

Abstract
The brain vasculature forms an immense network such that most neural cells are in contact with a microvessel. Here we tested the hypothesis that endothelia lining these vessels can be harnessed to create a cellular reservoir of enzyme replacement therapy to diseased brain. As a model system, we used animals with central nervous system (CNS) deficits due to lysosomal storage disease (LSD). The basic premise is that recombinant enzyme expressed in, and secreted from, the vascular endothelia will be endocytosed by underlying neurons and glia, decreasing neuropathology. We screened a phage library in vivo to identify peptides that bound the vascular endothelia1 in diseased and wildtype mice. Surprisingly, epitopes binding diseased brain were distinct from those panned from normal brain. Moreover, different epitopes were panned out of different disease models, implying a unique vascular signature imparted by the disease state. Importantly, presentation of these epitopes on the capsid of adeno-associated virus (AAV) expanded the biodistribution of IV-injected AAV from predominantly liver to include the CNS. Peripheral injection of the epitope-modified AAVs expressing the enzymes lacking in LSD mice reconstituted enzyme activity throughout the brain and improved disease phenotypes in two distinct models.
The vasculature of the mammalian brain provides a unique site for delivery of secreted proteins to underlying neural cells, since endothelial cells lining those vessels are in close apposition to most cells. Engineering brain endothelial cells to secrete a protein of interest, however, is problematic since no mechanism has been shown to functionally modify brain endothelia in situ. To address this, we injected a phage-display library intravenously into mice, similar to previous reports1 and subsequently isolated the brain microvasculature along with the bound phage. The isolated phage was then amplified, purified and injected again (referred to as phage panning). Unlike prior work, which was done in normal brain1 we also panned in diseased mice. After five rounds of panning in β-glucuronidase deficient mice, a LSD model of mucopolysaccharidosis type VII (MPS VII), or their normal littermates, DNA sequencing of the recovered phage revealed enrichment of peptide motifs from the initial phage library. Interestingly, the motifs enriched in wildtype mice were distinct from those in MPSVII mice, suggesting a vascular remodeling process in the diseased mice. Also, the motifs we identified in wild type mice were different than the epitope found previously after panning (3 rounds) in normal rats1.
In wildtype mouse brains, the peptide motifs, PxxPS, SPxxP, TLH and QSxY were identified (Fig. 1a). In panning MPS VII mice brain, the motifs LxSS, PFxG and SLxA were identified (Fig. 1b). Intravenous injection of selected, purified phage into naïve mice confirmed significantly improved homing to brain relative to insertless phage or phage representative of the unselected library (Fig. 1c, d).
Figure 1.




In vivo phage display panning to identify peptide motifs with high affinity for cerebral vasculature. (a,b) After in vivo phage display panning, phage with distinct peptide motifs were identified from wildtype (a) and MPS VII (b) mice. (c,d) Purified selected phage were individually injected via tail vein into naïve mice to validate enrichment in brain. Data show phage titers recovered from the cerebral vasculature of heterozygous (c) and MPS VII (d) mice. Data presented as mean ± SEM. One-way ANOVA revealed group (n=3 all groups) as a significant factor for both Het (F2,50 = 20; p<0.0001) and MPS VII (F2,38 = 251.7; p<0.0001) mice. For Het mice, 13 of 15 epitopes were significantly enriched in brain relative to mice injected with the unselected library. (Groups 1–3, p<0.05, Dunnett’s post hoc). For MPS VII mice, 9 of 11 epitopes were significantly enriched in brain relative to mice injected with the unselected library (for all but GSWAPA and SSIAASFS, p<0.05, Dunnett’s post hoc).
To test the utility of the epitope for brain endothelial targeting, sequences encoding them were inserted into the capsid of AAV22(Supplementary methods). Several modified AAVs were generated: AAV with a linker sequence cloned into site 587 (AAV-linker), AAV-TLH (contained the GWTLHNK epitope), AAV-PPS(epitope DSPAHPS), AAV-PFG (epitope WPFYGTP) and AAV-LSS (epitope LPSSLQK) (Supplementary Fig. 1). AAV-WT (no insert) and AAV-RGD (containing RGD epitope) served as additional controls. Four weeks after tail vein injections of AAV-WT, AAV-linker, AAV-RGD, AAV-PPS and AAV-TLH (wildtype mice) or AAV-WT, AAV-PFG and AAV-LSS (MPS VII mice), we assessed viral genomes in tissues. In wildtype mice, AAV-PPS was enriched in brain by several orders of magnitude. AAV-TLH had similarly improved brain targeting but retained liver uptake. In MPS VII mice, AAV-PFG was 35-fold higher in brain relative to liver, and like AAV-PPS had dramatically improved brain tropism relative to AAV-WT. AAV-LSS was also significantly enriched over AAV-WT in MPS VII mice brain, but similar to AAV-TLH, liver transduction was retained (Fig. 2a,b, and Supplementary Tables 1 and 2).
Figure 2.



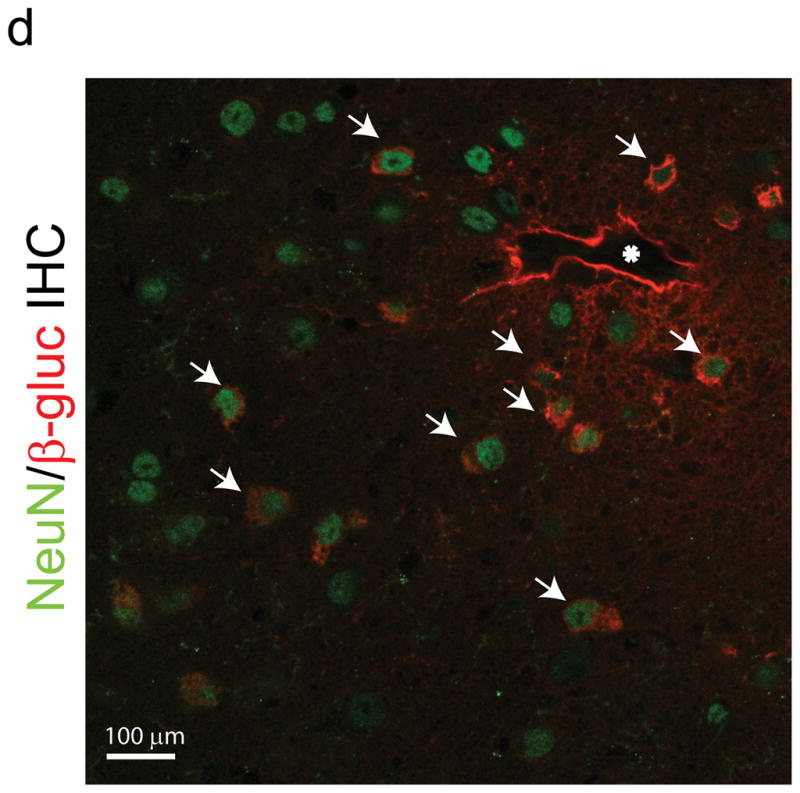
Peptide epitopes expand the tropism of AAV2. (a,b) Viral genomes in brain and liver of wildtype (a) and MPS VII (b) mice as quantified by Q-PCR after peripheral injection. Data are mean ± SEM. (a) AAV-PPS and AAV-TLH are significantly enriched in brain (p<0.001, students t-test) vs. AAV-WT. (b) AAV-PFG and AAV-LSS are significantly enriched in brain over AAV-WT (p<0.001, student’s t-test). (c) AAV-PFG.βgluc transduces the cerebral vasculature of MPS VII mice after tail vein injection. Representative sections were stained for β-glucuronidase activity in situ, which leaves a red precipitate. Representative photomicrographs from sections show that vessels throughout the brain display activity when MPS VII mice (left and middle panels), but not wildtype mice (upper right), were injected. (d) Confocal microscopy of sections stained with anti-β-glucuronidase (red) and anti-NeuN (green) reveals vascular and neuronal β-glucuronidase staining, respectively (denoted by arrows).
The extended tropism of AAV-PPS and AAV-PFG to include brain were confirmed using AAVs expressing the reporter protein eGFP (AAV-PPS.eGFP) or β-glucuronidase (AAV-PFG.βgluc). AAV-PPS.eGFP transduced brain microvessels in wildtype mice (data not shown). We used an in situ enzyme activity stain to assess β-glucuronidase activity after peripheral injection of AAV-PFG.βgluc. This stain is insensitive and requires high intracellular activity. Indeed, wildtype mice show limited endogenous activity using this assay (Fig. 2c, upper right panel). Notably, AAV-PFG.βgluc injection into MPS VII mice revealed overexpression of β-glucuronidase along large and small diameter vessels throughout the brain, in contrast to AAV-PFG.βgluc-injected wildtype mice (Fig 2c). As a result, enzyme activity was elevated in cortex and brain stem (Supplementary Fig. 2). Confocal microscopy of sections immunostained with anti-NeuN to detect neurons and anti-β-glucuronidase showed co-localization of enzyme in the underlying neuropil (Fig. 2d). Similar to AAV-WT, AAVs displaying epitopes panned from wildtype mice (AAV-TLH.βgluc and AAV-PPS.βgluc) did not transduce MPS VII mouse brain (data not shown). Thus, the phage epitopes retained their specificity in the context of the AAVs.
The region used for epitope insertion is important for AAV binding to its major receptor, heparan sulfate proteoglycan (HSPG), and insertion of peptides at this site often alters HSPG binding without compromising virus viability1,3,4. Our findings were similar. While the modified capsid proteins packaged AAV vector genomes with genomic titers comparable to those of wildtype virus (Supplementary Fig. 1), the AAVs did not retain HSPG binding (Supplementary Fig. 3).
We next tested the hypothesis that brain microvasculature endothelia can secrete recombinant enzymes sufficient to correct CNS deficits in the MPSV VII mice model. The β-glucuronidase-deficient mouse exhibits hallmarks of MPS VII disease including lysosomal storage and neurological dysfunction, and is a proven model for investigating novel therapies for lysosomal storage disorders5. AAV-PFG or AAV-WT expressing β-glucuronidase was injected via tail vein in the MPS VII mice at 6 weeks of age because disease onset and lysosomal storage deposits are apparent at this time. Four weeks later, we evaluated lysosomal storage and cellular distension in the brain. MPS VII mice treated with AAV-PFG.βgluc exhibited reduced levels of lysosomal storage relative to mice receiving AAV-WT.βgluc. Correction was noted in the olfactory bulb, cerebral cortex, hippocampus, striatum, and cerebellum (Fig. 3a). AAV-PFG.βgluc, but not AAV-WT.βgluc, significantly reduced the numbers of cells laden with storage vacuoles (Fig. 3b, p<0.001). Moreover, cellular dysmorphology improved after AAV-PFG.β-gluc treatment in contrast to control treated animals. The correction of pathology in multiple cell types, along with co-localization of enzyme in neurons, suggests that β-glucuronidase was secreted basolaterally by endothelial cells and subsequently cross-corrected adjacent neural cells. The correction of neuropathology in multiple structures throughout the entire rostral-caudal extent of the brain indicates broadly disseminated enzyme.
Figure 3.




Intravenous delivery of epitope-modified virus improves neuropathology in MPS VII mice. (a) Representative sections of cerebral cortex, hippocampus, striatum and cerebellum of MPS VII mice harvested after tail vein injection with either AAV-WT.βluc (left panels) or AAV-PFG.βluc (right panels) expressing β-glucuronidase. Yellow asterisk, denotes region magnified in inset (lower right, all panels) for better visualization of storage vacuoles. Arrows in inset point to vacuoles. Scale bar = 50 μm for all panels. (b) Quantitation of vacuolar storage in various brain regions. Tail vein injection of AAV-PFG.βluc but not AAV-WT.βluc significantly reduced lysosomal storage vacuoles in hippocampus, cortex and striatum (*p<0.001, Tukeys post hoc). (c) Binding of AAV-PFG to cerebral vasculature requires chondroitin sulfate. Purified brain microvessels from heterozygous or MPS VII mice were incubated with the reagents indicated, and bound viral particles quantified. Data presented as mean ± SEM. (*,p<0.001, Dunnett’s post hoc). (d) Binding of AAV-PFG to purified brain vasculature from MPS VII mice in the presence or absence of chondroitin sulfate. Data presented as mean ± SEM. *<0.01, student’s t-test.
β-Glucuronidase catalyzes the degradation of glycosaminoglycans (GAG’s) including heparan sulfate and chondroitin sulfate. In the disease state, catabolism of these molecules is blocked and causes lysosomal accumulation. We hypothesized that epitope-modified AAV might interact with GAG-containing glycoproteins accumulating on cell surfaces. Work with purified viral capsids indicate that endothelial cell binding of AAV-PFG is probably not mediated by heparan sulfate (Supplementary Fig. 3). To test a putative role for chondroitin sulfate, we measured the ability of AAV-PFG to bind to brain vasculature from MPS VII mice in the presence and absence of the enzyme chondroitinase ABC. Enzymatic treatment of the vasculature from MPS VII mice abolished AAV-PFG binding (Fig. 3c). Furthermore, an excess of chondroitin sulfate was sufficient to compete PFG-AAV binding (Fig. 3d). These data suggest that AAV-PFG binding to MPS VII mice brain vascular endothelia is due in part to accumulated chondroitin sulfate.
To investigate the broad utility of the approach, we repeated the in vivo panning in a mouse model of late infantile neuronal ceroid lipofuscinosis (LINCL). This model has mutations in Cln2, lacks expression of the lysosomal enzyme tripeptidyl peptidase I (TPP1), and recapitulates many pathological features of the human disease6,7. After five rounds of in vivo phage panning a single dominant peptide emerged—GMNAFRA. Interestingly, the epitope identified from panning in LINCL mice was distinct from those in wildtype or MPS VII mice, supporting a disease-specific vascular remodeling process. As before, an epitope-modified AAV expressing TPP-1 was produced (AAV-GMN). AAV-WT genomes were predominantly localized to liver after peripheral injection into LINCL mice or heterozygous littermates, (Fig. 4a and Supplementary Fig. 4). In contrast, AAV-GMN viral genomes were found at roughly equivalent levels in liver, cerebral cortex and cerebellum after tail vein injection into LINCL mice (Fig. 4a).
Figure 4.





An epitope panned from TPP-1 deficient mice extends AAV tropism to brain and allows correction of CNS deficits. (a)AAV-GMN vectors were significantly enriched in cortex, cerebellum and brainstem after tail vein injection, in contrast to those same regions harvested from LINCL injected with AAV-WT vectors (*,p<0.005). Vector genomes in spinal cord and liver were similar between the two groups (p=0.12 and 0.61, respectively; student’s t-test). NS, not significant. (b) Six weeks after tail vein injection, enzyme activity was significantly higher in all brain regions from AAV-GMN.TPP1 vs. AAV-WT.TPP1 treated LINCL mice (*, p<0.005; students t-test). (c) AAV-GMN.TPP1 but not AAV-WT.TPP1 improves glial activation in LINCL motor cortex. Representative photomicrographs of heterozygous mice or LINCL mice treated with AAV-WT.TPP1 (upper and middle panels) demonstrate the extent of glial activation in the model at P90. LINCL mice treated with AAV-GMN.TPP1 show dramatically reduced GFAP immunoreactivity. Scale bar = 100 μm. Quantitation using Image J indicates significant effects (*, p<0.001, Tukeys post hoc). Het vs. AAV-GMN-treated LINCL mice were not significantly different. (d) Tail vein injection of AAV-GMN.TPP1 prevents loss of deep cerebellar nuclei, in contrast to AAV-WT.TPP1. Scale bar = 500 μm. (e) AAV-GMN.TPP1, but not AAV-WT.TPP1 significantly improves the tremor phenotype in LINCL mice (* p<0.001, Dunnett’s post hoc). Tail vein injection of AAV-WT.TPP1 had no affect on the tremor phenotype of LINCL mice.
TPP-1 activity was detected in liver (Fig. 4b) and other peripheral tissues 6 weeks post-delivery of AAV-WT.TPP1 into tail veins of mutant or heterozygous mice (Supplementary Fig. 5). There was no detectable expression in brain above background levels (Fig. 4b). AAV-GMN.TPP1, which by viral genome analyses reached brain after tail vein injection (Fig. 4a), normalized TPP-1 activity to heterozygous levels in cerebral cortex, cerebellum, brain stem and spinal cord relative to AAV-WT.TPP1 injected LINCL mice (Fig. 4b). TPP-1 activity in the spinal cord was likely due to transduction elsewhere in the CNS, as vector copies in the spinal cord were low relative to other brain regions. As with AAV-PFG.βluc, AAV-GMN.TPP1 did not increase TPP-1 activity in brain when injected into heterozygous littermates (data not shown). Also, AAV-PFG-βgluc did not transduce LINCL brain (data not shown).
We next tested how peripheral delivery of AAV-GMN.TPP1 or AAV-WT.TPP1 to 8 wk old cln2−/− mice impacted disease readouts. Notable phenotypes in LINCL mice include progressive loss of the deep cerebellar nuclei (DCN), glial activation in the motor cortex as evidenced by enhanced glial fibrillary acidic protein (GFAP) immunoreactivity, and a progressive resting tremor7. Although both AAV-WT.TPP1 and AAV-GMN.TPP1 reconstituted peripheral enzyme activity in LINCL mice (Supplementary Fig. 5) only AAV-GMN.TPP1 improved CNS pathology, (Fig. 4c,d) and prevented the tremor phenotype (Fig. 4e). Together, these data present the CNS vasculature as a novel source for dissemination of therapeutic enzyme to the mammalian brain.
Prior work showed that organ specific epitopes could be isolated following in vivo panning after intravenous injection of phage libraries1,2. However, our identification of different epitopes after phage display panning in brains from wildtype and disease mouse models implies molecular differences in the various vascular beds that were previously unappreciated. In some instances, the epitopes panned from the brain expanded the tropism of the AAV to include the brain (PPS and TLH panned from WT mice, LSS panned from MPS VII mice, and GMN panned from TPP-1 deficient mice). Or, the tropism was shifted from predominately liver targeting to predominantly brain targeting (PFG panned from MPS VII mice). And although AAV.GMN and AAV-WT allowed transduction of peripheral organs, only the former corrected the CNS manifestations of the disease. Our data demonstrate that peripheral delivery of modified AAVs to adult mice can ameliorate brain phenotypes in LSD models.
Because the epitopes emerging from the in vivo panning of the MPS VII brain bound in part to accumulated chondroitin sulfate, they may be generally applicable for retargeting drugs, enzymes or vectors for other neuronopathic MPS. Finally, the implementation of vector libraries generated from sequences inserted into the viral capsid or capsid shuffling3,8,9,10 may provide another source for targeting brain endothelia. Such reagents will be generally useful for studying biological characteristics of this important tissue in situ, and how those properties are altered in disease states.
Materials and Methods
Experimental animals
MPS VII (B6.C-H-2bm1/byBir-gusmps/+) mice and heterozygous controls were obtained from the Jackson Laboratory, and subsequently bred and maintained at the University of Iowa animal facility. TPP1-deficient (LINCL) mice have been described previously6. All animal maintenance conditions and experimental protocols were approved by the University Of Iowa Animal Care and Use Committee.
In vivo biopanning
MPS VII, LINCL or heterozygous littermates (6–8 weeks of age) were each injected through the tail vein with 2 × 1010 PFU of phage from the Ph.D.7 phage display library (New England Biolabs) in 200 μl DMEM (Invitrogen) through the tail vein. After five min, mice were anesthetized and perfused transcardially with DMEM. Brains were extracted, and phage recovered and amplified. The amplified phage were then purified, titered, and re-injected in each of five consecutive rounds of panning. The input phage was kept at 2 × 1010 PFU/mice for each round. After the fifth round of panning, DNA from randomly selected clones was sequenced. To verify that selected phage bound to the brain vasculature, the selected phage from the firth round of panning were amplified individually, purified, and 2 × 1010 PFU injected through the tail vein (n=3 per phage motif). After 5 min, mice were perfused transcardially with DMEM, brains extracted, and the binding phage/mouse recovered and titered. Phage with no insert and the original unselected Ph.D.7 phage display library were used as controls.
In situ enzyme activity assay for β-glucuronidase activity and morphological assays
Mice injected with AAVs (1.0 × 1012 genome particles) were anesthetized and transcardially perfused with ice-cold 2% paraformaldehyde 4 weeks post-injection. Brains were harvested, embedded in OCT compound, and sectioned (16 μm) on a cryostat. For β-glucuronidase activity staining, sections from mice (n=3/group, 10 sections/mouse) were assayed using established methods11. For morphological assays, mice (n=3/group) were transcardially perfused with 2% paraformaldehyde and 2% glutaraldehyde in PBS, then post-fixed in the same fixative at 4°C overnight. Tissues were prepared and analyzed as previously described11. To quantify vacuolar storage, 200 cells per region over 3–4 sections per mouse were counted.
TPP-1 activity assay
LINCL(6–8 weeks ) mice and heterozygous littermates (n=3 per group) were injected intravenously with 1.0× 1012 genome particles of wild type AAV2 or epitope modified AAV2 via the tail vein. Six wks later, mice were transcardially perfused with ice-cold normal saline, and extracted tissues homogenized for enzyme assay using established methods7.
Quantitation of tremor in TPP1-deficient mice
CLN2−/− ( 6–8 weeks ) and age-matched CLN2+/− mice (n=5–8 per group) were injected intravenously with 1.0× 1012 genome particles of wild type AAV2 or epitope modified AAV2 via the tail vein. Six weeks later, mean tremor amplitudes were assessed with a tremor monitor (SD Instruments) using established methods7.
Immunohistochemistry
Mice injected with AAVs (1.0 × 1012 genome particles; n=5–8/group) were anesthetized and transcardially perfused with normal saline followed by 4% paraformaldehyde in normal saline. Brains were then extracted and post-fixed using standard methods. Sections (40 μM thick) were analyzed with goat anti-β-glucuronidase (a gift from Dr. William Sly, St. Louis University, 1:200), mouse anti- NeuN ( Chemicon, 1:500), and rabbit anti-GFAP (Dako, 1:2000). Biotinylated secondary antibody (Jackson, Immuno Research, 1:200) or fluorescent conjugated secondary antibody (InVitrogen, 1:500) were used subsequently. Where appropriate, slides were developed with DAB peroxidase substrate kit (Vector Laboratories).
Analysis of AAV binding
Mouse brain vasculature was isolated by centrifuging crude brain homogenate in 15% Dextran, and further purified by running through 105 μm and 70μm meshes, as previously described12. 50 mg of brain vasculature separated by the 70μm mesh was incubated with PBS alone, PNGase(100U/reaction), or chondroitinase ABC (2U/reaction) at 37°C × 1hr. The reaction was stopped by adding cold PBS and washed 3 times. The treated vasculatures were then incubated with virus (1.0×1011) in 500μl PBS with 0.1%BSA at 4°C × 1hr. After washing, the DNA was isolated and viral genomic particles were analyzed by real time PCR. For competitive binding of AAV-PFG to brain vasculature of MPS VII mice, 50 mg of brain vasculatures were incubated with AAV-PFG (1.0×1011) in the presence or absence of 2mg/ml chondroitin sulfate at 4°C × 1 hr. Bound virus was analyzed by real time PCR.
Statistics
Data were analyzed by paired student’s t-tests where appropriate. For Fig. 3b, one-way ANOVA revealed significant group effects for vacuolar storage in hippocampus (F2,11 = 2129; p<0.0001), cortex (F2,11 = 58.05; p<0.0001) and striatum (F2,11 = 27.62; p<0.0001). For Figs 3c, 4c and 4e one-way ANOVA revealed significant group effects (F2,17 = 103.3; p<0.0001), (F2,21 = 64.45; p<0.0001) and (F2,32 = 19.27; p<0.0001), respectively (StatView software; SAS Institute).
Supplementary Material
Acknowledgments
The authors thank Gumei Liu and Jessica Wilson for technical assistance, and the Central Microscopy Research Facility at the University of Iowa. This work was supported by the NIH (HD33531, NS34568 and DK54759), the Batten Disease Research and Support Association and the Roy J. Carver Trust.
Literature Cited
- 1.Work LM, et al. Vascular bed-targeted in vivo gene delivery using tropism-modified adeno-associated viruses. Mol Ther. 2006;13:683–693. doi: 10.1016/j.ymthe.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 2.Grifman M, et al. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Molecular Therapy. 2001;3(6):964–975. doi: 10.1006/mthe.2001.0345. [DOI] [PubMed] [Google Scholar]
- 3.Muller OJ, et al. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 4.Perabo L, et al. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J Virol. 2006;80:7265–7269. doi: 10.1128/JVI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogler C, et al. A novel model of murine mucopolysaccharidosis type VII due to an intracisternal a particle element transposition into the beta-glucuronidase gene: clinical and pathologic findings. Pediatr Res. 2001;49:342–348. doi: 10.1203/00006450-200103000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Sleat DE, et al. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang M, et al. Intraventricular enzyme replacement improves disease phenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. 2008;16:649–656. doi: 10.1038/mt.2008.9. [DOI] [PubMed] [Google Scholar]
- 8.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 9.Koerber JT, Maheshri N, Kaspar BK, Schaffer DV. Construction of diverse adeno-associated viral libraries for directed evolution of enhanced gene delivery vehicles. Nat Protoc. 2006;1:701–706. doi: 10.1038/nprot.2006.93. [DOI] [PubMed] [Google Scholar]
- 10.Grimm D, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu G, Martins I, Wemmie J, Chiorini J, Davidson B. Functional correction of CNS phenotypes in a lysosomal storage disease model using adeno-associated virus type 4 vectors. J Neurosci. 2005;25:9321–9327. doi: 10.1523/JNEUROSCI.2936-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song L, Pachter JS. Culture of murine brain microvascular endothelial cells that maintain expression and cytoskeletal association of tight junction-associated proteins. In Vitro Cell Dev Biol Anim. 2003;39:313–320. doi: 10.1290/1543-706X(2003)039<0313:COMBME>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.