

Abstract
Ataxia-telangiectasia mutated (ATM) encodes a nuclear serine/threonine protein kinase whose activity is increased in cells exposed to low doses of ionizing radiation (IR). Here we examine ATM kinase activation in cells exposed to either 32P- or 33P-orthophosphate under conditions typically employed in metabolic labelling experiments. We calculate that the absorbed dose of IR delivered to a 5 cm × 5 cm monolayer of cells incubated in 2 ml media containing 1 mCi of the high-energy (1.70 MeV) β-particle emitter 32P-orthophosphate for 30 min is ~1 Gy IR. The absorbed dose of IR following an otherwise identical exposure to the low-energy (0.24 MeV) β-particle emitter 33P-orthophosphate is ~0.18 Gy IR. We show that low-energy β-particles emitted by 33P induce a greater number of ionizing radiation-induced foci (IRIF) and greater ATM kinase signaling than energetic β-particles emitted by 32P. Hence, we demonstrate that it is inappropriate to use 33P-orthophosphate as a negative control for 32P-orthophosphate in experiments investigating DNA damage responses to DNA double-strand breaks (DSBs). Significantly, we show that ATM accumulates in the chromatin fraction when ATM kinase activity is inhibited during exposure to either radionuclide. Finally, we also show that chromosome aberrations accumulate in cells when ATM kinase activity is inhibited during exposure to ~0.36 Gy β-particles emitted by 33P. We therefore propose that direct cellular exposure to 33P-orthophosphate is an excellent means to induce and label the IR-induced, ATM kinase-dependent phosphoproteome.
Keywords: ATM, DNA damage signaling, p53
1. Introduction
Ataxia-telangiectasia mutated (ATM) encodes a 350 kDa nuclear serine/threonine protein kinase whose activity is increased in cells exposed to ionizing radiation (IR) [1–4]. Biallelic mutations in ATM cause the devastating childhood disorder ataxia telangiectasia (AT) that is characterized by neurodegeneration, predisposition to cancers and clinical radiosensitivity [5]. Cells derived from A-T patients exhibit defective cell cycle checkpoint responses to IR, profound radiosensitivity and high levels of chromosome aberrations, indicating the importance of ATM for the maintenance of chromosome stability [5]. A considerable body of literature documents the ATM-dependent mobilization, modification and upregulation of proteins critical for the induction of cell cycle checkpoints, DNA repair mechanisms and apoptosis following IR [5–7].
The kinetics and sensitivity of ATM kinase activation following IR are extraordinary. We have previously shown that ATM kinase activation is associated with autophosphorylation on serine-1981 [4]. We generated antibodies that recognize ATM only when it phosphorylated on serine-1981 and showed that ATM kinase activity is maximal within 15 min following 0.4 Gy IR, at which point over 50% of ATM is phosphorylated [4]. We also showed that ATM kinase activity is increased in cells exposed to as little as 0.05 Gy IR and following the introduction of just 2 double strand breaks (DSBs) [4,8]. Consistent with this exquisite sensitivity of ATM kinase activation, it was evident in our experiments that metabolic labelling using 32P-orthophosphate was sufficient to induce ATM kinase activity (CJB, unpublished observations). While this was expected, since it had previously been shown that metabolic labelling using 32P-orthophosphate is sufficient to induce a p53-mediated cellular response [9], the apparent sensitivity of ATM kinase activation to cellular exposure to 32P-orthophosphate was surprising. Since 32P- or 33P-orthophosphate are frequently used in metabolic labelling experiments to identify ATM kinase-dependent phosphorylations in γ-irradiated, but not mock-irradiated cells, it is important to establish whether direct cellular exposure to either 32P- or 33P-orthophosphate induces biologically significant ATM kinase-dependent signaling.
Here we show that the low-energy β-particles emitted by 33P induce a greater number of ionizing radiation-induced foci (IRIF) and greater ATM kinase signaling than the energetic β-particles emitted by 32P. Unexpectedly, we also show that ATM accumulates in the chromatin fraction when ATM kinase activity is inhibited during exposure to β-particles emitted by either 33P or 32P. This suggests that an ATM kinase-dependent phosphorylation in the chromatin is essential for ATM mobility in cells exposed to β-particles. Finally, we show that chromosome aberrations accumulate when ATM kinase activity is inhibited during exposure to the β-particles emitted by 33P.
2. Materials and methods
2.1. Dosimetry
The calculations assumed that the radionuclide uniformly distributed in an area of L and W, where L is the length and W is the width. The height (H) of the radioactive liquid was determined with the equation H = V/(LW), where V is the total volume of the media. The dose was calculated using kernel integration [10–12] where k was the dose kernel per unit activity for the radionuclide, ρm was the activity density, v was the volumetric location of the radioactive liquid, was the point of interest, which was where the cell was (in this case, the cells were under the media), and was a point inside the space occupied by radioactive source. Integration was performed over the volumetric space occupied by the radioactive source. In the calculation, several assumptions were made: (1) the calculation was done numerically, meaning a finite grid resolution was used, and (2) the radionuclide self-attenuation was not taken into account.
2.2. Cultivation, metabolic labelling and irradiation
The normal diploid fetal human lung fibroblast cell line IMR90 (ATCC, Manassas, VA) was cultured in DMEM (Lonza, Basel, Switzerland) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA). The specific activity of 32P-orthophosphate (#NEX053; Perkin-Elmer, Waltham, MA) and 33P-orthophosphate (#NET080; Perkin-Elmer) was determined in a scintillation counter immediately before each experiment. Activity was determined as average counts/counting efficiency (100% for 32P-orthophosphate and 94% for 33P-orthophosphate)/2.22 × 106 dpm. The difference between the measured specific activity and that stated by the manufacturer was as high as ~30%. For exposure to β-particle emitters, exponentially growing IMR90 cells were cultured in 25 cm2 (5 cm × 5 cm) flasks for <24 h and then exposed to 2 ml preconditioned DMEM (Lonza) supplemented with 10% FBS containing 0.5 mCi/ml orthophosphate for the time indicated (30 or 60 min). For these experiments, cells were not incubated in phosphate-free media and the cells were washed 3 times with PBS following exposure to the radionuclide. Following such exposures, we determined that the uptake of 33P-orthophosphate into cells is negligible (<0.000001%). For exposure to γ-rays, cells grown under identical conditions were irradiated in a Shepherd Mark I Model 68 [137Cs] irradiator (J.L. Shepherd & Associates, San Fernando, CA) at a dose rate of 0.783 Gy/min.
2.3. Ionizing radiation-induced foci (IRIF) analyses
IMR90 fibroblasts were cultured in single-well chamber slides (Nalge Nunc, Rochester, NY) and exposed to ~2Gy β-particles emitted by 32P, ~0.36 Gy β-particles emitted by 33P or 2 Gy γ-rays. Fibroblasts were fixed with 2% paraformaldehyde (Sigma-Aldrich) for 30 min and permeabilized in 0.2% Triton X-100/1× PBS. Permeabilized fibroblasts were blocked in 5% donkey serum/1× PBS and incubated with anti-phospho H2AX (Ser 139) mouse monoclonal, clone JBW301 (05–636), or rabbit polyclonal (07–164) or anti-53BP1 mouse monoclonal, clone BP13 (05–736), Upstate Biotechnology, Waltham, MA for 1 h. The primary antibody was detected with donkey anti-mouse Alexa 488 (Molecular Probes, Carlsbad, CA) for 1 h. Fibroblasts were counterstained with Vectashield mounting medium containing DAPI (Vector Laboratories, Burlingame, CA) and analyzed with an epifluorescence microscope. A minimum of 200 fibroblasts was scored for each set of conditions, and each experiment was repeated three times. Results were reported as percent positive or the mean number of foci, and error was reported as standard error of the mean.
2.4. Cell fractionation
To minimize the exposure of equipment to high levels of radionuclides, cell fractionation was performed chemically using the 2D Sample Prep for Nuclear Proteins preparation kit (Pierce Biotechnology #89863, Thermo Fisher Scientific, Waltham, MA). The fractionation protocol consisted of an initial incubation of intact cells in a hypotonic buffer (CERI) on ice. Detergent was then added (CERII) and the cells were rapidly lysed in a vortex. The nuclei were collected by centrifugation and the cytoplasmic protein fraction was removed. The nuclei were extracted with a high salt buffer (NER) on ice. The insoluble nuclear fraction containing chromatin was collected by centrifugation and the soluble nuclear fraction was removed. The chromatin-bound fraction was digested in 20 mM Tris-HCl pH 7.5, 100 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 0.3 M sucrose, 0.1% Triton X-100 with 1 unit of micrococcal nuclease (#4797, Worthington Biochemical, Lakewood, NJ) at room temperature. The remaining insoluble nuclear material was collected by centrifugation and the micrococcal nuclease-digested chromatin fraction was removed. The remaining insoluble nuclear material was extracted in 200 mM HCl. Insoluble material was collected by centrifugation (and discarded) and the acid-extracted chromatin fraction was removed and neutralized by the addition of HEPES pH 8.8–200 mM.
2.5. Immunoblotting
Protein extracts were resolved in 3–8% Tris-acetate gels (Invitrogen, Carlsbad, CA). Rabbit monoclonal anti-ATM 1981S-P antisera (EP1890Y, Epitomics, Burlingame, CA), generic mouse monoclonal anti-ATM antisera (MAT3-4G10/8, Sigma-Aldrich, St. Louis, MO), rabbit anti-P53 15S-P (9284, Cell Signaling Technology, Danvers, MA), generic goat anti-p53 (sc6243-G, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-CHK2 68T-P (2661, Cell Signaling, Danvers, MA) and generic mouse anti-CHK2 (Ab5/DCS270, Neomarkers, Fremont, CA) were used in immunoblotting.
2.6. Enumerating chromosome aberrations
Exponentially dividing IMR90 were cultured in 25 cm2 (5 cm × 5 cm) for <24 h and exposed directly to 2 ml preconditioned DMEM (Lonza) supplemented with 10% FBS containing 0.5 mCi/ml 33P-orthophosphate for 30 min or indirectly to a 137Cs source for ~3 min. After 30 min, the 33P-orthophosphate containing media was removed and the cells were washed 5 times in preconditioned media to remove traces of 33P-orthophosphate. Cells were harvested at 48 h with 50 nM calyculin A (Calbiochem, Gibbstown, NJ) for 30 min or 250 nM colcemid (Irvine Scientific, Santa Ana, CA) for 2–4 h. Harvested cells were harvested and dropped onto slides using conventional methods, and then solid-stained for 8 min in 10% Giemsa. Catalogued chromosome aberrations included: chromosome breaks, chromatid gaps/breaks and acentric fragments. No quadriradials, triradials, giants, rings, minutes or dicentrics were observed following these exposures. Total aberrations (TA) per cell were calculated for each treatment. An unpaired t-test was used for statistical comparison at specified cellular exposures.
3. Results
3.1. Dosimetry
We used kernel integration [10–12] to calculate the absorbed dose of ionizing radiation delivered to a monolayer of cells in a common metabolic labelling experiment. A 30 min exposure of a 5cm × 5 cm (25cm2) monolayer of cells to 2 ml media containing 1 mCi of the low-energy (0.24 MeV) β-particle emitter 33P-orthophosphate results in an absorbed dose of approximately 0.18 Gy IR (Fig. 1A). A 30 min exposure of a 5 cm × 5cm (25 cm2) monolayer of cells to 2 ml media containing 1 mCi of the high-energy (1.70 MeV) β-particle emitter 32P-orthophosphate results in an absorbed dose of approximately 1 Gy IR (Fig. 1B).
Fig. 1.

Calculated dose of IR absorbed by a 25 cm2 monolayer of cells incubated in 2ml of media containing 1 mCi of a β-particle. (A) Incubation of a 25 cm2 monolayer of cells in 2ml media containing 1 mCi 33P-orthophosphate for 30 min results in cellular exposure to approximately ~0.18 Gy IR. (B) In contrast, incubation of a 25 cm2 monolayer of cells in 2ml media containing 1 mCi 32P-orthophosphate for 30 min results in cellular exposure to approximately 1 Gy IR. The z-axis is the dose, which varies with location (designated with coordinates (x, y)).
3.2. Ionizing radiation-induced foci (IRIF) in cells exposed to β-particles emitted by 32P or 33P
In order to determine the number of DNA double-strand breaks (DSBs) induced in IMR90 primary fibroblasts following exposure to the β-particles emitted by either 32P or 33P, we examined γH2AX and 53BP1 which coincide at sites of DSBs following exposure to IR [13,14]. Cells were exposed to 2 Gy γ-rays, ~2Gy β-particles emitted by 32P (1 mCi/2 ml for 1 h), or ~0.36 Gy β-particles emitted by 33P (1 mCi/2 ml for 1 h). While γH2AX foci were evident in cells exposed to either γ-rays or β-particles emitted by 33P, high levels of pan-nuclear γH2AX were seen in all cells exposed to the β-particles emitted by 32P (Fig. 2A). Approximately 40 γH2AX foci were seen in cells exposed to 2 Gy γ-rays while approximately 20 γH2AX foci were seen in cells exposed to ~0.36 Gy β-particles emitted by 33P (Fig. 2B).
Fig. 2.

The β-particles emitted by 32P induce pan-nuclear γH2AX while the β-particles emitted by 33P and γ-rays induce foci. IMR90 fibroblasts were exposed to the β-particles emitted by 32Por 33Por γ-rays and analyzed at 1 h following the initial exposure. (A) Representative images are presented for each condition. (B) The numbers of γH2AX foci were greatest in cells exposed to 2 Gy γ-rays. High numbers of γH2AX foci were seen in cells exposed to ~0.36 Gy β-particles emitted by 33P. These experiments were performed two times.
53BP1 foci were evident in cells exposed to either γ-rays or β-particles emitted by 33P and in a sub-population of cells exposed to β-particles emitted by 32P (Fig. 3A). In three experiments, approximately 90% of cells exposed to ~2Gy β-particles emitted by 32P contained 10 or less 53BP1 foci (Fig. 3B). The 53BP1 foci observed in cells exposed to β-particles emitted by 32P were smaller and less distinct than those in cells exposed to either γ-rays or β-particles emitted by 33P. Approximately 30 53BP1 foci were seen in cells exposed to 2 Gy γ-rays while approximately 5 53BP1 foci were seen in cells exposed to ~2Gy β-particles emitted by 32P and approximately 19 53BP1 foci were seen in cells exposed to ~0.36 Gy β-particles emitted by 33P (Fig. 3C). These data are similar to the recent observation that high levels of pan-nuclear γH2AX are induced by UV irradiation in the absence of 53BP1 foci [15].
Fig. 3.

The β-particles emitted by 33P induce greater numbers of 53BP1 foci than the β-particles emitted by 32P. IMR90 fibroblasts were exposed to the β-particles emitted by 32Por 33Por γ-rays and analyzed at 1 h following the initial exposure. (A) Representative images are presented for each condition. (B) >10 53BP1 foci were seen in approximately 100% of cells exposed to either the β-particles emitted by 33Por γ-rays. Approximately 90% of cells exposed to the β-particles emitted by 32P contained less than 10 53BP1. (C) The numbers of γH2AX foci were greatest in cells exposed to 2 Gy γ-rays. High numbers of 53BP1 foci were seen in cells exposed to ~0.36 Gy β-particles emitted by 33P. These experiments were performed three times.
3.3. ATM kinase-dependent signaling in cells exposed to β-particles emitted by 32P or 33P
In order to examine the ATM kinase-dependent signaling induced in IMR90 primary following exposure to the β-particles emitted by either 32P or 33P we examined ATM kinase-dependent phosphorylations on p53, ATM and CHK2 as well as total protein levels. The accumulation of p53 is a rapid response that is mediated by mechanisms that increase p53 translation and disrupt p53 degradation [16–18]. The accumulation of p53 is defective in A–T cells exposed to agents that damage DNA including IR [19]. ATM phosphorylates a p53 peptide on serine-15 in vitro and this phosphorylation correlates with p53 protein stabilization in cells exposed to IR [2,3,20].
p53 protein and p53 serine-15 phosphorylation were evident in the soluble nuclear fraction generated from cells exposed to either ~1Gy β-particles emitted by 32P (1 mCi/2 ml for 30 min) or ~0.18 Gy β-particles emitted by 33P (1 mCi/2 ml for 30 min) (Fig. 4A - middle panels). p53 protein and p53 serine-15 phosphorylation were greater in the soluble nuclear fraction generated from cells exposed to either ~2Gy β-particles emitted by 32P (1 mCi/2 ml for 1h)or ~0.36 Gy β-particles emitted by 33P (1 mCi/2 ml for 1 h). Further, p53 protein and p53 serine-15 phosphorylation were greater in the soluble nuclear fraction generated from cells exposed to 33P-orthophosphate than 32P-orthophosphate. p53 protein and p53 serine-15 phosphorylation were induced in the cytoplasmic and micrococcal nuclease-digested chromatin fractions generated from cells exposed to either ~2Gy β-particles emitted by 32P or ~0.36 Gy β-particles emitted by 33P (Fig. 4A - upper and lower panels). In all samples the increase in p53 protein and p53 serine-15 phosphorylation was prevented by concurrent exposure to the selective inhibitor of ATM kinase activity KU55933 [21].
Fig. 4.

DNA damage signaling is induced in IMR90 cells exposed to the β-particles emitted by 33Por 32P. ATM kinase-dependent signaling is identified by concurrent treatment with the selective ATM kinase inhibitor 10 μM KU55933. Cells were exposed to 1 mCi/2 ml 33P- or 32P-orthophoshate, harvested at either 30 min or 1 h and cytoplamic, soluble nuclear and micrococcal nuclease-digested chromatin fractions were prepared. Protein extracts were resolved and immunoblotted for (A) p53, (B) ATM and (C) CHK2 using both phosphospecific and generic antisera.
ATM kinase activity correlates with ATM serine-1981 phosphorylation [4]. ATM serine-1981 phosphorylation was evident in the micrococcal nuclease-digested chromatin fraction generated from cells exposed to either ~1Gy β-particles emitted by 32Por ~0.18 Gy β-particles emitted by 33P (Fig. 4B). ATM serine-1981 phosphorylation was greater in cells exposed to 33P-orthophosphate than in cells exposed to 32P-orthophosphate. ATM serine-1981 phosphorylation was prevented by concurrent exposure to KU55933. ATM protein accumulated in the micrococcal nuclease-digested chromatin fraction generated from cells exposed to either 33P-orthophosphate or 32P-orthophosphate and concurrently exposed to the selective inhibitor of ATM kinase activity KU55933.
The protein kinase CHK2 is phosphorylated and activated in response to DNA damage by ionizing radiation [22]. Phosphorylation on CHK2 threonine-68 is ATM kinase-dependent in response to IR and CHK2 threonine-68 phosphorylation is essential for activation of CHK2 kinase activity [23]. CHK2 threonine-68 phosphorylation was induced in cells exposed to ~2Gy β-particles emitted by 32P or ~0.36 Gy β-particles emitted by 33P (Fig. 4C). The increase in CHK2 threonine-68 phosphorylation was greater in cells exposed to β-particles emitted by 33P than in cells exposed to β-particles emitted by 32P. Further, ATM-dependent CHK2 threonine-68 phosphorylation was prevented by concurrent exposure to KU55933.
3.4. Accumulation of ATM in the chromatin fraction in cells exposed to β-particles emitted by 32P or 33P
We determined the relative amount of ATM in cell fractions generated from IMR90 treated with either vehicle or KU55933 during an exposure to ~0.36 Gy β-particles emitted by 33P (1 mCi/2 ml for 1 h). This was because our observation that ATM protein accumulated in the micrococcal nuclease-digested chromatin fraction generated from cells exposed to either 33P-orthophosphate or 32P-orthophosphate and the selective inhibitor of ATM kinase activity KU55933 was unexpected (Fig. 4B). ATM serine-1981 phosphorylation was evident in IMR90 exposed to ~0.36 Gy β-particles emitted by 33P and ATM serine-1981 phosphorylation was prevented by concurrent treatment with KU55933 (Fig. 5 - upper panel). ATM protein again accumulated in the micrococcal nuclease-digested chromatin fraction generated from cells exposed to β-particles emitted by 33P and concurrently exposed to the selective inhibitor of ATM kinase activity KU55933 (Fig. 5 - lower panel). The ATM protein levels in the soluble nuclear fraction generated from IMR90 treated with either vehicle or KU55933 were equal. However, ATM protein was decreased in the cytoplasm fraction generated from cells exposed to β-particles emitted by 33P and concurrently exposed to the selective inhibitor of ATM kinase activity KU55933 (Fig. 5 - lower panel). No ATM protein was evident in the acid-extracted chromatin fraction.
Fig. 5.
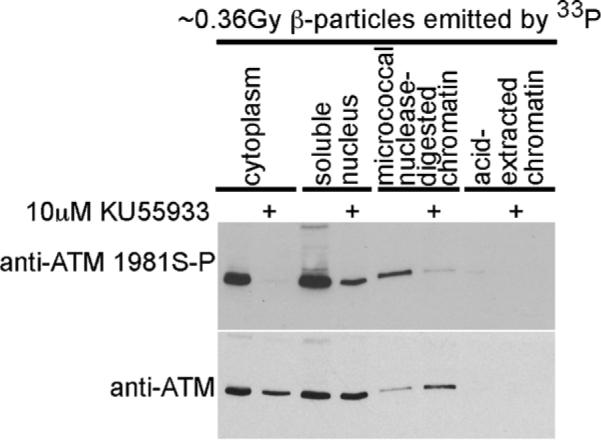
ATM protein accumulates in the micrococcal nuclease-digested chromatin fraction generated from cells exposed to 33P-orthophosphate and the selective inhibitor of ATM kinase activity KU55933. Cells were exposed to 1 mCi/2 ml 33P-orthophosphate and either vehicle or 10 μM KU55933. Cells were harvested at 1 h and cytoplasmic, soluble nuclear, micrococcal nuclease-digested chromatin and acid-extracted fractions were prepared. Protein extracts were resolved and immunoblotted for ATM using both phosphospecific and generic antisera.
3.5. Chromosome aberrations in cells exposed to β-particles emitted by 33P
In order to determine whether the ATM kinase-dependent signaling that we observed in IMR90 exposed to ~0.36 Gy β-particles emitted by 33P has biological significance, we enumerated chromosome aberrations in cells exposed to 33P-orthophosphate and either vehicle or KU55933 for 1 h. We have previously shown that chromosome aberrations accumulate in IMR90 exposed to 2Gy γ-rays when ATM kinase activity is inhibited from +15 to +75 min [8,24]. Cells were harvested 48 h following an exposure to 250 nM colcemid, a microtubule inhibitor that allows visualization of M-phase cells, or 50 nM calyculin A, which prematurely condenses chromatin allowing visualization of late-S-, G2- and M-phase cells.
In the M-phase IMR90 cells, less than 1 chromosome aberration per cell was observed irrespective of the exposure to IR or the ATM kinase inhibitor KU55933 (Fig. 6). In mock-irradiated and irradiated late-S- and G2-phase IMR90 cells exposed to vehicle, and in mock-irradiated late-S- and G2-phase IMR90 cells exposed to KU55933, the average number of chromosome aberrations per cell was also less than 1. We were unable to record observations from 50 cells in colcemid-harvested cells exposed to ~0.36 Gy β-particles emitted by 33P. We believe that this is the result of residual DNA damage preventing these cells from entering mitosis and/or reaching M-phase. We believe that the residual damage induces an ATM kinase-dependent G2/M-phase checkpoint. Consistent with this hypothesis, while we were only able to generate 16 mitotic spreads from cells exposed to 33P and vehicle, we were able to generate 42 mitotic spreads from cells exposed to ~0.36 Gy β-particles emitted by 33P and the selective ATM kinase inhibitor KU55933.
Fig. 6.

The frequency of chromosome aberrations is increased 48 h following irradiation in KU55933 treated late-S and G2 cells. Cells were treated with 10 μM KU55933 from +15 min to +75 min following exposure to 2 Gy IR or ~0.36 Gy β-particles emitted by [33P]. The total aberrations per cell (TA/cell ± SEM) increased from 0.28 to 2.13 (p < 0.001) when KU55933 was added to the cells following exposure to 2 Gy γ-rays. Similarly, the total aberrations per cell (TA/cell ± SEM) increased from 0.28 (±0.10) to 1.81 (p < 0.001) when KU55933 was added to the cells concurrently with a 1 h exposure to 33P-orthophosphate (~0.36 Gy). There was not a significant difference between the total aberrations per cell observed when KU55933 was added to the cells following exposure to 2 Gy IR or ~0.36 Gy β-particles emitted by [33P].
In late-S- and G2-phase IMR90 exposed to 2 Gy γ-rays and treated with KU55933 from +15 to +75 min following irradiation there were approximately 2 chromosome aberrations per cell. Approximately 75% of these chromosome aberrations were chromatid breaks (Table 1). Similarly, in late-S- and G2-phase IMR90 exposed to ~0.36 Gy β-particles emitted by 33P (1 mCi/2 ml for 1 h) and concurrently treated with KU55933 there were approximately 2 chromosome aberrations per cell. Approximately 80% of these chromosome aberrations were chromatid breaks.
Table 1.
Chromosome aberrations induced by 2 Gy γ-IR or ~0.36Gy β-particles in IMR90 fibroblasts when ATM was inhibited for 1 h.
| TA/cella | Percent positive | Chromosome breaks | Chromatid breaks | Acentric fragments | Gaps | |
|---|---|---|---|---|---|---|
| Colcemid | ||||||
| Veh +15 to +75 | 0.06 | 6 | 0 | 3 | 0 | 0 |
| KU55933 +15 to +75 | 0.23 | 16 | 1 | 7 | 3 | 0 |
| 2Gy + veh +15 to +75 | 0.14 | 6 | 0 | 7 | 0 | 0 |
| 2Gy + KU55933 +15 to +75 | 0.2 | 16 | 0 | 6 | 2 | 2 |
| 33P + vehb | 0.19 | 18.8 | 0 | 3 | 0 | 0 |
| 33P + KU55933c | 0.19 | 16.7 | 0 | 7 | 0 | 1 |
| Calyculin A | ||||||
| Veh +15 to +75 | 0.28 | 20 | 1 | 12 | 0 | 1 |
| KU55933 +15 to +75 | 0.63 | 44 | 1 | 29 | 0 | 2 |
| 2Gy + veh +15 to +75 | 0.61 | 40 | 0 | 30 | 0 | 1 |
| 2Gy +KU55933 +15 to +75 | 2.13 | 78 | 5 | 79 | 12 | 10 |
| 33P + veh | 0.68 | 36 | 1 | 28 | 6 | 0 |
| 33P + KU55933 | 1.81 | 70 | 6 | 73 | 10 | 2 |
TA/cell represents the total number of aberrations per cell.
This data represents 16 cells.
This data represents 42 cells.
In IMR90 cells, the total aberrations per cell (TA/cell) increased from 0.28 to 2.13 (p < 0.001) and 1.81 (p < 0.001) when KU55933 was added to the cells following exposure to 2 Gy γ-rays and ~0.36 Gy β-particles emitted by 33P, respectively. There was no significant difference between the total aberrations per cell observed when KU55933 was added to cells exposed to 2 Gy γ-rays and ~0.36 Gy β-particles emitted by 33P(p = 0.36). In addition, the total number of chromosome aberrations increased from 14 (20% of cells positive) to 106 (78%) and 91 (70%) when KU55933 was added to the cells following exposure to 2 Gy γ-rays and ~0.36 Gy β-particles emitted by 33P, respectively.
4. Discussion
Here we show that direct cellular exposure to the low-energy (0.24 MeV) β-particles emitted by 33P induce a greater number of ionizing radiation-induced foci (IRIF) and greater ATM kinase signaling than direct cellular exposure to the energetic (1.70 MeV) β-particles emitted by 32P. ATM kinase-dependent p53 stabilization, p53 serine 15 phosphorylation and p53 DNA binding were greater in cells exposed to the β-particles emitted by 33P than in cells exposed to the β-particles emitted by 32P. ATM serine 1981 phosphorylation and, most strikingly, CHK2 threonine-68 phosphorylation was also greater in cells exposed to 33P-orthophosphate than in cells exposed to 32P-orthophosphate. Although a 30 min exposure of a 5 cm × 5 cm (25 cm2) monolayer of cells to 1 mCi/2 ml of the low-energy β-particles emitted by 33P results in an absorbed dose of approximately 0.18 Gy IR, while an otherwise identical exposure to the energetic β-particles emitted by 32P results in an absorbed dose of approximately 1 Gy IR, it is clear that the β-particles emitted by 33P induce greater levels of ATM kinase-dependent substrate phosphorylation than those emitted by 32P. Our analyses of IRIF suggest that this may be a consequence of differences in the frequency and density of clusters of ionizations produced along single tracks of 0.24 MeV β-particles and 1.70 MeV β-particles.
ATM kinase activity is increased in cells exposed to agents that induce DSBs, including ionizing radiation (IR) [2–4]. However, the DSBs induced by IR are a wide variety of multiple damaged sites. That is, the DSBs induced by IR frequently arise when two (or more) DNA lesions that give rise to single-strand breaks (SSBs) are within approximately 10 base pairs of each other and on opposite DNA strands [25–27]. DSBs induced by IR may arise following the localized attack of a sugar and cleavage of the backbones of each DNA strand by two or more hydroxyl radicals generated from the ionization of water [27]. Alternatively, DSBs induced by IR may arise following the localized attack of a sugar and cleavage of the backbone of one DNA strand and the attack of a base in the opposite strand. Base excision repair requires the cleavage of that opposite strand to excise the damaged base and this can result in two juxtaposed SSBs on opposite DNA strands [28]. In this regard, it is significant that the majority of SSBs induced by IR have unusual or damaged termini that preclude repair by a simple DNA ligation step [27,29]. DSBs induced by IR may also arise if a SSB, a base lesion that impedes the replication fork, or an interstrand crosslink is not repaired prior to the G1/S-phase transition [30]. Such lesions induce stalled and collapsed replication forks that are susceptible to single-strand nucleases whose activity would generate a DSB [30].
Because the density of energy deposition (frequency and density of clusters of ionizations produced along single tracks) of α-particles, β-particles, γ-rays and X-rays varies considerably, the density of DNA lesions, and therefore the number of DSBs that arise following cellular exposure to these different types of IR, also varies considerably [27]. The number of DSBs that arise following cellular exposure to different energy β-particles is also predicted to vary considerably. One parameter that is used to describe the density of energy deposition by various kinds of IR is linear energy transfer (LET) [31]. LET is the energy transferred per unit length of an ionizing track. Low-LET radiation such as 60Co γ-rays or X-rays has typical LET values of 0.3 and 2 keV/μM, respectively [32]. Higher LET radiation such as α-particles loses about 1000 times as much energy in a given length of track, with a typical LET of 250 keV/μM [32]. Consequently, α-particles have a very limited range but have much more concentrated energy deposition. Here we suggest that (0.24 MeV) β-particles emitted by 33P have higher LET than the high-energy (1.70 MeV) β-particles emitted by 32P. As such we suggest that the frequency and density of clusters of ionizations produced in a monolayer of cells exposed to 33P-orthophosphate is greater than that produced in a monolayer of cells exposed to 32P-orthophosphate. Consistent with this hypothesis we observed a greater number of 53BP1 foci in cells exposed to the β-particles emitted by 33P than an otherwise identical exposure to the β-particles emitted by 32P. This may explain our observation that cellular exposure to the low-energy (0.24 MeV) β-particle emitter 33P-orthophosphate induces more ATM kinase signaling than direct cellular exposure to the high-energy (1.70 MeV) β-particle emitter 32P-orthophosphate.
While γH2AX foci were evident in cells exposed to either γ-rays or the β-particles emitted by 33P, high levels of pan-nuclear γH2AX were seen in all cells exposed to the β-particles emitted by 32P. The significance of the 32P-induced pan-nuclear γH2AX is not clear. However, high levels of pan-nuclear γH2AX are also induced in the absence of 53BP1 foci in S-phase cells exposed to by UV irradiation [15]. We do not believe that the pan-nuclear γH2AX seen in cells exposed to ~2Gy β-particles emitted by 32P is an artifact since it was observed using both monoclonal and polyclonal anti-phospho H2AX (Ser 139) antisera that revealed IRIF in control cells exposed to γ-rays. We also do not believe that the pan-nuclear γH2AX seen in cells exposed to 2Gy β-particles emitted by 32P (1 mCi/2 ml for 1 h) is a preapoptotic signal since cells survived when the 32P-orthophosphate was removed and the cells were washed three times with PBS. In addition, the absence of 53BP1 foci and the reduced ATM kinase-dependent signaling at the culmination of a ~2 Gy exposure to β-particles emitted by 32P over the course of 1 h suggests that the pan-nuclear γH2AX is not the result of a rapid incidence of very high levels of DSBs. Rather, the pan-nuclear γH2AX staining may be a consequence of signaling induced by stress response pathways that are distinct to those that generate γH2AX foci in response to IR. This hypothesis is further supported by preliminary experiments in which we observed pan-nuclear γH2AX in cells exposed to 1Gy β-particles emitted by 32P (1 mCi/2 ml for 30 min) and in cells exposed to ~0.5 Gy β-particles emitted by 32P (1 mCi/2 ml for 15 min) (data not shown).
Our observations are generally significant for the interpretation of metabolic labelling experiments that employ either 33P-orthophosphate or 32P-orthophosphate. An important aspect of our work is that for metabolic labelling experiments investigating DNA damage responses it is inappropriate to use exposure to 33P-orthophosphate as a negative control for exposure to 32P-orthophosphate. Rather, for metabolic labelling experiments investigating DNA damage responses, it is likely more appropriate to suggest that there is no truly appropriate negative control. Metabolic labelling with either 33P-orthophosphate or 32P-orthophosphate activates ATM kinase. We suggest that exposing cells to 10 Gy γ-rays prior to metabolic labelling with 33P-orthophosphate is unlikely to significantly change the induction of ATM kinase signaling by more than a few-fold at best. In retrospect, we were fortunate to identify the ATM serine-1981 phosphorylation by metabolic labelling using 32P-orthophosphate [4].
Unexpectedly, we show that ATM accumulates in the micrococcal nuclease-digested chromatin fraction when ATM kinase activity is inhibited using KU55933 during cellular exposure to either 33P-orthophosphate or 32P-orthophosphate, but not γ-rays. This may be a consequence of the greater number of DSBs and/or the complexity of DSBs, that may delay DSB repair, in cells exposed to β-particles rather than γ-rays. The increased level of ATM protein observed in the micrococcal nuclease-digested chromatin fraction correlates with a decreased level of ATM protein in the cytoplasmic fraction purified from cells exposed to 33P-orthophosphate and KU55933. This suggests that an ATM kinase-dependent phosphorylation in the chromatin is essential for ATM mobility in cells exposed to the β-particles. We hypothesize that the best candidate substrates include MRE11, RAD50 and NBS1 (the MRE11 complex). ATM is known to bind the C-terminus of NBS1 [33,34] and this binding is required for ATM kinase activation [34–36]. The NBS1 hub appears suitable to promote integration of repair sensing and effector activities of the MRE complex by interface exchange and handoff interactions with multiple partners [37]. ATM kinase-dependent phosphorylations have been identified on MRE11, RAD50 and NBS1 [6]. ATM kinase activity and mobilization may be required for MRE11 complex conformational changes and ATM kinase inhibition may disrupt MRE11 activity.
Finally, we show that chromosome aberrations accumulate when ATM kinase activity is inhibited during direct cellular exposure to 33P-orthophosphate. This is significant because it indicates that the ATM kinase signaling induced by the β-particles emitted by 33P is biologically relevant. The number of chromosome aberrations in IMR90 cells is significantly increased in late-S- and G2-, but not M-phase, cells when ATM kinase is inhibited during an exposure to ~0.36 Gy β-particles emitted by 33P. Approximately 80% of these aberrations are chromatid breaks. The number of chromosome aberrations in IMR90 cells was also significantly increased in late-S- and G2-, but not M-phase, cells when ATM kinase was inhibited during an exposure to 2 Gy γ-rays. Approximately 75% of these aberrations are chromatid breaks. Since inhibition of ATM kinase activity in cells exposed to either the β-particles emitted by 33P or γ-rays emitted by 137Cs results in the same spectrum of chromosome aberrations, the ATM kinase-dependent mechanism that suppresses these chromosome aberrations, the majority of which are chromatid breaks, is likely to be identical. We therefore propose that direct cellular exposure to 33P-orthophosphate is an excellent means to induce and label the IR-induced, ATM kinase-dependent phosphoproteome.
Acknowledgements
We thank Graeme C. Smith, Mark O'Connor and Stephen P. Jackson for providing the ATM and DNA-PK kinase inhibitors. We thank Kevin Bohner from the Radiation Safety Office, University of Pittsburgh for assistance in determining the specific activity of radionuclides. This work was supported by CA148644 (CJB), the Lung Cancer Research Foundation (CJB), the Frieda G. and Saul F. Shapira BRCA Cancer Research Program (David C. Whitcomb), the Lung Cancer SPORE grant P50 CA090440 (Jill Siegfried) and the Breast Cancer Research Foundation (Nancy E. Davidson).
Footnotes
Conflict of interest None.
References
- [1].Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- [2].Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- [3].Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- [4].Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- [5].McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5:772–776. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- [7].Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- [8].White JS, Choi S, Bakkenist CJ. Irreversible chromosome damage accumulates rapidly in the absence of ATM kinase activity. Cell Cycle. 2008;7:1277–1284. doi: 10.4161/cc.7.9.5961. [DOI] [PubMed] [Google Scholar]
- [9].Bond JA, Webley K, Wyllie FS, Jones CJ, Craig A, Hupp T, Wynford-Thomas D. p53-Dependent growth arrest and altered p53-immunoreactivity following metabolic labelling with 32P ortho-phosphate in human fibroblasts. Oncogene. 1999;18:3788–3792. doi: 10.1038/sj.onc.1202733. [DOI] [PubMed] [Google Scholar]
- [10].Carlsson AK, Ahnesjo A. Point kernels and superposition methods for scatter dose calculations in brachytherapy. Phys. Med. Biol. 2000;45:357–382. doi: 10.1088/0031-9155/45/2/308. [DOI] [PubMed] [Google Scholar]
- [11].Janicki C, Duggan DM, Coffey C, Fischell DR, Fischell TA. Radiation dose from a phosphorous-32 impregnated wire mesh vascular stent. Med. Phys. 1997;24:437–445. doi: 10.1118/1.597910. [DOI] [PubMed] [Google Scholar]
- [12].Prestwich WV, Kennet TJ, Kus FW. The dose distribution produced by a 32P coated stent. Med. Phys. 1995;22:313–320. doi: 10.1118/1.597455. [DOI] [PubMed] [Google Scholar]
- [13].Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- [14].Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000;151:1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].de Feraudy S, Revet I, Bezrookove V, Feeney L, Cleaver JE. A minority of foci or pan-nuclear apoptotic staining of gammaH2AX in the S phase after UV damage contain DNA double-strand breaks. Proc. Natl. Acad. Sci. U.S.A. 2010;107:6870–6875. doi: 10.1073/pnas.1002175107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- [17].Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- [18].Fu L, Benchimol S. Participation of the human p53 3'UTR in translational repression and activation following gamma-irradiation. EMBO J. 1997;16:4117–4125. doi: 10.1093/emboj/16.13.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- [20].Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–3481. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- [22].Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- [23].Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].White JS, Choi S, Bakkenist CJ. Transient ATM, kinase inhibition disrupts DNA damage-induced sister chromatid exchange. Sci. Signal. 2010;3:ra44. doi: 10.1126/scisignal.2000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hutchinson F. Chemical changes induced in DNA by ionizing radiation. Prog. Nucleic Acid Res. Mol. Biol. 1985;32:115–154. doi: 10.1016/s0079-6603(08)60347-5. [DOI] [PubMed] [Google Scholar]
- [26].Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- [27].Ward JF. The yield of DNA double-strand breaks produced intracellularly by ionizing radiation: a review. Int. J. Radiat. Biol. 1990;57:1141–1150. doi: 10.1080/09553009014551251. [DOI] [PubMed] [Google Scholar]
- [28].Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst.) 2007;6:695–711. doi: 10.1016/j.dnarep.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Obe G, Johannes C, Schulte-Frohlinde D. DNA double-strand breaks induced by sparsely ionizing radiation and endonucleases as critical lesions for cell death, chromosomal aberrations, mutations and oncogenic transformation. Mutagenesis. 1992;7:3–12. doi: 10.1093/mutage/7.1.3. [DOI] [PubMed] [Google Scholar]
- [30].Cimprich CA, Cortez D. ATR: and essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Goodhead DT. The initial physical damage produced by ionizing radiations. Int. J. Radiat. Biol. 1989;56:623–634. doi: 10.1080/09553008914551841. [DOI] [PubMed] [Google Scholar]
- [32].Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Herndon, VA, USA: 2006. [Google Scholar]
- [33].Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- [34].You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 2005;25:5363–5379. doi: 10.1128/MCB.25.13.5363-5379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, Tainer JA. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]