

Abstract
Recently, M3-muscarinic receptor (M3R) has been identified as the bona fide receptor responsible for the cholinergic regulation of glucose-induced insulin release. The molecular mechanisms of such regulation have also begun to be unravelled. These include the conventional G protein-dependent pathways involving calcium mobilization and activation of protein kinase C. In addition, recent studies also provided evidence for G protein-independent pathways in the regulation of insulin secretion by M3R. These include phosphorylation/arrestin-dependent activation of protein kinase D1, Src family kinase-dependent activation of the sodium channel NALCN and the involvement of regulator of G protein signaling (RGS)-4. Time has now come to extend these studies which were done mainly in rodents to human and explore the potential for targeting such pathways at different levels for the treatment of diseases with impaired insulin secretion such as type II diabetes.
Key words: M3-muscarinic receptor, insulin secretion, glucose homeostasis, protein kinase D1, ankyrin-B, RGS4, NALCN, biased ligands
Insulin secretion by β cells of the islets of Langerhans in the pancreas is a process tightly regulated by glucose and other circulating nutrients. It is also modulated by many other factors, including hormones and neurotransmitters. One of the most prominent of these regulatory mechanisms is mediated by acetycholine (Ach) originating from the parasympathetic cholinergic input.1,2 Although cholinergic regulation of insulin release has been known for many years, the mechanism of regulation and in particular the identity of the subtype of cholinergic receptor responsible for this regulation has only recently been established. There are five cholingeric muscarinic receptor subtypes (M1–M5) and the work of Gautam and colleagues using transgenic and gene knockout technology have determined that the M3-muscarinic receptor (M3R) is the bona fide acetylcholine receptor that is responsible for enhancing glucose-dependent insulin release in β cells.3
Molecular Mechanisms for M3R-Mediated Insulin Secretion
The mechanism by which M3R regulates insulin release was thought to be primarily via G-protein depending signaling to the calcium and PKC pathways. As a prototypical Gq/11-coupled receptor, activation of M3R induces the hydrolysis of membrane phospholipid phosphatidylinositol-4,5-biphosphate (PIP2), catalyzed by phospholipase C (PLC). This generates two second messengers, inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 in turn mobilizes calcium from the IP3-sensitive stores while DAG activates PKC. Both of these pathways have been thought to play an important role in M3R-mediated insulin secretion.1
However, recent molecular and genetic studies have pointed to further mechanisms. In particular, sustained insulin release associated with the enteric phase appears to be mediated by a process that is independent of G-protein signaling. This is evidenced by studies from our laboratory and others which demonstrated that protein kinase D1 (PKD1) as one of the key components by which M3R regulates glucose-dependent insulin release.4,5 PKD1 is activated by the phosphorylated form of the M3R via a G-protein-independent, β-arrestin-dependent process that results in secretary vesicle priming.4 In addition, M3R has also been shown to stimulate insulin release by inhibiting the mitogen activated protein kinase p38δ activity, which has inhibitory effects on PKD1 in β cells.5
Other studies have, however, returned to the importance of calcium signaling. Healy and colleagues demonstrated that the expression level of IP3-receptors in β cells is crucial for M3R-mediated insulin release.6 Binding of the adaptor protein ankyrin-B to the IP3-receptors in β cells stabilizes the receptors and thus enhances the calcium signal in the cells.6 Pancreatic islets from heterozygous ankyrin-B mutant (ankB+/−) mice exhibited a reduction in both basal and carbachol-stimulated intracellular calcium release,6 suggesting that the IP3-receptor is stabilized in the open state.
In addition, the sodium channel designated NALCN, which is short for sodium leak channel non-selective, has also been demonstrated to play a role in M3R-mediated insulin release.7 This channel, formerly named Rb21 then VGCNL1, belongs to the four domain ion channel family. M3R has been shown to activate this channel in the model pancreatic β cell line, MIN-6, via the Src family of tyrosine kinases (SFKs).7 In addition, one more piece of the jigsaw puzzle is the regulators of G-protein signaling protein, RGS4. Ruiz de Azua and colleagues have demonstrated that RGS4 negatively modulate M3R-mediated insulin secretion in β cells due to its selective inhibition of M3R signaling in β cells.8
Hence, there appears to be a number of possible in vivo mechanisms that act in concert to regulate the early and late phases of insulin secretion by M3R (Fig. 1).
Figure 1.
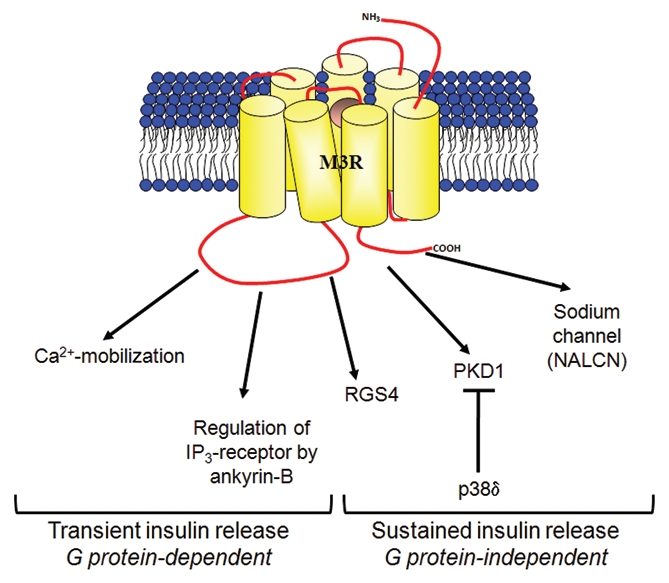
The possible mechanisms of M3-muscarinic receptor (M3R)-mediated insulin secretion. Conventional studies have provided evidence that the transient, early phase of insulin secretion is mediated by M3R via G protein-dependent signaling that results in increase in intracellular calcium and activation of protein kinase C.1 Recent studies have shown that the sustained, late phase of insulin secretion is enhanced by M3R via G protein-independent pathways which requires receptor phosphorylation/arrestin-dependent signaling to PKD1 4 and Src family tyrosine kinases signaling to sodium channel NALCN.7
Physiological Implication of M3R-Mediated Insulin Secretion
Though the cellular mechanisms by which the M3R mediates insulin release have only recently been elucidated, the in vivo effects of muscarinic receptor agonists in regulating glucose-stimulated insulin secretion have been first examined over a decade ago.9 Ahren and colleagues demonstrated that the non-specific muscarinic receptor agonists, carbachol, when administered intravenously, not only potentiated glucose-stimulated insulin secretion in mice fed with either a control or a high-fat diet, but also normalized glucose-stimulated insulin secretion and glucose tolerance in mice subjected to a high-fat diet.9 This study led to the proposal that the development of islet-specific muscarinic agonists, with lesser general muscarinic activity, might be a feasible target to improve insulin secretion in type II diabetes.9
This proposal has only been put to vigorous investigation in recent years. After identifying M3R as the bona fide acetylcholine receptor that is responsible for enhancing glucose-dependent insulin release, Gautam and colleagues performed a series of studies which concluded with the generation of transgenic mice that express a constitutively active mutant of M3R specifically in pancreatic β cells in order to mimick the effects of a drug that chronically activates β cell M3Rs.10 These mutant mice exhibited markedly improved glucose tolerance and increased serum insulin levels as well as resistance to diet-induced glucose intolerance and hyperglycemia.10 These studies strongly supported the hypothesis that chronic, sustained activation of β cell M3Rs might produce beneficial effects to glucose homeostasis, and further established the therapeutic potential for M3R selective agonists for the treatment of type II diabetes. However, due to the multiple peripheral actions of M3Rs such as smooth muscle contraction and saliva secretion, the use of such agonists may be limited by their possible side effects.
Clinical Targeting of M3R Signaling in Insulin Secretion in Disease
The notion of targeting M3Rs in disorders where insulin secretion is de-regulated such as type II diabetes runs into the difficulty of developing ligands that are selective to the M3R subtype and that do not also interact with the other muscarinic receptor subtypes, namely M1, M2, M4 and M5. The problems associated with developing subtype selective ligands centre on the fact that the acetylcholine binding site on muscarinic receptors is very conserved between the five receptor subtypes. However, hope for subtype-selective targeting of the muscarinic receptors has recently emerged with the discovery of ligands that interact with allosteric sites on the receptors.11,12 These so called allosteric modulators target variant regions of the receptor and therefore can show subtype selectivity. With the use of positive allosteric modulators (PAMs), which increase the affinity and/or efficacy of acetylcholine to the M3R, the effects of the natural ligand acetylcholine can be enhanced selectively at the M3R.
In addition to allosteric modulators the discovery that the M3R can potentiate insulin release through an arrestin-dependent mechanism suggests that biased-agonists that direct muscarinic receptor signaling to arrestin-dependent pathways would be of therapeutic benefit.4 The effectiveness of such biased-agonists has been demonstrated in the case of the β-adrenoceptor agonist carvedilol for the treatment of heart disease. The therapeutic efficacy of carvedilol in the treatment of heart disease has been attributable to the fact that this ligand can direct signaling of the β-adrenoceptor to the arrestin-dependent pathways.13 Similar biased ligands that direct signaling of the M3R via arrestin signaling would be expected to potentiate insulin release at β-islets in a manner that results in reduced side effects since G-protein-dependent calcium signaling would be minimized.
It is also possible to target the signaling proteins downstream of M3Rs. In particular, PKD1 and ankyrin-B are two potentially promising targets as evidenced by both in vitro and in vivo data. Though homozygous PKD1-knockout (PKD1−/−) mice are embryonic lethal,14 deletion of PKD1 in INS1 insulinoma cells completely abolished the insulin release induced by glucose and carbachol.5 siRNA-knockdown of PKD1 in mouse islets also impaired the M3R-mediated augmentation of glucose-induced insulin secretion.4 Carbachol augmentation of glucose-induced insulin secretion was significantly impaired in islets prepared from ankB+/− mice or in rat islets following siRNA-knockdown of ankyrin-B.6 In addition, ankB+/− mice exhibited hyperglycemia after oral ingestion of glucose and the R1788W mutation of ankyrin-B impaired its function in islets and is associated with type II diabetes in Caucasians and Hispanics.6 These studies demonstrate that targeting the signaling pathways downstream of the M3R can effectively modulate insulin release.
Concluding Remarks
It is now an exciting time to put these hypotheses to test and devise further therapeutic treatments for type II diabetes. Though much is now known about the M3R-mediated insulin secretion pathway, much still needs to be done.
The next step forward is to determine the effectiveness of β cell M3Rs and/or downstream signaling components as drug targets for the treatment of type II diabetes and in particular it will be important to extend the studies that have been largely conducted in rodent models to humans.
References
- 1.Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr Rev. 2001;22:565–604. doi: 10.1210/edrv.22.5.0440. [DOI] [PubMed] [Google Scholar]
- 2.Malaisse WJ. Stimulus-secretion coupling in the pancreatic B-cell: the cholinergic pathway for insulin release. Diabetes Metab Rev. 1986;2:243–259. doi: 10.1002/dmr.5610020303. [DOI] [PubMed] [Google Scholar]
- 3.Gautam D, Han SJ, Hamdan FF, Jeon J, Li B, Li JH, et al. A critical role for beta cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo. Cell Metab. 2006;3:449–461. doi: 10.1016/j.cmet.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 4.Kong KC, Butcher AJ, McWilliams P, Jones D, Wess J, Hamdan FF, et al. M3-muscarinic receptor promotes insulin release via receptor phosphorylation/arrestin-dependent activation of protein kinase D1. Proc Natl Acad Sci USA. 2010;107:21181–21186. doi: 10.1073/pnas.1011651107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumara G, Formentini I, Collins S, Sumara I, Windak R, Bodenmiller B, et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell. 2009;136:235–248. doi: 10.1016/j.cell.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Healy JA, Nilsson KR, Hohmeier HE, Berglund J, Davis J, Hoffman J, et al. Cholinergic augmentation of insulin release requires ankyrin-B. Sci Signal. 2010;3:19. doi: 10.1126/scisignal.2000771. [DOI] [PubMed] [Google Scholar]
- 7.Swayne LA, Mezghrani A, Varrault A, Chemin J, Bertrand G, Dalle S, et al. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic beta-cell line. EMBO Rep. 2009;10:873–880. doi: 10.1038/embor.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruiz de Azua I, Scarselli M, Rosemond E, Gautam D, Jou W, Gavrilova O, et al. RGS4 is a negative regulator of insulin release from pancreatic beta-cells in vitro and in vivo. Proc Natl Acad Sci USA. 2010;107:7999–8004. doi: 10.1073/pnas.1003655107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahren B, Sauerberg P, Thomsen C. Increased insulin secretion and normalization of glucose tolerance by cholinergic agonism in high fat-fed mice. Am J Physiol. 1999;277:93–102. doi: 10.1152/ajpendo.1999.277.1.E93. [DOI] [PubMed] [Google Scholar]
- 10.Gautam D, Ruiz de Azua I, Li JH, Guettier JM, Heard T, Cui Y, et al. Beneficial metabolic effects caused by persistent activation of beta-cell M3 muscarinic acetylcholine receptors in transgenic mice. Endocrinology. 2010;151:5185–5194. doi: 10.1210/en.2010-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 13.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fielitz J, Kim MS, Shelton JM, Qi X, Hill JA, Richardson JA, et al. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci USA. 2008;105:3059–3063. doi: 10.1073/pnas.0712265105. [DOI] [PMC free article] [PubMed] [Google Scholar]