

Abstract
Conventionally, C—H oxidation reactions are used to install functional groups. The use of C—H oxidation to transform simple starting materials into highly versatile intermediates, which enable rapid access to a range of complex target structures is a new area with tremendous potential in synthesis. Herein we report a Pd(II)/sulfoxide-catalyzed allylic C—H oxidation to form anti-1,4-dioxan-2-ones from homoallylic oxygenates. These versatile building blocks are rapidly elaborated to differentiated syn-1,2-diols, stereodefined amino-polyols, and syn-pyrans, structures ubiquitous in medicinally important complex molecules found in Nature. We also demonstrate that a C—H oxidation approach to the synthesis of these motifs is orthogonal and complementary to other state-of-the-art methods.
Introduction
Selective C—H oxidation reactions have emerged as powerful methods for rapidly introducing functional groups onto preformed carbon skeletons,1 often enabling a significant streamlining of the synthesis of complex molecules.2 Tremendous untapped opportunity also exists to use C—H oxidation as a means of transforming simple starting materials into versatile synthetic intermediates en route to more complex products.3 Ideally, these pluripotent intermediates could rapidly be transformed into a range of structurally and functionally diverse products of high synthetic value. Herein we describe the realization of this concept for the straightforward synthesis of anti-1,4-dioxan-2-ones from simple olefins using Pd(II)/sulfoxide catalysis. These highly versatile synthetic intermediates are rapidly elaborated to varied motifs prevalent in biologically active, oxygenated natural products: differentiated syn-1,2-diol functionality, polyfunctionalized acyclic structures, as well as syn-pyran skeletons.
Design Principles
In addition to being present in some classes of natural products,4 dioxanones are a particularly valuable target because of their potential for high synthetic versatility. Enolization of anti-dioxanones is known to promote skeletal rearrangement with relay of stereochemistry to furnish syn-pyrans.5 Furthermore, the development of a method for selective cleavage of the dioxanone C—O bonds (ethereal vs. acyl) would enable efficient installation of differentially protected syn-1,2-diol functionality.6 In considering C—H activation platforms to access dioxanones, Pd(II)/sulfoxide catalysis presents unique opportunties for efficiency and selectivity.7–9 Given our success in using tethered nucleophiles for diastereoselective allylic C—H aminations,8a, d we postulated that an intramolecular process would provide rapid access to stereochemically-defined 1,4-dioxan-2-ones via allylic C—H oxidation of readily available chiral homoallylic alcohols with a carboxylic acid tether. Moreover, given the proven levels of broad functional group tolerance and high chemoselectivity for α-olefins versus more electron rich π-systems, a palladium(II)/sulfoxide catalyzed allylic C—H oxidation reaction would provide complementary reactivity to other olefin oxidation methods. The terminal olefin in these products may also be utilized for iterative C—H oxidation sequences, providing a new approach for retrosynthetic simplification of polyfunctionalized targets.10 The ability to selectively transform simple homoallylic oxygenates to differentiated diols, polyfunctionalized acyclic structures, or stereochemically defined pyrans presents a unique opportunity to use C—H oxidation as a means of rapidly constructing diverse and biologically privileged oxygenated motifs.
Results and Discussion
Reaction Development
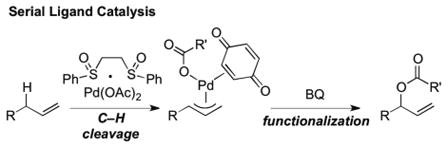
Despite the success of using carboxylic acid nucleophiles for intermolecular allylic C—H esterifications with a wide range of substrates,7 we were surprised to find that substrates having oxygenation in the homoallylic position afforded only poor yields of 1,2-dioxygenation products with poor regioselectivities (Table 1, linear(L):branched(B) ratio). This low reactivity persists even when p-nitrobenzoic acid (previously shown to be the most effective nucleophile in intermolecular C—H esterifications) is used as a functionalization reagent and is unaffected by changing the nature of the homoallylic ether substituent (Table 1, R = CH2CO2Me or Bn). This result stands in stark contrast to the same reaction with alkyl homoallylic substitution, which proceeds in good yield (73%) and outstanding regioselectivity (>20:1 B:L) favoring the branched product, albeit with no diastereoselectivity.2a Intermolecular C—H esterification is thought to proceed via a serial ligand catalysis mechanism involving Pd(II)/sulfoxide promoted allylic C—H cleavage to generate a π-allylPd-carboxylate followed by a benzoquinone (BQ) mediated C—O bond forming event from within the sphere of the metal (eq. 1).7b,11 We hypothesize that homoallylic oxygen substituents may chelate to the palladium and occupy a requisite site for inner-sphere functionalization thereby forcing functionalization to proceed via an outer-sphere pathway. In support of this, the large amount of linear regioisomer formed (Table 1) is consistent with an outer-sphere, non-BQ dependent functionalization pathway.8b, c,12
Table 1.
Intermolecular C—H Esterification with Homoallylic Oxygen Substitution
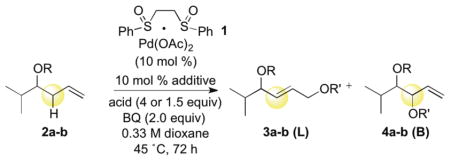 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R | R′a | additive | isolated yieldb | L:Bc 3/4 | E:Z (3)d | dr (4)e |
| 1 | CH2CO2Me | Ac | none | 11 | 1:2 | >20:1 | 16:1 |
| 2 | CH2CO2Me | Ac | Cr(salen)Cl | 10 | >20:1 | >20:1 | N/A |
| 3 | CH2CO2Me | p-NO2Bz | none | 16 | 1:2 | nd | >20:1 |
| 4 | CH2CO2Me | p-NO2Bz | Cr(salen)Cl | >5 | nd | nd | nd |
| 5 | Bn | p-NO2Bz | none | >5 | nd | nd | nd |
AcOH (4.0 equiv) or p-NO2BzOH (1.5 equiv) used as the acid nucleophile.
Average of 2 runs at 0.5 mmol.
Linear(L):Branched(B) ratio determined by GC of the crude reaction after workup.
E:Z ratio determined by GC of the crude reaction after workup.
Diastereomeric ratio determined by GC of the crude reaction after workup. BQ = p-benzoquinone, salen = 1,2-cyclohexanediamine-N, N′-bis(3,5-di-tert-butylsalicylidine).
 |
(1) |
We hypothesized that by tethering the carboxylic acid nucleophile to the homoallylic oxygen substituent, a productive Pd-carboxylate chelate would be formed, which upon BQ activation, would lead to the desired C—O bond formation. In addition to overcoming reactivity and selectivity issues inherent to the intermolecular process, we considered that such an intramolecular process would also expand the synthetic utility of this method beyond formation of 1,2-dioxygenation patterns. We therefore examined the reaction of 2-((2-methylhex-5-en-3-yl)oxy)acetic acid 5a, containing a carboxylic tether, which could fulfill these goals (Table 2). Under standard allylic C—H oxidation conditions,7b dioxanone 6a was produced in improved yield (38%), compared to intermolecular oxidation, and good diastereoselectivity favoring the anti-stereoisomer (Table 2, entry 1). We next examined diisopropylethylamine (DIPEA) and Cr(salen)Cl as catalytic additives due to their known ability to promote functionalization of carboxylic acid and carbamate nucleophiles without attenuating C—H cleavage.8b, c,13 While addition of 10 mol % DIPEA provided only a modest increase in yield (entry 2), 10 mol % Cr(salen)Cl markedly improved the yield to 83% with identical stereoselectivity (entry 3).14 Interestingly, Cr(salen)Cl did not improve reactivity in the intermolecular C—H esterification system (Table 1, entries 2 and 4). We also performed a series of experiments to probe if this reaction, like the intermolecular version,7b proceeds via a serial ligand catalysis mechanism. Use of 2,6-dimethyl-1,4-benzoquinone (2,6-Me2BQ), a sterically encumbered quinone, which does not coordinate well to the π-allylPd intermediate, failed to produce the dioxanone (Table 2, entry 4). Moreover, Pd(OAc)2 in the absence of bis-sulfoxide ligand also failed to furnish significant quantities of the desired product (entry 5). Finally, Cr and oxidant alone fail to promote the reaction (entry 6).15 These experimental findings are consistent with the restoration of a serial ligand catalysis mechanism in the intramolecular C—H esterification reaction where both sulfoxide and quinone ligands working sequentially with the palladium are required to furnish the C—H oxidation product.
Table 2.
Optimization of the Intramolecular C—H Oxidation
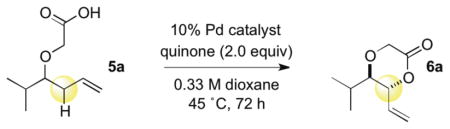 | |||||
|---|---|---|---|---|---|
| entry | additive | quinone | Pd catalyst | isolated yield | dr (anti:syn)a |
| 1 | none | BQ | 1 | 38 | 9:1 |
| 2 | DIPEA | BQ | 1 | 46 | 7:1 |
| 3 | Cr(salen)Cl | BQ | 1 | 83 | 9:1 |
| 4 | none | 2,6-Me2BQ | 1 | 0 | nd |
| 5 | none | BQ | Pd(OAc)2 | >5 | nd |
| 6 | Cr(salen)Cl | BQ | none | 0 | nd |
Diastereomeric ratio determined by 1H NMR of the crude reaction after workup. DIPEA = diisopropylethylamine.
Scope
We next evaluated a series of substrates to examine the effects of neighboring substituents (R group, Table 3) on reactivity and selectivity. Proximal aryl moieties are well tolerated (entry 2). Reactions with unbranched substrates proceeded in good yield, but diminished stereoselectivity (entry 3); whereas a substrate with a bulky quaternary alkyl substituent gave poor yields, but excellent diastereoselectivity (entry 4). Despite the apparent sensitivity to sterics, tertiary centers are well tolerated (entries 5 and 6). Notably, competing methods for forming these diol products would require setting the geometry of a tri-substituted olefin.16 Additionally, α-stereocenters influence the magnitude of the diastereoselectivity (entries 7 and 8), but the overall diastereomeric outcome of the reaction favoring the anti-dioxanone is relayed exclusively from the stereocenter bearing the carboxylic acid tether. Significantly, these reactions feature a high level of operational simplicity. The reactions are run with no precautions taken to exclude O2 or water and the analytical scales directly translate to preparative scales with no diminishment in yield or selectivity (10 mmol scale, 80% yield, entry 1). Moreover, the major anti-dioxanone diastereomer can generally be isolated in pure form using standard chromatography techniques.
Table 3.
Scope of the Intramolecular C—H Oxidation
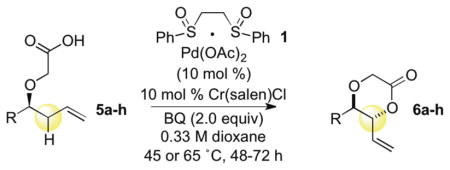 | ||||
|---|---|---|---|---|
| entry | R | product | isolated yielda | dr (anti:syn)b |
| 1 | i-Pr | (−)-6a | 83 (80)c | 9:1d |
| 2 | 4-BrPh | 6b | 57 | 3:1 |
| 3 | n-Pent | 6c | 82 | 2:1 |
| 4 | t-Bu | 6d | 22 | 11:1 |
| 5 |
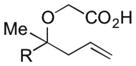
|
6e R=Ph | 57 | 3:1 |
| 6 | 6f R=n-Bu | 60 | 3:1 | |
| 7 |
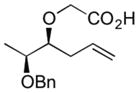
|
(−)-6g | 62 | 8:1 |
| 8 |
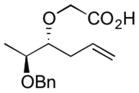
|
(+)-6h | 62 | 4:1 |
Average of 2 runs at 0.3 mmol.
Diastereomeric ratio determined by GC of the crude reaction after workup.
Average of 2 runs at 10 mmol.
Diastereomeric ratio determined by 1H NMR of the crude reaction after workup.
Chemoselectivity
The C—H oxidation approach to these 1,2-dioxygenated motifs also provides orthogonal and complementary selectivity to the Sharpless asymmetric dihydroxylation (SAD).17 When multiple olefins are present in a molecule, SAD generally selects for the more electron rich olefin with variable chemoselectivity. For example, SAD of a terminal diene to generate the vinylic diol products produced from C—H oxidation gives mixtures of regioisomeric diols.17b Consequently, SAD is often not used to install diols when multiple olefins are present and alternate, lengthy routes must be devised, reducing overall synthetic efficiency.18 In contrast, the allylic C—H oxidation reaction gives complete selectivity for the terminal olefin allowing a diol to be installed in the presence of tetra-substituted, tri-substituted, and trans- and cis-di-substituted olefins (Table 4).
Table 4.
Chemoselectivity of the Intramolecular C—H Oxidation
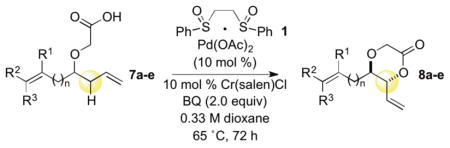 | |||
|---|---|---|---|
| entry | product | isolated yielda | dr (anti:syn)b |
| 1 |
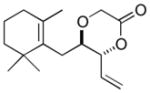 8a 8a
|
74 | 4:1 |
| 2 |
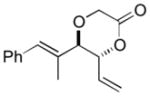 8b 8b
|
77 | 6:1 |
| 3 |
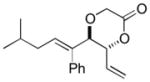 8c 8c
|
53 | 7:1 |
| 4 |
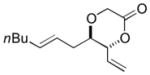 8d 8d
|
72 | 3:1 |
| 5 |
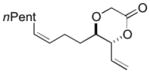 8e 8e
|
52 | 2:1 |
Average of 2 runs at 0.3 mmol.
Diastereomeric ratio determined by GC of the crude reaction after workup.
Differentiated 1,2-Diols
We first sought to use this intramolecular C—H esterification reaction to develop a streamlined route to differentiated, chiral syn-1,2-diols (Scheme 1); compounds, which generally require extensive functional group manipulations to access. In order achieve this goal, we needed to: 1) evaluate the feasibility of generating optically enriched dioxanones using this method, and 2) develop a novel deprotection sequence for converting the chemically inequivalent oxygens in the dioxanone products to differentiated, chiral syn-1,2-diols. Starting from a simple aldehyde, asymmetric allylation affords an enantioenriched homoallylic alcohol. Alkylation with bromoacetic acid followed by C—H oxidation catalyzed by Pd(II)/bis-sulfoxide 1 gives anti-dioxanone (−)-6a with no erosion in enantioenrichment. The anti-dioxanone is rapidly unmasked via base-promoted lactone opening to generate a hydroxyacid, which can be protected to afford (−)-9. The α–alkoxy ester moiety in (−)-9, is then cleaved under mild reducing conditions using SmI219 to furnish a free alcohol, which may be protected as desired. Benzyloxymethyl protection gave differentiated diol (−)-10, a compound that is challenging to obtain via other oxidation methods, like the Sharpless asymmetric dihydroxylation (SAD), which directly afford unprotected syn-1,2-diol motifs.
Scheme 1. Access to Differentiated syn-1,2-Diols.

aConditions: (a) 10% 1, 10% Cr(salen)Cl, BQ (2.0 equiv), dioxane, 45 °C (72% of >20:1 anti-diastereomer; 83%, 9:1 crude dr); (b)(1) LiOH (2.0 equiv), 3:1 THF:H2O, 0 °C, (2) TBSOTf (3.0 equiv), 2,6-lutidine (6.0 equiv), CH2Cl2 0 °C, (3) MeI (3.0 equiv), K2CO3 (3.0 equiv, DMF, RT (89%, 3 steps); (c) SmI2 (3.0 equiv), ethylene glycol (1.2 equiv), THF/HMPA, RT (61%, 12% rsm); (d) BOMCl (1.5 equiv), iPr2NEt (1.75 equiv, CH2Cl2, 0 °C to RT (79%).
Polyoxidized Motifs via Iterative C—H Oxidation
The α-olefin moiety of the differentiated syn-1,2-diol products (e.g. (−)-10) can be used as a functional handle to iterate this and other allylic C—H oxidation processes for the purpose of rapidly constructing densely functionalized chains from simple starting materials (Scheme 2).10 A hydroboration-oxidation sequence affords an intermediate aldehyde that can undergo asymmetric allylation to generate a homoallylic alcohol as one diastereomer. After carbamate formation to yield (−)-11, a masked syn-1,2-amino alcohol moiety can be installed in good yields via Pd(II)/bis-sulfoxide 1 catalyzed allylic C—H amination.8a Amino-polyols like (−)-12, which are generated in optically pure form via this sequence, are found in several classes of natural products,20 such as bengazole A (a potent antifungal agent) and AAL Toxin TA. Traditional approaches to these polyoxidized motifs often rely on chiral relay strategies, making the synthesis of multiple stereoisomeric compounds challenging. This sequence highlights the power of diastereoselective C—H oxidation and amination reactions, when used in combination with powerful reagent controlled alkylation methodology, to facilitate the synthesis of a wide assortment of diastereomers with varied oxygen and nitrogen motifs.
Scheme 2. Iterative C—H Oxidation for the Synthesis of Polyoxidized Motifs.

aConditions: (a) (1) BH3-Me2S (2.4 equiv), THF; 2-methyl-2-butene (4.7 equiv); 7, 0 to 45 °C; 3.0 M NaOH, 30% wt. H2O2, (2) PCC (1.3 equiv), CH2Cl2, RT (74%, 2 steps); (b) (+)-Ipc2B-allyl, Et2O, −78 °C; 3.0 M NaOH, 30% wt. H2O2 (95%); (c) NsNCO (1.5 equiv), THF, RT (90%); (d) 10% 1, 5% l,2-bis(phenylsulfinyl)ethane, PhBQ (1.05 equiv), THF, 45 °C (65%, 1.6:1 crude dr).
Syn-Pyrans
Efficient generation of dioxanones via C—H oxidation additionally allows for rapid structural diversification from topologically simple starting materials. 1,4-dioxan-2-ones undergo stereoselective Ireland-Claisen5d rearrangements to form syn-pyran skeletons.5,21 We present two case studies illustrating the utility and advantages of C—H oxidation for the synthesis of syn-pyrans. First, bifunctional pyrans like (−)-16 are powerful building blocks used in a number of natural product syntheses. For example, dioxanone 14, readily formed via C—H oxidation, undergoes facile rearrangement to give dihydropyran 15 (Scheme 3). Hydrogenation, reduction of the ester, benzylation and desilylation gives (−)-16, an intermediate in the synthesis of SCH 351448.22 Our intramolecular C—H oxidation not only sets a stereocenter selectively, which is relayed to the pyran 2-position, but the tether can also be incorporated into the final product, eliminating extraneous masking or directing groups.
Scheme 3. Synthesis of a Useful Bifunctional Pyran.

aConditions: (a) NaH (3.0 equiv), BrAcOH (1.1 equiv), THF/DMF 0 °C to RT (70%); (b) 10% 1, 10% Cr(salen)Cl, BQ (2.0 equiv), dioxane, 65 °C (83%), 3:1 crude dr, mixture of diastereomers taken forward); (c) (1) LiHMDS (2.0 equiv), 1:1 v:v TMSCl:Et3N, THF, −78 °C then reflux in toluene, (2) MeI (3.0 equiv), K2CO3 (3.0 equiv), DMF, RT (83%); (d) 10% wt. of 5% Pd/C, H2 (1 atm), EtOAc, RT (68% of >20:1 syn-diastereomer, 3:1 crude dr); (e) LiAlH4 (2.0 equiv), THF, 0 °C; (f) BnBr (2.0 equiv), NaH (2.0 equiv), DMF, 0 °C to RT; (g) 3 M HCl, EtOH, RT (74%, 3 steps).
Particularly in the context of complex molecule synthesis, C—H oxidation methods must be effective in the presence of dense functionality. Scheme 4 shows the rapid synthesis of highly oxygenated dioxanone (+)-18 and subsequent rearrangement to dihydropyran (+)-19. Reduction of the ester, benzyl protection and hydrolysis of the acetonide gives diol (+)-20, an intermediate in the synthesis of ent-goniodomin A, a potent antifungal agent.23 This route compares favorably to the reported synthesis of this pyran (7 versus 10 steps from commercial material) and demonstrates the ability of C—H oxidation to enable the rapid construction of densely functionalized pyran skeletons.
Scheme 4. Synthesis of a Densely Functionalized Pyran.

aConditions: (a) NaH (3.0 equiv), BrAcOH (1.1 equiv), THF/DMF 0 °C to RT (54%); (b) 10% 1, 10% Cr(salen)Cl, BQ (2.0 equiv), dioxane, 65 °C (56% of >20:1 dr anti-diastereomer; 73%, 3:1 crude dr); (c) (1) LiHMDS (2.0 equiv), 1:1 v:v TMSCl:Et3N, THF, −78 °C then reflux in toluene, (2) MeI (3.0 equiv), K2CO3 (3.0 equiv), DMF, RT (82%); (d) LiAlH4 (2.0 equiv), THF, 0 °C; (e) BnBr (2.0 equiv), NaH (2.0 equiv), DMF, 0 °C to RT; (f) 1N HCl, THF, RT (60%, 3 steps).
Conclusion
We have developed a novel approach to differentiated polyoxygenated motifs and bifunctional syn-pyrans from homoallylic alcohols using Pd(II)/bis-sulfoxide C—H oxidation catalysis. This work underscores the power of selective C—H oxidation reactions for installing versatile functionality that enables rapid access to functionally and topologically diverse structures from simple starting materials.
Supplementary Material
Figure 1.

Rapid Access to Complex Structures via Allylic C—H Oxidation
Acknowledgments
Financial support was provided by NIH/NIGMS (GM07615). P.E.G. is the recipient of the University of Illinois Synthetic Organic and NSF Graduate Research Fellowships. We thank TCI and Aldrich Chemical Company for generous gifts of commercial bis-sulfoxide/Pd(OAc) catalyst (1). We also thank Mr. Marinus Bigi for checking the experimental procedure in Table 3, entry 1.
Footnotes
Supporting Information. Complete experimental procedures, compound characterization data and NMR spectra for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For examples of C—H activation reactions to introduce oxygen see: Chen MS, White MC. Science. 2010;327:566. doi: 10.1126/science.1183602.Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597.Groves JT, Nemo TE. J Am Chem Soc. 1982;105:6243.Chen K, Que L., Jr J Am Chem Soc. 2001;123:6327. doi: 10.1021/ja010310x.Mello R, Fiorentino M, Fusco C, Curci R. J Am Chem Soc. 1989;111:6749.Brodsky BH, Du Bois J. J Am Chem Soc. 2005;127:15391. doi: 10.1021/ja055549i.Zhou M, Schley ND, Crabtree RH. J Am Chem Soc. 2010;132:12550. doi: 10.1021/ja1058247.Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300. doi: 10.1021/ja031543m.Zhang YH, Yu JQ. J Am Chem Soc. 2009;131:14654. doi: 10.1021/ja907198n.
- 2.For examples of late-state C—H oxidations in total synthesis see: Stang EM, White MC. Nat Chem. 2009;1:547. doi: 10.1038/nchem.351.Hinmann A, DuBois J. J Am Chem Soc. 2003;125:11510.Davies HML, Dai X, Long MS. J Am Chem Soc. 2006;128:2485. doi: 10.1021/ja056877l.Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238. doi: 10.1126/science.1170777.Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claibourne CF, Reynaud J, Couladouros EA, Paulvannan K, Sorenson EJ. Nature. 1994;367:630. doi: 10.1038/367630a0.Wender PA, Jesudason CD, Nakhira H, Tamura N, Tebe AL, Ueno YJ. J Am Chem Soc. 1997;119:12976.
- 3.For an example of C—H activation/Cope rearrangement for forming carbocyclic skeletons see: Davies HML, Jin Q. Proc Nat Acad Sci. 2004;101:5472. doi: 10.1073/pnas.0307556101.Hansen J, Gregg TM, Ovalles SR, Lian Y, Autschbach J, Davies HML. J Am Chem Soc. 2011;133:5076. doi: 10.1021/ja111408v.
- 4.Washida K, Abe N, Sugiyama Y, Hirota A. Biosci Biotechnol Biochem. 2009;73:1355. doi: 10.1271/bbb.90015. [DOI] [PubMed] [Google Scholar]
- 5.Burke SD, Armistead DM, Schoenen FJ, Fevig JM. Tetrahedron. 1986;42:2787.For the dioxanone to pyran approach previously applied to total synthesis see: Burke SD, Sametz GM. Org Lett. 1999;1:71. doi: 10.1021/ol9905506.Burke SD, Piscopio AD, Kort ME, Matulenko MA, Parker MH, Armistead DM, Shankaran K. J Org Chem. 1994;59:332.For the seminal report of the Ireland-Claisen rearrangement see: Ireland RE, Mueller RH. J Am Chem Soc. 1972;94:5897. doi: 10.1021/ja00765a079.
- 6.For an example of the synthesis of differentiated cyclic 1,2-diols see: Lim SM, Hill N, Myers AG. J Am Chm Soc. 2009;131:5763. doi: 10.1021/ja901283q.
- 7.For C—H esterification see: Chen MC, White MC. J Am Chem Soc. 2004;126:1346. doi: 10.1021/ja039107n.Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198.Fraunhoffer KF, Prabagaran N, Sirois LE, White MC. J Am Chem Soc. 2006;128:9032. doi: 10.1021/ja063096r.
- 8.For C—H amination see: Fraunhoffer KJ, White MC. J Am Chem Soc. 2007;129:7274. doi: 10.1021/ja071905g.Reed SA, White MC. J Am Chem Soc. 2008;130:3316. doi: 10.1021/ja710206u.Reed SA, Mazzotti AR, White MC. J Am Chem Soc. 2009;131:11701. doi: 10.1021/ja903939k.Rice GT, White MC. J Am Chem Soc. 2009;131:11707. doi: 10.1021/ja9054959.
- 9.For C—H alkylation see: Young AJ, White MC. J Am Chem Soc. 2008;130:14090. doi: 10.1021/ja806867p.Young AJ, White MC. Angew Chem Int Ed. 2011;50 doi: 10.1002/anie.201101654.Lin S, Song CX, Cai GX, Wang WH, Shi ZJ. J Am Chem Soc. 2008;130:12901. doi: 10.1021/ja803452p.
- 10.For examples of an iterative approach to polyol synthesis see: Zacuto MJ, Leighton JL. J Am Chem Soc. 2000;122:8587.Iwata M, Yazaki R, Suzuki Y, Kumagai N, Shibasaki M. J Am Chem Soc. 2009;131:18244. doi: 10.1021/ja909758e.Menz H, Krisch SF. Org Lett. 2009;11:5634. doi: 10.1021/ol902135v.Hassan A, Lu Y, Krische MJ. Org Lett. 2009;11:3112. doi: 10.1021/ol901136w.
- 11.An analogous intermediate has been proposed for the Pd(OAc)2-catalyzed 1,4-diacetoxylation of 1,3-dienes and stereochemical evidence supports an inner-sphere C—O bond forming process. See: Bäckvall JE, Byström SE, Nordberg RE. J Org Chem. 1984;49:4619.
- 12.For example, intermolecular C—H amination, thought to proceed via outer-sphere C—N bond formation, forms predominantly the linear regioisomer and is tolerant of homoallylic oxygen functionality. See ref. 8b.
- 13.For a detailed exploration of the role of Cr(salen)X in allylic C—H oxidations see: Covell DJ, White MC. Angew Chem, Int Ed. 2008;47:1. doi: 10.1002/anie.200802106.For other applications of Cr(salen)Cl in C—H oxidations see references 8b and 8c and: Qi X, Rice GT, Lall MS, Plummer MS, White MC. Tetrahedron. 2010;66:4816. doi: 10.1016/j.tet.2010.04.064.Wu L, Qiu S, Liu G. Org Lett. 2009;11:2707. doi: 10.1021/ol900941t.
- 14.The use of lower palladium loadings for C—H dioxanone formation led to significantly diminished yields. For a discussion on the need for high catalyst loadings in Pd(II)-catalyzed oxidations see: Steinhoff BA, King AE, Stahl SS. J Org Chem. 2006;71:1861. doi: 10.1021/jo052192s.For recent C—H oxidation processes used in drug discovery that operate at 10 mol% Pd(II) catalyst loadings see: ref. 13b and Dai HX, Stepan AF, Plummer MS, Zhang YH, Yu JQ. J Am Chem Soc. 2011;133:7222. doi: 10.1021/ja201708f.
- 15.For PhI(OAc)2-mediated benzylic C—H lactonizations see: Dohi T, Takenage N, Goto A, Maruyama A, Kita Y. Org Lett. 2007;9:3129. doi: 10.1021/ol071315n.In contrast, BQ and PhI(OAc)2 alone failed to promote this reaction: see Supporting Information S8–9.
- 16.For an example in total synthesis see: Taillier C, Gille B, Bellosta V, Cossy J. J Org Chem. 2005;70:2097. doi: 10.1021/jo048115z.
- 17.(a) Kolb HC, VanNieuwenhze MS, Sharpless BK. Chem Rev. 1994;94:2483. [Google Scholar]; (b) Xu D, Crispino GA, Sharpless KB. J Am Chem Soc. 1992;114:7570. [Google Scholar]
- 18.For an example of installation of a diol motif in the presence of unsaturated functionality see:. Sabitha G, Gopal P, Reddy N, Yadav JS. Tetrahedron Lett. 2009;50:6298.
- 19.For general applications of SmI2 see: Molander GA, Harris CR. Chem Rev. 1996;96:307. doi: 10.1021/cr950019y.For cleavage of α-alkoxy esters see: Kasuda K, Inanaga J, Yamaguchi M. Tetrahedron Lett. 1989;30:2945.
- 20.(a) Chandrasekhar S, Sudhakar A. Org Lett. 2010;12:236. doi: 10.1021/ol9024138. [DOI] [PubMed] [Google Scholar]; (b) Shier WT, Abbas HK, Badria FA. Tetrahedron Lett. 1995;36:1571. [Google Scholar]
- 21.For selected examples of pyran synthesis in complex molecules see: Smith AB, III, Verhoest PR, Minbiole KP, Schelhaas M. J Am Chem Soc. 2001;123:4834. doi: 10.1021/ja0105055.Simpson GL, Heffron TP, Merino E, Jamison TF. J Am Chem Soc. 2006;128:1056. doi: 10.1021/ja057973p.Keck GE, Kraft MB, Truong AP, Li W, Sanchez CC, Kedei N, Lewin NE, Blumberg PM. J Am Chem Soc. 2008;130:6660. doi: 10.1021/ja8022169.
- 22.Cheung LL, Marumoto S, Anderson CD, Rychnovsky SD. Org Lett. 2008;10:3101. doi: 10.1021/ol8011474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito T, Fuwa H, Sasaki M. Org Lett. 2009;11:5274. doi: 10.1021/ol902217q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.