a dose 1 × 104/mouse also died within 81 days after tumor challenge. The data indicate that the minimal tumor dose for WF-3 tumor cells to develop a tumor and kill the tumor-challenged mice is 5 × 104/mouse.
WF-3 Tumor Cells Express Higher Levels of Mesothelin Compared With WF-0 Tumor Cells
To evaluate the gene expression profile between the WF-0 and WF-3 tumor cells, microarray analysis using gene chips was performed. Table 1 summarizes the elevated gene expression in WF-3 tumor cells as compared with the parental WF-0 tumor cells. Several genes were highly expressed in WF-3 tumor cells compared with WF-0 cells, including lipocalin 2, clusterin, and mesothelin.
TABLE 1.
Elevated Gene Expression in WF-3 Virulent Tumor Cells Compared With the Maternal WF-0 Tumor Cells
| Gene name | Mouse access no. | WF-0 | WF-3 | |
|---|---|---|---|---|
| 1 | Lipocalin 2 | AA087193 | 1 | 9.9 |
| 2 | Clusterin | AA210481 | 1 | 8.4 |
| 3 | Mesothelin | AA673869 | 1 | 8.3 |
| 4 | Ankyrin-like repeat protein | AA792499 | 1 | 6.7 |
| 5 | Epidermal growth factor-containing fibulin-like extracellular matrix protein 1 | AI156278 | 1 | 6.5 |
| 6 | ESTs | AA607132 | 1 | 5.8 |
| 7 | Calmodulin 2 | AA575501 | 1 | 5.7 |
| 8 | Lectin, galactose-binding, soluble 3 | AA066647 | 1 | 5.6 |
| 9 | RIKEN cDNA 1300019I03 gene | AA600596 | 1 | 4.7 |
| 10 | ESTs | AA106833 | 1 | 4.6 |
| 11 | High-mobility group AT-hook 1 | AA067083 | 1 | 4.5 |
| 12 | ESTs | AA546740 | 1 | 4.5 |
| 13 | Inter-α trypsin inhibitor, heavy chain 2 | W83984 | 1 | 4.4 |
| 14 | Potassium intermediate/small conductance calcium-activated channel, subfamily N, member 4 | AA185547 | 1 | 4.4 |
| 15 | Angiopoietin-like 2 | AA755981 | 1 | 4.3 |
| 16 | ESTs | AA793588 | 1 | 4.3 |
| 17 | ESTs | AA475611 | 1 | 4.3 |
| 18 | Insulin-like growth factor binding protein 4 | AA108417 | 1 | 4.1 |
| 19 | ESTs | AA760260 | 1 | 4.0 |
| 20 | Myeloid differentiation primary response gene 116 | AA684447 | 1 | 3.8 |
| 21 | Glutathione S-transferase like | W16059 | 1 | 3.8 |
ESTs indicates mouse-expressed sequence tags.
The representative 21 genes whose expression is consistently enhanced at least 3.8-fold in WF-3 compared with WF-0. Data are presented as fold expression compared with WF-0 tumor cells. ESTs were named according to the gene to which they demonstrated the greatest sequence similarity. The accession number is the GenBank entry from which the oligonucleotide probe sequences were drawn.
Because mesothelin has been found to be highly expressed in the majority of human intraperitoneal tumors such as ovarian cancer and malignant mesothelioma, but not in normal tissue,15 it has been implicated as a potential tumor marker for the screening and diagnosis of intraperitoneal tumors as well as a potential target for developing cancer vaccines and immunotherapeutic strategies.13–15 We therefore initially focused on the characterization of the mesothelin expression in the WF-3 tumor model. As shown in Table 1, the expression levels of mesothelin in the WF-3 cells were 8.3-fold higher than those in the WF-0 cells. Furthermore, Northern blot analysis confirmed that the expression level of mesothelin in the WF-3 cells was significantly higher than that in the WF-0 cells (Fig. 7). Our data indicate that mesothelin is highly expressed in WF-3 tumor cells but not in WF-0 cells.
FIGURE 7.

Northern blot analysis to characterize mesothelin expression in WF-0 and WF-3 cells. Total cellular RNA from cultured WF-0 and WF-3 tumor cells were isolated. A 20-μg portion of total RNA was resolved on a 1.5% formaldehyde-agarose gel (B) and blotted onto a nylon membrane (A). The hybridization was then performed with 32P-labeled mouse mesothelin DNA fragment probe generated by the random-priming method. Note: Equal amounts of RNA from WF-0 and WF-3 tumor cells were loaded onto the wells as indicated by the same amounts of 28S and 18S RNA (indicated by arrows, B). The expression level of mesothelin in WF-3 cells is greater than that of the WF-0 cells (indicated by arrow, A).
Mesothelin Levels in Serum and Ascites Fluids Correlate With Tumor Burden
The mesothelin levels of the serum and ascites in the WF-0- or WF-3-challenged mice were assayed over time using ELISA with specific antibodies against murine mesothelin. As shown in Figure 8, the levels of mesothelin in both serum and ascites fluids derived from the WF-3 tumor-challenged mice were significantly higher than those derived from the WF-0 tumor-challenged mice, particularly 14 days after tumor challenge (P <.05, 1-way ANOVA). These data indicate that the mesothelin levels in serum and ascites fluids increased over time in WF-3 tumor-challenged mice but not in WF-0 tumor-challenged mice. In addition, the levels of mesothelin in the ascites were significantly higher than those of the serum.
FIGURE 8.
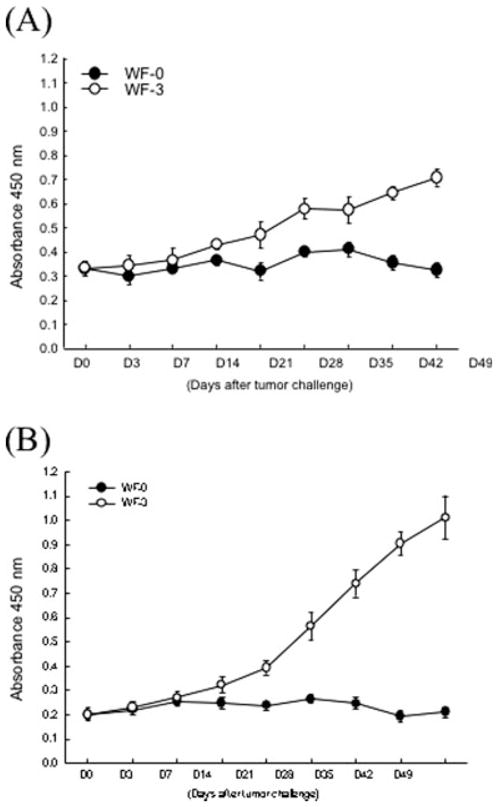
Titers of mesothelin in sera and ascites of mice challenged with WF-0 and WF-3 cells. To evaluate the levels of mesothelin in the sera or ascites of mice challenged with WF-0 or WF-3 cells, we performed enzyme-linked immunoadsorbent assay (ELISA). (A) ELISA assay characterizing serum mesothelin levels in WF-0 (closed circle) or WF-3 (open circle) tumor challenged mice. (B) ELISA assay characterizing ascites mesothelin levels in WF-0 (closed circle) or WF-3 (open circle) tumor-challenged mice. Note that both the serum and ascites mesothelin levels in WF-3 tumor-challenged mice were significantly higher than those of WF-0 tumor-challenged mice at 14 days after tumor challenge (P < .05, 1-way analysis of variance [ANOVA]). nm indicates nanometers.
We further evaluated the body weight changes between mice injected with WF-0 or WF-3 tumor cells and the correlations between body weight and mesothelin level. As shown in Figure 9A, the body weights were significantly heavier in the WF-3-injected mice than those in the WF-0-injected mice, particularly 15 days after tumor injection. We also noted that the mesothelin levels, in both serum and ascites fluids, correlated with the body weights of WF-3-challenged mice more obviously when tumor-challenged mice weigh more than 25 g (Fig. 9B). Taken together, our data suggest that mesothelin can be detected in the serum and ascites fluid of WF-3-challenged mice and that it may be used to reflect the tumor burden and the severity of intraperitoneal diseases of WF-3-challenged mice.
FIGURE 9.

Characterization of body weights and mesothelin level in serum and ascites after WF-3 tumor cell challenge. Mice were challenged with WF-3 tumor or 1× phosphate-buffered saline (PBS). The body weight of the challenged mice was followed over time. Furthermore, the serum and ascites mesothelin levels of the WF-3 tumor-challenged mice were characterized by enzyme-linked immunoadsorbent assay (ELISA). (A) Body weight measurements after WF-3 tumor (open circle) or PBS (closed circle) challenge. Note that the WF-3 tumor-challenged mice gained significant weight compared with PBS-challenged mice starting 15 days after challenge. (B) Correlations between body weights and mesothelin levels in serum (closed circle) or ascites (open circle) of WF-3-injected mice. Note that the body weights appear to correlate with the serum or ascitic mesothelin levels in WF-3 tumor-challenged mice more obviously when the mice weight is >25 g. SD indicates standard deviation; nm, nanometers.
DISCUSSION
We created a mesothelin-expressing ascitogenic malignant tumor model that demonstrates morphological features of intraperitoneal tumorigenesis. The tumor model (WF-3) also demonstrated relatively high proliferation and migration rates compared with the parental cell line (WF-0). Our tumor model was also capable of implanting into multiple organs including the diaphragm, intestines, and peritoneal wall. Furthermore, the tumor expressed high levels of mesothelin, commonly observed in intraperitoneal tumors such as malignant tumors and ovarian cancers. In addition, we found that the serum and ascites mesothelin levels correlated with tumor loads in tumor-challenged mice.
The current ascitogenic malignant tumor model serves as an excellent preclinical model for investigating the malignant progression of the intraperitoneal tumors because the availability of both the parental cell line (WF-0) and the cell lines from different stages of the malignant progression (WF-1, -2, -3). Using the microarray analysis, we obtained several candidate genes that are up-regulated in the later stages of the tumor progression (WF-3). With the availability of the cell lines at different stages of malignant progression, we will be able to test the contribution of candidate genes to the malignant progression using functional assays such as migration assays, proliferation assays, and tumorigenicity assays. We will be able to introduce the candidate gene(s) into WF-0 and compare the function phenotype (such as migration, proliferation, and tumorigenicity) with WF-0 cells transduced with control vector. Alternatively, we can use RNA interference technology to knockdown the candidate gene(s) in WF-3 tumor cells and compare them with the WF-3 without candidate gene knockdown. Thus, the current model serves as a potentially excellent preclinical model for us to test the clinical relevant candidate gene(s) that are important for tumor progression of intraperitoneal tumors such as malignant mesothelioma and ovarian cancer tumors.
The mesothelin-expressing WF-3 tumor model also serves as a suitable preclinical model for developing mesothelin-specific cancer immunotherapy.3,4,13 It is now clear that the majority of ovarian cancer, malignant mesothelioma, and pancreatic cancer express high levels of mesothelin.14,15,20,21 Furthermore, low levels or no expression of mesothelin was identified in normal tissues.15 Thus, cancer immunotherapy targeting mesothelin may potentially lead to control of mesothelin-expressing malignant tumors without damaging normal tissue. Because advanced-stage ovarian cancer, mesothioma, and pancreatic cancer represent very serious illnesses and current therapies such as surgery, chemotherapy, and radiotherapy usually fail to control these diseases,3,4,22–24 the development of cancer immunotherapy represents a plausible alternative approach to control such dreadful diseases. The availability of a WF-3 preclinical model in immunocompetent mice serves as an important foundation for the development of effective cancer vaccines and immunotherapeutic strategies for these peritoneal malignant tumors.
In the current study, we observed that the level of mesothelin expressed in the serum and ascites fluid reflected the level of tumor burden in tumor challenged mice. Hassan et al.13 reported that the serum mesothelin levels also correlated with the tumor loads in patients with ovarian cancer and malignant mesothelioma. They found that there is a significant reduction in mesothelin serum levels after surgery of malignant peritoneal mesothelioma and advanced ovarian cancer. Therefore, the WF-3 tumor model serves as an excellent preclinical tumor model to study clinically relevant issues, such as screening and evaluating the effectiveness of therapeutic intervention by monitoring mesothelin levels.
Acknowledgments
Supported by grants from the National Science Committee of Taiwan (NSC 90-2314-B-002-157 and 91-2314-B-002-315) and by ovarian cancer grants from the Alliance for Cancer Gene Therapy (ACGT) and the NCDGG (1U19 CA113341-01).
References
- 1.Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA Cancer J Clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- 2.Schink JC. Current initial therapy of stage III and IV ovarian cancer: challenges for managed care. Semin Oncol. 1999;26:2–7. [PubMed] [Google Scholar]
- 3.Zellos LS, Sugarbaker DJ. Multimodality treatment of diffuse malignant pleural mesothelioma. Semin Oncol. 2002;29:41–50. doi: 10.1053/sonc.2002.30230. [DOI] [PubMed] [Google Scholar]
- 4.Nowak AK, Lake RA, Kindler HL, Robinson BW. New approaches for mesothelioma: biologics, vaccines, gene therapy, and other novel agents. Semin Oncol. 2002;29:82–96. doi: 10.1053/sonc.2002.30234. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Yang G, Thompson-Lanza JA, et al. A genetically defined model for human ovarian cancer. Cancer Res. 2004;64:1655–1663. doi: 10.1158/0008-5472.can-03-3380. [DOI] [PubMed] [Google Scholar]
- 6.Lichtenbeld HC, Ferarra N, Jain RK, Munn LL. Effect of local anti-VEGF antibody treatment on tumor microvessel permeability. Microvasc Res. 1999;57:357–362. doi: 10.1006/mvre.1998.2140. [DOI] [PubMed] [Google Scholar]
- 7.Rancourt C, Rogers BE, Sosnowski BA, et al. Basic fibroblast growth factor enhancement of adenovirus-mediated delivery of the herpes simplex virus thymidine kinase gene results in augmented therapeutic benefit in a murine model of ovarian cancer. Clin Cancer Res. 1998;4:2455–2461. [PubMed] [Google Scholar]
- 8.Mesiano S, Ferrara N, Jaffe RB. Role of vascular endothelial growth factor in ovarian cancer: inhibition of ascites formation by immunoneutralization. Am J Pathol. 1998;153:1249–1256. doi: 10.1016/S0002-9440(10)65669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roby KF, Taylor CC, Sweetwood JP, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–591. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Yang N, Garcia JR, et al. Generation of a syngeneic mouse model to study the effects of vascular. endothelial growth factor in ovarian carcinoma. Am J Pathol. 2002;161:2295–2309. doi: 10.1016/s0002-9440(10)64505-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conejo-Garcia JR, Benencia F, Courreges MC, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10:950–958. doi: 10.1038/nm1097. [DOI] [PubMed] [Google Scholar]
- 12.Davis MR, Manning LS, Whitaker D, Garlepp MJ, Robinson BW. Establishment of a murine model of malignant mesothelioma. Int J Cancer. 1992;52:881–886. doi: 10.1002/ijc.2910520609. [DOI] [PubMed] [Google Scholar]
- 13.Hassan R, Bera T, Pastan I. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 2004;10:3937–3942. doi: 10.1158/1078-0432.CCR-03-0801. [DOI] [PubMed] [Google Scholar]
- 14.Robinson BW, Creaney J, Lake R, et al. Mesothelin-family proteins and diagnosis of mesothelioma. Lancet. 2003;362:1612–1616. doi: 10.1016/S0140-6736(03)14794-0. [DOI] [PubMed] [Google Scholar]
- 15.Scholler N, Fu N, Yang Y, et al. Soluble member(s) of the mesothelin/megakaryocyte potentiating factor family are detectable in sera from patients with ovarian carcinoma. Proc Natl Acad Sci U S A. 1999;96:11531–11536. doi: 10.1073/pnas.96.20.11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behrens CK, Igney FH, Arnold B, Moller P, Krammer PH. CD95 ligand-expressing tumors are rejected in anti-tumor TCR transgenic perforin knockout mice. J Immunol. 2001;166:3240–3247. doi: 10.4049/jimmunol.166.5.3240. [DOI] [PubMed] [Google Scholar]
- 18.Andoh A, Takaya H, Saotome T, et al. Cytokine regulation of chemokine (IL-8, MCP-1, and RANTES) gene expression in human pancreatic periacinar myofibroblasts. Gastroenterology. 2000;119:211–219. doi: 10.1053/gast.2000.8538. [DOI] [PubMed] [Google Scholar]
- 19.Cheng WF, Hung CF, Chai CY, et al. Enhancement of Sindbis virus self-replicating RNA vaccine potency by linkage of Mycobacterium tuberculosis heat shock protein 70 gene to an antigen gene. J Immunol. 2001;166:6218–6226. doi: 10.4049/jimmunol.166.10.6218. [DOI] [PubMed] [Google Scholar]
- 20.Argani P, Iacobuzio-Donahue C, Ryu B, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE) Clin Cancer Res. 2001;7:3862–3868. [PubMed] [Google Scholar]
- 21.Yen MJ, Hsu CY, Mao TL, et al. Diffuse mesothelin expression correlates with prolonged patient survival in ovarian serous carcinoma. Clin Cancer Res. 2006;12:827–831. doi: 10.1158/1078-0432.CCR-05-1397. [DOI] [PubMed] [Google Scholar]
- 22.Ozols RF. Systemic therapy for ovarian cancer: current status and new treatments. Semin Oncol. 2006;33:S3–11. doi: 10.1053/j.seminoncol.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Bhoola S, Hoskins WJ. Diagnosis and management of epithelial ovarian cancer. Obstet Gynecol. 2006;107:1399–1410. doi: 10.1097/01.AOG.0000220516.34053.48. [DOI] [PubMed] [Google Scholar]
- 24.Pfisterer J, Ledermann JA. Management of platinum-sensitive recurrent ovarian cancer. Semin Oncol. 2006;33:S12–16. doi: 10.1053/j.seminoncol.2006.03.012. [DOI] [PubMed] [Google Scholar]