

Abstract
17-β-Estradiol (E2) is a steroid hormone involved in numerous bodily functions, including several brain functions. In particular, E2 is neuroprotective against excitotoxicity and other forms of brain injuries, a property that requires the extracellular signal-regulated kinase (ERK) pathway and possibly that of other signaling molecules. The mechanism and identity of the receptor(s) involved remain unclear, although it has been suggested that E2 receptor α (ERα) and G proteins are involved. We, therefore, investigated whether E2-mediated neuroprotection and ERK activation were linked to pertussis toxin (PTX)-sensitive G-protein-coupled effector systems. Biochemical and image analysis of organotypic hippocampal slices and cortical neuronal cultures showed that E2-mediated neuroprotection as well as E2-induced ERK activation were sensitive to PTX. The sensitivity to PTX suggested a possible role of G-protein- and β-arrestin-mediated mechanisms. Western immunoblots from E2-treated cortical neuronal cultures revealed an increase in phosphorylation of both G-protein-coupled receptor-kinase 2 and β-arrestin-1, a G-protein-coupled receptor adaptor protein. Transfection of neurons with β-arrestin-1 small interfering RNA prevented E2-induced ERK activation. Coimmunoprecipitation experiments indicated that E2 increased the recruitment of β-arrestin-1 and c-Src to ERα. These findings suggested that ERα is regulated by a mechanism associated with receptor desensitization and downregulation. In support of this idea, we found that E2 treatment of cortical synaptoneurosomes resulted in internalization of ERα, whereas treatment of cortical neurons with the ER agonists E-6-BSA-FITC [β-estradiol-6-(O-carboxymethyl)oxime-bovine serum albumin conjugated with fluorescein isothiocyanate] and E-6-biotin [1,3,5(10)-estratrien-3,17β-diol-6-one-6-carboxymethloxime-NH-propyl-biotin] resulted in agonist internalization. These results demonstrate that E2-mediated neuroprotection and ERK activation involve ERα activation of G-protein- and β-arrestin-mediated mechanisms.
Introduction
Neuronal death induced by excitotoxicity is triggered by increased intracellular calcium ion concentration and activation of a number of death signaling pathways. The NMDA receptor, an ionotropic glutamate receptor, is involved in regulating intracellular calcium levels and plays a pivotal role in regulating neuronal death as well as synaptic plasticity. We previously showed that 17-β-estradiol (E2) reduced NMDA-induced neuronal death and facilitated synaptic plasticity by activating the extracellular signal-regulated kinase (ERK) pathway (Bi et al., 2000, 2003). The mechanism linking neuroprotection against NMDA-induced toxicity and ERK activation is still poorly understood, although it has been proposed to involve G-protein-coupled signaling (Alexaki et al., 2006; Kumar et al., 2007).
It has been reported that E2 stimulates a membrane-localized E2 receptor α (ERα) with features resembling those of G-protein-coupled receptors (GPCRs) (D'Souza et al., 2004; Evinger and Levin, 2005; Kumar et al., 2007). Others have shown that some of ERs actions are mediated by GPCR transactivation (Boulware et al., 2005; Dewing et al., 2007; Kuo et al., 2008). Although it is not well understood how intracellular ERs become inserted into membranes and use GPCR systems (Lannigan, 2003; Song et al., 2005; Pedram et al., 2007), it has been proposed that post-transcriptional modifications of ERs is responsible (Acconcia et al., 2005; Boulware et al., 2007). Despite this uncertainty, immunohistochemistry experiments as well as the use of ER agonists and antagonists have provided evidence for a membrane-localized ERα/GPCR-like mechanism responsible for many of E2's rapid effects in different cell types (Pappas et al., 1995; Toran-Allerand, 2005; Pedram et al., 2006; Kumar et al., 2007).
GPCR functions are regulated by a number of mechanisms. The first step after ligand binding to a GPCR is the activation and dissociation of G proteins. G proteins are heterotrimeric proteins comprising α, β, and γ subunits, and during their activation, Gα dissociates from Gβγ, and both complexes trigger activation of downstream effector systems. Several GPCRs are coupled to pertussis toxin (PTX)-sensitive G proteins, and the in vitro use of this toxin has helped to elucidate and identify Gαiβγ-coupled effector systems. A major Gβγ-regulatory pathway involves the activation of receptors by GPCR kinases (GRKs, a.k.a. β-adrenergic receptor kinase) and recruitment of arrestin proteins, such as β-arrestins, to GPCRs. The phosphorylation of activated GPCRs by GRKs and the recruitment of activated β-arrestins induce ERK pathway activation. This regulatory pathway is involved in desensitization and downregulation of GPCRs, a critical step to adapt the responsiveness of GPCRs to levels of receptor stimulation.
The goals of the present study were first to determine the effects of PTX on E2-mediated neuroprotection and ERK activation to re-evaluate the potential role of Gαi and Gβγ in these effects. Second, to further evaluate the participation of GPCR-mediated effector systems in E2 action, we evaluated the role of GRKs and β-arrestins in E2-mediated responses in cultured neurons. Finally, we determined whether ERs were downregulated by internalization after E2 activation. Our results indicate that E2-mediated neuroprotection and ERK activation involve G-protein- and β-arrestin-coupled mechanisms.
Materials and Methods
Animals.
Animals were treated in accordance with the principles and procedures of the National Institutes of Health Guide for the Care and Use of Laboratory Animals; all protocols were approved by the Institutional Animal Care and Use Committee of the University of Southern California. Timed pregnant Sprague Dawley rats were obtained from Harlan and kept in the vivarium in a temperature- and light-controlled environment with a 12 h light/dark cycle. On the day of experimentation, rats were removed from their home cage and anesthetized using halothane.
Organotypic slice cultures.
Slices were prepared from postnatal day 8–10 Sprague Dawley rat pups. After decapitation, brains were removed from the cranium and placed into chilled Hanks balanced salt solution. Brains were placed into chilled cutting medium (Earle's MEM, 25 mm HEPES, 10 mm Tris-base, 10 mm d-glucose, 3 mm MgCl2, pH 7.2) to isolate hippocampi; transverse hippocampal slices (400 μm thick) were prepared with a McIlwain tissue chopper and placed in inserts (Millipore Bioscience Research Reagents). Cultures were maintained in basal medium eagle (BME) containing charcoal-stripped horse serum (20%) (Earle's balanced salts, glutamine 2 mm, HEPES 25 mm, 7.5% NaHCO3, d-glucose 25 mm, ascorbic acid 0.333 mm, 1% penicillin/streptomycin, insulin, pH 7.3; Sigma). Slices were kept at 35°C with 5% CO2 and maintained for 7 d with complete medium exchange every 2 d.
Primary cortical cultures.
Cortical neurons were extracted from prenatal pups from embryonic day 18 pregnant female rats. Cultures were prepared by the methods described by Brewer et al. (1993) and plated on poly-l-lysine (0.1 mg/ml) coated 6-well culture dishes, at a density of 2 × 106 cells/ml for biochemical experiments and 0.5 × 106 cells/ml for molecular and immunohistochemistry experiments. Cultures were maintained in serum-free neurobasal (NB) medium (B27, 2 mm glutamine, 1% penicillin/streptomycin; Invitrogen) and were kept in a constant environment at 37°C and 5% CO2. Fifty percent of the medium was exchanged with fresh medium every other day for 2 weeks. On the day before experimentation, culture medium was completely replaced with fresh medium for accurate dilution of chemical treatments. After treatment, cells were lysed (1% SDS plus 100 mm Tris, pH 7.4, 95–100°C) and briefly sonicated. The homogenate was boiled for 5 min, clarified by centrifugation at 14,000 × g for 5 min at 4°C, the supernatant collected, and the pellet discarded. An aliquot was taken for protein determination by the BCA method (Pierce).
Cell death assays.
Hippocampal slices were maintained for 7 d and then switched to serum-free BME medium and allowed to recover for 24 h before experimental treatments. At the end of treatments, slices were washed with serum-free BME and underwent a 24 h recovery period in serum-free BME medium containing propidium iodide (PI; 3 μm; Calbiochem). Cell death was measured as described previously (Bruce et al., 1995). After recovery, the medium was collected to measure the amount of lactate dehydrogenase (LDH) released and replaced with serum-free BME, and the slices were imaged using epifluorescence. Slices were viewed using an inverted Leica DMIRB with a 560/630 dichromic filter and fitted with a 5× phase contrast objective and a Spot RT slider color charge-coupled device (CCD) camera (Roper Scientific). Viewed images were captured using Photoshop CS, the images inverted, and a threshold was applied to eliminate background fluorescent intensity uniformly throughout all images. Arbitrary fluorescent intensity was established by generating pixel values using Scion Image software.
Immunohistochemisrty and cell counts.
After day 14 in vitro, cortical neuronal cultures were subjected to various pharmacological treatments, washed once with NB, and then fixed in 3% paraformaldehyde for 20 min at 4°C. After fixation, neurons were washed three times with PBS plus 0.05% Tween, pH 7.4, at room temperature. Unspecific binding sites on cells were blocked using 3% horse serum, and cells were incubated with rabbit polyclonal anti microtubule-associated protein-2 (MAP2) antibody (AB6522; Millipore Bioscience Research Reagents) in 1% bovine serum albumin (BSA) overnight at 4°C. The following day, neurons were washed three times in PBS plus 0.05% Tween, pH 7.4, and the binding of anti-MAP2 binding was detected using an anti-rabbit secondary antibody labeled with Alexa488 (in 1% BSA) (JacksonImmuno Research) for 1 h at room temperature. Glass slides were placed into wells with ProLong Gold antifade mounting medium plus 4′,6-diamidino-2-phenylindole (DAPI) nucleic acid stain (Invitrogen). Digital images were acquired using an inverted Leica DMIRB with 480/535 and 360/460 dichromic filters and fitted with a 40× phase-contrast objective and a Spot RT slider color CCD camera (Roper Scientific). Viewed images were captured and processed using Photoshop CS (Adobe). The number of MAP2-positive(+) neurons and DAPI(+) cells was counted for each image using ImageJ software.
Western immunoblot analysis.
At the end of all experimental treatments, samples were prepared for immunoblotting by dilution 1:1 in Laemmli buffer with 5% β-mercaptoethanol (Bio-Rad). Twenty micrograms of proteins were loaded onto 10% SDS-PAGE gels along with precision unstained molecular weight markers to approximate protein molecular weight (Bio-Rad). After electrophoresis, proteins were electroblotted onto NitroPure nitrocellulose membranes (Osmonics). Western blot membranes were washed with TBS-Tween (0.1%) and blocked with 5% nonfat milk (Carnation). Each experiment was performed in duplicate.
Antibodies.
Immunodetection of proteins was performed using anti-diphosphorylated ERK1/2 (pThr202/pTyr204; 1:1000) and anti-ERK1/2 (1:2000) (Cell Signaling Technology) antibodies, polyclonal anti-phosphorylated c-Src antibody (pTyr139; 1:250), and anti-c-Src (N-19; 1:250; Santa Cruz Biotechnology) antibodies, polyclonal anti-phosphorylated GRK2 (pSer370; 1:1000) and anti-GRK2 (1:2000) (Sigma) antibodies, monoclonal anti-phosphorylated β-arrestin-1 (pSer412; 1:1000) (Cell Signaling Technology), and polyclonal anti-β-arrestin-1 (K16; 1:2000; Santa Cruz Biotechnology) antibodies, a polyclonal anti-ERα (MC-20) antibody (Santa Cruz Biotechnology), and peroxidase-labeled secondary antibodies (Jackson ImmunoResearch). Unlabeled standards were tagged using peroxidase labeled StrepTactin (Bio-Rad). Immunoblots (IBs) were visualized autoradiographically using enhanced chemiluminescence (Pierce).
Densitometric analysis.
To quantify protein levels, films were scanned (CanoScan N656U scanner; Canon) at 300 dpi using a grayscale and analyzed. Band intensities were calculated using Scion Image software. Total band intensity values were calculated by subtracting the background for each film to account for any variation in background intensity across films. Ratios of optical density (OD) for phosphorylated (ODphospho) proteins over the corresponding OD for total (ODtotal) proteins were calculated (ODphospho/ ODtotal), and expressed as means ± SEM for the indicated number of experiments. For coimmunoprecipitation and synaptoneurosome experiments (see below), immunoblot ratios were normalized by dividing them by the mean ratio value for the vehicle control calculated across independent experiments [(ODprotein-of-interest/ODcontrol) individual values · 1/(ODprotein-of-interest/ODcontrol) vehicle average value]; this was done to minimize variation across experiments. Data were then expressed as mean ± SEM for the indicated number of experiments.
Small interfering RNA transfection.
A pool of four small interfering RNA (siRNA) duplexes of 20–25 nt were purchased from Santa Cruz Biotechnology. The siRNAs were designed to target the sequences 5′-GCAUCAUCGUUUCCUACAAAG-3′, 5′-CUAGUCAGCUUAUCUGAUAUC-3′, 5′-CAUGCCCAGUGUUUGUUGUUA-3′, 5′-UGAACCUCCUACAUUUCAUAA-3′ of human and mouse β-arrestin-1. A mixture of scrambled nonsilencing RNA duplexes was used as a negative control (Santa Cruz Biotechnology). After day 9 in vitro, cells were washed once and maintained in antibiotic-free NB medium overnight. On the following day, cells were transfected with 25 nm siRNA or scrambled control sequences using 5 μl HiPerFect Transfection Reagent (TR; Qiagen) for every 1 ml of antibiotic-free NB medium. After 24 h, antibiotics were added back to NB medium and the cultures maintained for 120 h at 37°C with 5% CO2. Western immunoblot analysis was used to verify β-arrestin-1 and ERK protein expression.
Confocal microscopy.
Cortical cultures were prepared and maintained as described above and plated onto acid-washed glass coverslips coated with 0.1 mg/ml poly-l-lysine. After 2 weeks, cultures were treated with 1 μg/ml of β-estradiol-6-(O-carboxymethyl)oxime-bovine serum albumin conjugated with fluorescein isothiocyanate (E-6-BSA-FITC; Sigma) or with 50 nm 1,3,5(10)-estratrien-3,17β-diol-6-one-6-carboxymethloxime-NH-propyl-biotin (E-6-biotin; Steraloids) for 60 min at 37°C with 5% CO2. E2-BSA-FITC and E-6-biotin were prepared using an ultracentrifugation protocol to reduce free E2 (Taguchi et al., 2004). Neurons were placed on ice and fixed with 3% formaldehyde in PBS, pH 7.4, washed with TBS, pH 7.6, and then fixed with methanol for 10 min at −20°C. To visualize E-6-biotin, neuronal membranes were permeabilized with 0.1% Tween 20, treated with Image-iT FX (Invitrogen), and labeled with 1 mg/ml streptavidin-Alexa488 for 1 h at room temperature. Profiles were illuminated with an Argon2 laser (BP505-550) to visualize E-6-BSA-FITC and fluorescein-labeled E-6-biotin (LP505). Lipid rafts were identified by fluorescently labeling ganglioside GM1 (Vybrant Lipid Raft, Alexa Fluor 555; Invitrogen) and illuminated with a helium-neon 1 laser (LP 550). Coverslips were mounted with ProLong Gold antifade mounting medium (Invitrogen) onto glass slides. Images were obtained with a Zeiss 510 META laser-scanning confocal microscope fitted with a Zeiss Plan-Apochromat 100× oil objective. The detector gain and amplifier offset were adjusted to vehicle control values between experimental sets to eliminate variation in autofluorescence, and z-stacks were acquired, reconstructed, and analyzed using the Zeiss META 510 software.
Synaptoneurosome preparation.
Synaptoneurosomes were prepared from postnatal day 10 Sprague Dawley rats after a previously described protocol (Hollingsworth et al., 1985; Dominguez et al., 2007). After halothane (Sigma) anesthesia, rats were decapitated and brains were removed from the cranium and placed into chilled modified Krebs solution buffered with NaHCO3 (mKrebs/NaHCO3) (118.5 mm NaCl, 4.7 mm KCl, 1.18 mm KH2PO4, 10 mm d-glucose, 24.9 mm NaHCO3, pH 7.3) and equilibrated with O2/CO2 (95:5). Brains were placed into equilibrated and chilled mKrebs/NaHCO3 containing low calcium and high magnesium (1 mm CaCl2 and 1.5 mm MgSO4) and cortex was dissected out and homogenized in a 7 ml Kontes tissue dounce homogenizer in mKrebs buffered with HEPES (20 mm) (mKrebs/HEPES; 1 mm CaCl2 and 1.5 mm MgSO4, pH 7.3) with five passes. Homogenized tissue was filtered through three layers of 100 μm nylon mesh filter, and the resulting suspension was filtered again through a 5 μm pore-size acrodisc syringe filter with a supor membrane (Pall Life Sciences). The filtrate was then centrifuged at 1000 × g for 15 min at 4°C, washed once, centrifuged again, and the pellet was resuspended and adjusted to ∼250 μg/ml of protein in mKrebs/HEPES with high calcium (2 mm CaCl2) and low magnesium (1 mm MgSO4); synaptoneurosome suspension were kept on ice until experimental treatments took place.
Coimmunoprecipitation assays.
Cortical neuronal cultures were collected in radioimmunoprecipitation assay lysis buffer [Halt protease inhibitor cocktail (Pierce), 1% Triton X-100, 50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1 mm NaF, 1 mm NaVO4, pH 7.4]. Equal amounts of lysates were incubated with 40 μg precipitating antibody (ERα, MC-20) overnight at 4°C with end-over-end rocking. A 1:1 protein A-agarose slurry (New England Biolabs) was added and samples rolled end-over-end at 4°C overnight. The samples were then pelleted, washed, and proteins were eluted using Laemmli buffer with 5% mercaptoethanol (Bio-Rad), boiled for 5 min, and prepared for immunoblot analysis as described above using 4–15% gradient gels (Pierce). Anti c-Src (N-19; 1:250), anti β-arrestin-1 (K-16; 1:2000) (Santa Cruz Biotechnology), and anti ERα (MC-20; 1:1000) antibodies were used for immunoblot analysis.
Statistical analysis.
One-way ANOVA followed by a Tukey's post hoc test were used for pairwise comparisons between experimental treatments. Significant interactions between vehicle control and E2 treatment were assessed with a two-tailed Student's unpaired t test, when applicable. Data were analyzed using GraphPad Prism 4 software, and significance level was set at 0.05 for all experiments.
Results
E2-mediated neuroprotection in organotypic hippocampal slice and cortical neuronal cultures is sensitive to PTX
We previously showed that stimulation of organotypic hippocampal slice cultures with E2 prevented NMDA-induced neuronal death (Bi et al., 2000). In vivo and in vitro experiments have also indicated that E2-mediated neuroprotection in various protocols required the rapid and transient activation of the ERK pathway (Singer et al., 1999; Wu et al., 2005; Bryant et al., 2006; Xu et al., 2006). The mechanism linking E2 to its cellular effects remains unclear, but current evidence supports the idea that membrane-localized ERα receptors and PTX-sensitive heterotrimeric G proteins are involved (Evinger and Levin, 2005; Song et al., 2005).
To further investigate this issue, we tested whether E2-mediated neuroprotection against NMDA-induced excitotoxicity in organotypic hippocampal slice cultures was sensitive to PTX treatment (Fig. 1). Analysis of PI uptake in slices (Fig. 1A,B) (ANOVA, F(7,122) = 13.5, p < 0.0001) and of LDH release in medium (Fig. 1C) (ANOVA, F(7,70) = 18.11, p < 0.0001), two widely used markers of cell death, indicated that E2-mediated neuroprotection against NMDA-induced cell death was reversed after pretreatment of cultured slices with PTX. When PTX binds to Gαi subunits, it catalyzes ADP-ribosylation of the subunit, thereby inhibiting GDP for GTP exchange and preventing activation of the GPCR and the downstream effector systems coupled to Gαi and Gβγ (Cabrera-Vera et al., 2003). Post hoc analysis of LDH release revealed that PTX alone did not alter the viability of slices nor NMDA toxicity (p < 0.01); similar results were obtained with analysis of PI uptake. Immunohistochemistry was used to verify whether a similar phenomenon was also present in primary cortical neuronal cultures. Quantitative analysis of MAP2-positive neurons indicated that E2-mediated neuroprotection against NMDA-induced cell death was also prevented by PTX treatment (ANOVA, F(7,16) = 50.17, p < 0.0001) (Fig. 2). The sensitivity of E2-mediated neuroprotection to PTX suggests that the hormone's neuroprotective effects are associated with Gαi- and Gβγ-linked systems.
Figure 1.

E2-mediated neuroprotection against NMDA-induced excitotoxicity in hippocampal slice cultures is sensitive to PTX. Organotypic hippocampal slice cultures (n = 3–8) were pretreated with 500 ng/ml PTX before E2 and NMDA treatments. On the day of treatment, slices were treated with 10 nm E2 for 3 h and then with 50 μm NMDA for an additional 3 h; after washing, slices were maintained in fresh hormone-free medium containing PI. A, B, Analysis of arbitrary fluorescent values from PI-stained slices 24 h after treatment. C, LDH release in culture medium (activity is expressed as mg protein · s−1 · mg−1). Results are means ± SEM (Tukey's post hoc test, *p < 0.05; **p < 0.01; ***p < 0.001). Veh, Vehicle (for this and all other applicable figures); int., intensity.
Figure 2.

E2-mediated neuroprotection in cortical neuronal cultures is PTX sensitive. Primary cortical neuronal cultures were pretreated with 500 ng/ml PTX before E2 and NMDA treatments. On the day of treatment, cultures were stimulated with 10 nm E2 for 3 h and then with 50 μm NMDA for an additional 18 h. After treatments, cells were fixed, neurons labeled using an anti-MAP2 antibody and an Alexa488-labeled secondary antibody, and coverslips mounted using medium containing DAPI fluorescent nucleic acid stain (top). Neuronal survival was calculated by dividing the number of MAP2-positive neurons by the number of DAPI-labeled cells; these values were then normalized (percentage) by the mean value for the vehicle control (bottom). Results are means ± SEM of three experiments. Scale bar, 100 μm (Tukey's post hoc test; *p < 0.05; **p < 0.01; ***p < 0.001).
E2-induced activation of ERK pathway is PTX sensitive
The observation that E2-mediated neuroprotection was sensitive to PTX suggested that E2-mediated activation of the ERK pathway might also be sensitive to the toxin. We observed that E2-induced ERK activation (Fig. 3A) (ANOVA, F(3,16) = 7.83, p = 0.0019), as well as activation of its upstream kinase, c-Src (Fig. 3B) (ANOVA, F(3,18) = 16.37, p = 0.0009), were sensitive to PTX, in good agreement with previously reported data (Wyckoff et al., 2001; D'Souza et al., 2004; Belcher et al., 2005; Kumar et al., 2007). In addition, pretreatment with PTX for 2 or 24 h was sufficient to block E2-induced ERK activation, whereas PTX alone did not significantly alter ERK basal level of activation (data not shown). Interestingly, in cortical neuronal cultures treated with E2 plus PTX, c-Src phosphorylation decreased below basal levels (Tukey's post hoc test, p < 0.05); similar observations have been reported and proposed to be the results of the activation by E2 of protein phosphatase 2A, in a PTX-independent manner (Lu et al., 2004; Belcher et al., 2005). Thus, like E2-mediated neuroprotection, E2-mediated c-Src and ERK activation is sensitive to PTX, suggesting these effects of E2 are mediated through Gαi- and Gβγ-coupled mechanisms.
Figure 3.
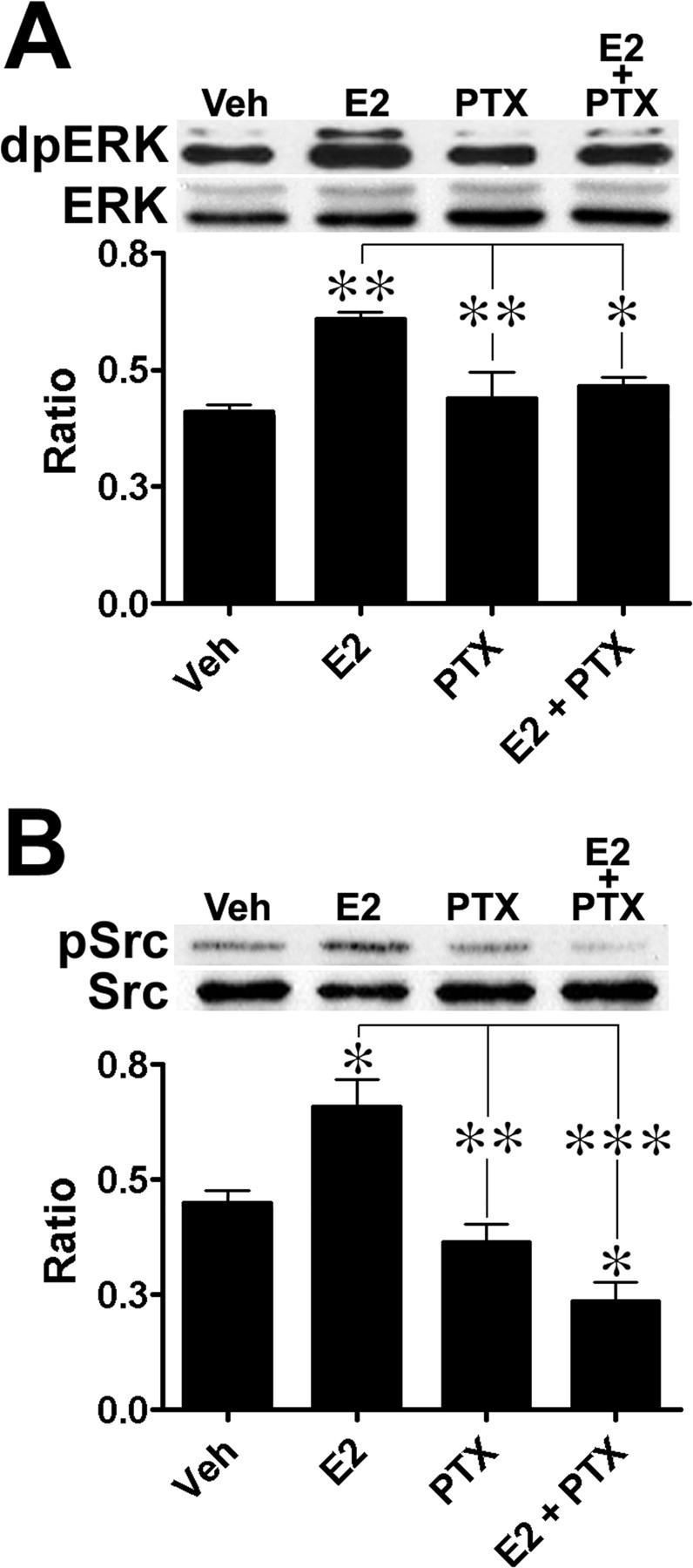
E2-induced ERK and c-Src activation in cortical neuronal cultures is sensitive to PTX. Cortical neuronal cultures were pretreated with 500 ng/ml PTX and then treated with 10 nm E2 for 30 min (n = 4) and prepared for Western blot analysis. A, E2-induced ERK1/2 activation; top, representative Western blot; bottom, quantification of Western blots. B, E2-induced c-Src activation; top, representative Western blot; bottom, quantification of Western blots. Results are expressed as ratio values (means ± SEM) of dpERK over ERK or pSrc over Src (see Materials and Methods; Tukey's post hoc test; *p < 0.05; **p < 0.01; ***p < 0.001).
E2-induced GRK2 and β-arrestin-1 activation
GPCRs are regulated by G-protein- and β-arrestin-dependent pathways. These pathways control desensitization and downregulation of GPCRs and involve GRK2 and β-arrestin-1, as well as c-Src, activation (Waters et al., 2004). Because of the existence of interactions between ERα and Gαi and Gβγ proteins (Kumar et al., 2007), we evaluated whether E2 stimulation of cortical neuronal cultures activated β-arrestin-dependent pathways. Immunoblot analysis indicated that E2 stimulation of cortical neuronal cultures caused a rapid (∼5 min) increase in GRK2 phosphorylation (Fig. 4A). Activation of GRK2 reached a maximum after 60 min of exposure to the hormone and remained elevated at this level with continuous treatment (ANOVA, F(4,15) = 3.287, p = 0.0401). Rapid E2-dependent activation of GRK2 was also observed in acute and cultured hippocampal slices; moreover, the time period for maximal GRK2 activation (60 min) coincided with the termination of E2-induced ERK activation (data not shown). E2-induced GRK2 activation (Fig. 4B) (ANOVA, F(3,16) = 6.473, p = 0.0045), and β-arrestin-1 activation (Fig. 4C) (ANOVA, F(3,8) = 13.8, p = 0.0016), were also completely prevented by PTX treatment. Together, these findings indicate that E2 rapidly triggers GRK2 and β-arrestin-1 activation suggesting that some of E2 actions are linked to β-arrestin-coupled mechanisms.
Figure 4.
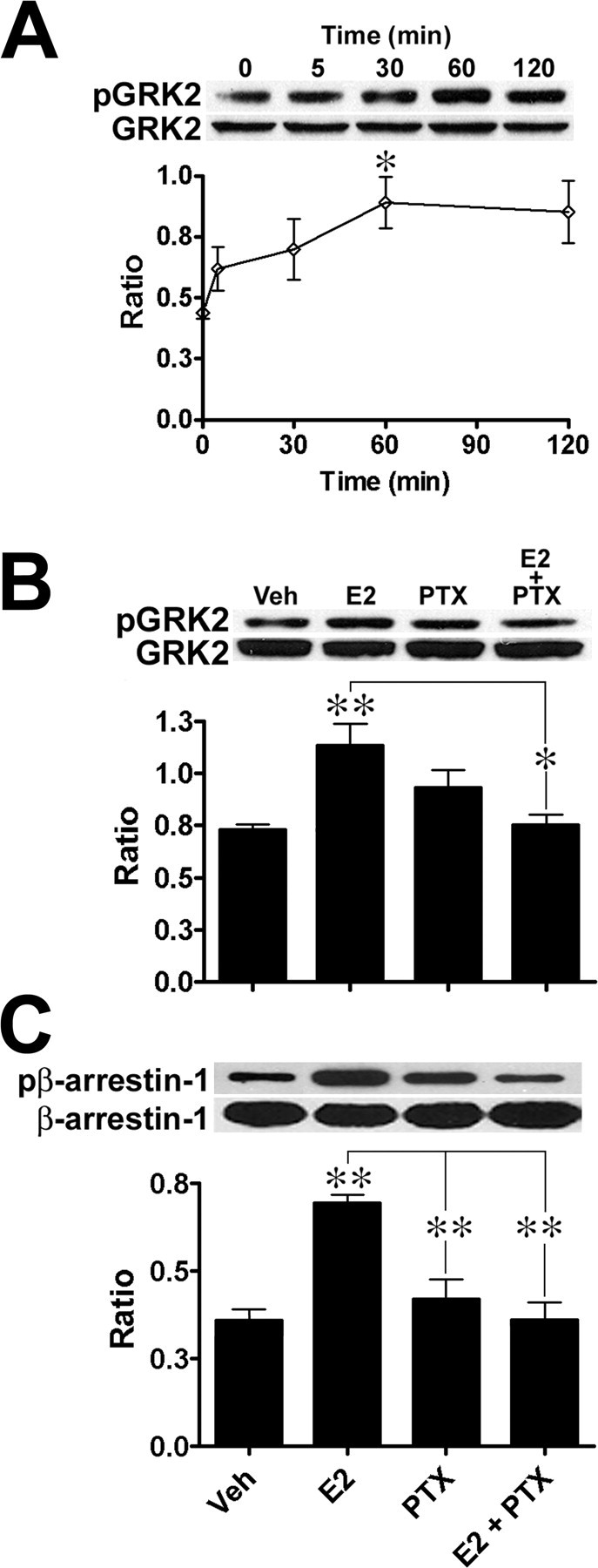
E2 treatment rapidly induces GRK2 and β-arrestin-1 activation in cortical neuronal cultures. A, Cortical neuronal cultures were treated with 10 nm E2 for the indicated times (n = 5): top, representative Western immunoblot; bottom, quantitative analysis. Activation reached a maximum after 60 min of incubation. B, E2-mediated GRK2 activation is PTX sensitive; top, representative Western immunoblot; bottom, quantitative analysis. C, E2-mediated β-arrestin-1 activation is PTX sensitive (top, representative Western immunoblot; bottom, quantitative analysis). Cortical neuronal cultures were pretreated with 500 ng/ml PTX and then treated with 10 nm E2 for 30 min (n = 4). For immunoblot analysis, pGRK2/GRK2 and pβ-arrestin-1β-arrestin-1 ratios were calculated, and results are expressed as means ± SEM (see Materials and Methods; Tukey's post hoc test; *p < 0.05; **p < 0.01).
Effect of β-arrestin-1 siRNA on E2-induced ERK activation
We hypothesized that E2-induced ERK activation was mediated by β-arrestin mechanisms. To test this hypothesis, primary cortical cultures were transfected with siRNAs designed to reduce endogenous levels of β-arrestin-1. Treatments with siRNA for 120 h resulted in a reduction in β-arrestin-1 protein levels to ∼63.3 ± 3.4% (Fig. 5A) (ANOVA, F(3,11) = 8.455, p = 0.0034, Tukey's post hoc, p < 0.05) to those of control values; reduction in β-arrestin-1 protein levels by treatment with siRNA for 24 h (92.8%) and 96 h (81.6%) were also tested but were not significant (data not shown). The reduction of β-arrestin-1 levels prevented the activation of the ERK pathway after stimulation with 10 nm E2 for 30 min; however, transfection of cultured neurons with the scrambled nonsilencing RNA sequences did not prevent E2-induced ERK activation (Student's t test, p = 0.0128) (Fig. 5B). The parallel reduction in β-arrestin-1 protein expression and ability of E2 to activate the ERK pathway support the notion that E2-induced ERK activation involves β-arrestin-mediated mechanisms.
Figure 5.
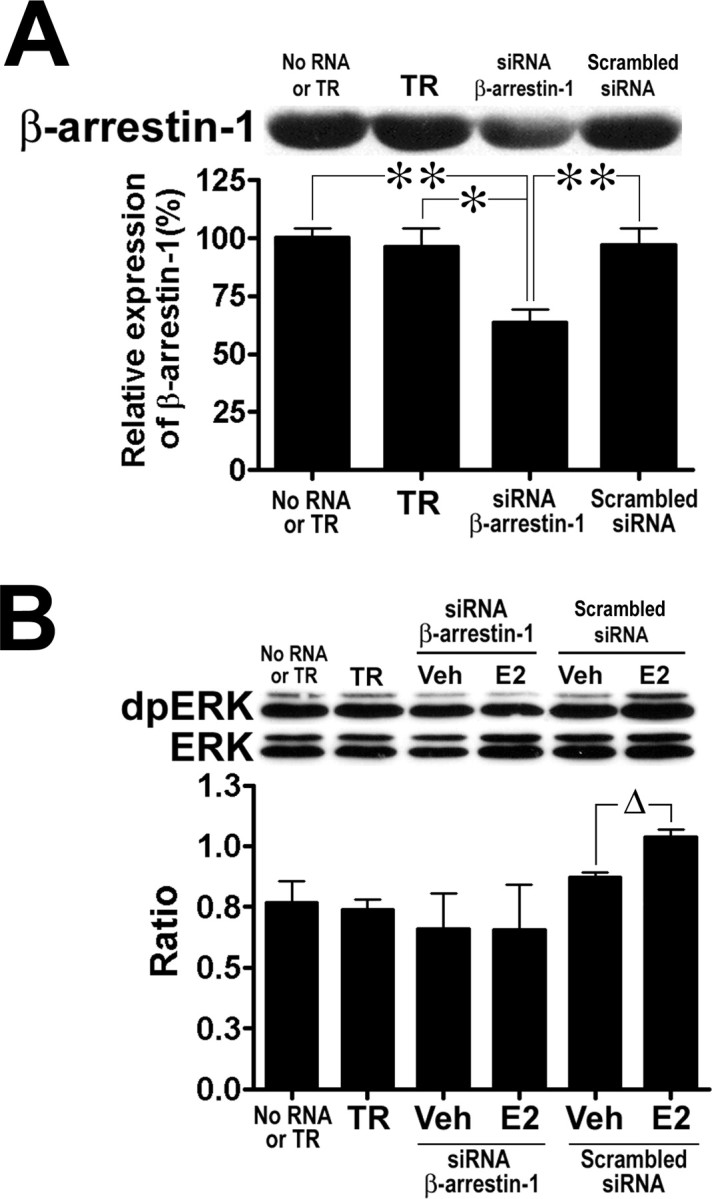
Effects of β-arrestin-1 siRNA on E2-stimulated ERK activation in cortical neuronal cultures. Cultures were transfected with 25 nm siRNA or scrambled siRNA controls for 120 h; TR alone was also tested. After experimental treatments with E2, cultures were prepared for Western immunoblot analysis (see Materials and Methods). A, Depletion of β-arrestin-1 levels after treatment with siRNA. Equal amounts of proteins were loaded (20 μg) on 10% SDS-PAGE gels, and membranes were probed with a polyclonal anti-β-arrestin-1 antibody (top) to examine protein expression. The bar graph represents values expressed as percentage of the level of β-arrestin-1 in mock cultures (no RNA or TR; Tukey's post hoc test; *p < 0.05; **p < 0.01). B, Effects of treatment with β-arrestin-1 siRNA on E2-induced ERK activation. Transfected cultures were treated with 10 nm E2 or vehicle for 30 min (n = 3) and prepared for Western immunoblot analysis. Top, Representative immunoblot; bottom, quantitative analysis indicating that E2-induced ERK phosphorylation was prevented by depletion of β-arrestin-1, whereas cultures transfected with the scrambled siRNA sequences still exhibited E2-induced ERK activation (Student's t test, *p = 0.0034). Results are expressed as ratios (means ± SEM) of dpERK over ERK (see Materials and Methods).
β-Arrestin-1 is rapidly and transiently recruited to ERα after E2 stimulation
To further examine the links between β-arrestins and E2 signaling, we used coimmunoprecipitation to determine whether ERα could form a complex with β-arrestin-1. After acute E2 stimulation (0–120 min), primary neuronal cultures were collected and an antibody against the ligand binding domain of ERα (MC-20), previously shown to label membrane-localized receptors in neurons, was used to immunoprecipitate ERα-like receptors (Clarke et al., 2000; Marin et al., 2003; Toran-Allerand, 2004). The amount of β-arrestin-1 coimmunoprecipitated along with ERs was determined by Western IB analysis, as was the amount of immunoprecipitated (IP) ERα (Fig. 6). In cultures stimulated with 10 nm E2 for 30 min, the levels of β-arrestin-1 bound to ERα receptors significantly increased compared with those found in vehicle-treated cultures (ANOVA, F(4,15) = 3.992, p = 0.0212). This time period corresponds with the time when E2-induced ERK activation reaches its maximum in cortical neurons and synaptoneurosomes (Singer et al., 1999; Dominguez et al., 2007). Several studies have reported that ERα forms a receptor-signaling complex with Src kinase to activate the ERK pathway (Evinger and Levin, 2005; Song et al., 2005). Therefore, we also determined whether immunoprecipitated ERα was bound to c-Src (Fig. 6). Immunoblot analysis indicated that E2 stimulation increased the amount of c-Src bound to ERα in a time-dependent manner, with a maximum at 60 min, a time course that matches that for GRK2 activation (Fig. 4A). These results indicate that ERα receptors form a signaling complex with c-Src and β-arrestin-1 and provide further evidence that E2 treatment leads to rapid β-arrestin-1 activation in cortical neurons. Whether ERα is directly bound to β-arrestin-1 or is the result of the formation of a ERα/GPCRβ-arrestin complex is unknown and should be addressed in future studies.
Figure 6.

E2 treatment increases ERαβ-arrestin-1 interactions in cortical neuronal cultures. Cortical neuronal cultures were treated with 10 nm E2 for the times indicated and collected. An antibody raised against ERα (MC-20) was used to immunoprecipitate (IP) receptors from cellular extracts and determine the levels of coimmunoprecipitated β-arrestin-1 and c-Src using Western IB analysis. Top, Representative Western IBs; bottom, quantitative analysis. IB of ERα was used to verify equal loading. Normalized ratios were calculated as β-arrestin-1 or c-Src values over ERα values (means ± SEM; Tukey's post hoc test; *p < 0.05).
Stimulation with E2 initiates ERα internalization
We recently reported that E2-induced ERK activation was mediated by a membrane-localized ERα-like receptor in cortical synaptoneurosomes (Dominguez et al., 2007). In view of the above results, we investigated whether E2 stimulation resulted in the internalization of membrane-localized ERα receptors in synaptoneurosomes. Such internalization has previously been reported in crude membrane preparations from goat uterine tissue (Karthikeyan and Thampan, 1996). Treatment of cortical synaptoneurosomes resulted in the internalization of ERα receptors as evidence from a shift in localization of the receptor from the mitochondrial to the postmitochondrial fraction (Fig. 7). Immunoblot analysis of the 67 kDa immunoreactive band demonstrated that the ratio of the levels of ERα receptors in the mitochondrial fraction over those in the postmitochondrial fraction and vehicle control almost doubled after E2 stimulation of synaptoneurosomes (E2-treated ratio, 0.71 ± 0.1; vehicle-treated ratio, 0.39 ± 0.02; Student's t test, t(4) = 3.105; p = 0.036; R2 = 0.7068). We also identified other immunoreactive bands (∼55, 63, ∼80, 120 kDa) in our cortical synaptoneurosome preparation, although it is unclear whether these represent functional estrogen-binding proteins or are the result of aggregation or degradation (Toran-Allerand, 2005; Marin et al., 2006; Gorosito et al., 2008; Raz et al., 2008).
Figure 7.

E2 treatment of cortical synaptoneurosomes results in ERα internalization. Cortical synaptoneurosomes were prepared and treated with 10 nm E2 for 60 min at 32°C (n = 3). At the end of treatment, synaptoneurosomes were collected and homogenized, and the mitochondrial and postmitochondrial fractions were separated by centrifugation at 14,000 rpm for 20 min at 4°C and were processed for Western blot with ERα antibodies (MC-20). Top, Representative Western blots indicating the existence of several bands reacting with ERα antibodies. Bottom, Quantitative analysis. Values for the 67 kDa band were normalized with actin levels.
To evaluate whether ERα internalization also occurred in intact neurons, we treated cortical neuronal cultures with the fluorescently labeled, BSA-conjugated estrogen, E-6-BSA-FITC. We used the BSA-conjugated estrogen because it has previously been shown that different conjugated estrogen compounds can be visualized within the cytoplasmic space, which suggests that membrane-bound ERs could be internalized (Moats and Ramirez, 1998; Benten et al., 2001). Examination of labeled neurons was performed using z-stack slices and reconstructed z-stack images to determine whether the fluorescently labeled hormone was internalized. Analysis of neuronal profiles indicated that E-6-BSA-FITC was associated with neuronal membranes (Fig. 8). More importantly, E-6-BSA-FITC was also found within the subcellular space, suggesting that the ligand-receptor complex was sequestered by cortical neurons. Neurons were double-stained with the lipid raft marker GM1, a glycosphingolipid expressed on cell surfaces and participating in endocytosis, to investigate whether this marker was associated with E-6-BSA-FITC binding (Chinnapen et al., 2007). Although the overall localizations of GM1 and ERα were clearly different, several profiles indicated their colocalization, and more importantly, the internalization of E-6-BSA-FITC. Similar findings were also observed when cortical cultures were treated with E-6-biotin (Fig. 9); in this case, intracellular biotin labeling with avidin after incubation of neurons with the ligand indicated that the ligand was internalized. Although these results did not directly indicate internalization of ERα, it certainly supports the idea that ERα receptors are present in lipid rafts and could be internalized by a mechanism associated with endocytosis.
Figure 8.

E-6-BSA-FITC is internalized along with the lipid raft marker GM1 in primary cortical neurons. Cortical neuronal cultures were prepared on glass coverslips and treated with 1 μg/ml E-6-BSA-FITC (green) for 60 min at 37°C, fixed, and prepared for confocal microscopy. Lipid rafts were labeled by adding fluorescently labeled cholera toxin subunit B (CTX-B) 10 min before fixation; the CTX-B (red) conjugate binds to the plasma membrane ganglioside GM1, which selectively partitions into lipid rafts. Analysis of z-stack slices (through a 5-μm-thick optical section) showed that E-6-BSA-FITC binding was localized on neuritic processes and cell bodies (arrows) as well as within subcellular compartments (arrowheads) in several neuronal profiles. Binding of E-6-BSA-FITC was also found colocalized with GM1 (yellow). Reconstruction of z-stacks was used to create orthogonal views (side panels) to help visualize internalization of E-6-BSA-FITC. Scale bar, 10 μm.
Figure 9.

E-6-biotin is internalized into cortical neurons. Cortical neuronal cultures were prepared on glass coverslips and treated for 60 min at 37°C under various conditions. A, Treatment with 50 nm E-6-biotin; at the end of incubation, cells were fixed, permeabilized, and prepared for confocal microscopy. Internalized E-6-biotin was labeled with Alexa488-streptavidin. B, Vehicle control cultures were processed as in A. C, Treatment with E-6-biotin only. D, Treatment with Alexa488-streptavidin only. Analysis of z-stack slices (through a 6-μm-thick optical section) showed that E-6-biotin/Alexa488-streptavidin binding was localized within subcellular compartments in several neuronal profiles. Reconstruction of z-stacks were used to create orthogonal views (A; side panels) to help visualize internalization of E-6-biotin/Alexa488-streptavidin. Scale bar, 10 μm.
Discussion
E2-mediated protection against different neuronal death-inducing events has been well documented both in vivo and in vitro (Singer et al., 1999; Raval et al., 2006; Bains et al., 2007). The detailed mechanisms involved in this action of E2 remain widely unknown, although the use of ERα- and ERβ-specific agonists has indicated that both types of ERs may be involved (Zhao et al., 2004; Cordey and Pike, 2005). In contrast, accumulating evidence suggests that neuroprotection is mediated by the activation of a subpopulation of membrane-localized ERα receptors associated with the rapid activation of several prosurvival signaling cascades, including the induction of anti-apoptotic genes (Honda et al., 2000; Wu et al., 2005; Alexaki et al., 2006). Hippocampal CA1 pyramidal neurons have been repeatedly reported to be protected by E2, and experiments with ER knock-out mice have shown that ERα is the critical receptor involved in neuroprotection against ischemia (Dubal et al., 2001; Sato et al., 2001; Merchenthaler et al., 2003). Furthermore, we, as well as others, have demonstrated that E2-mediated neuroprotection against NMDA-induced cell death required the activation of the ERK pathway (Weaver et al., 1997; Bi et al., 2000; Sato et al., 2001). In the present study, we show that E2-mediated neuroprotection and ERK activation are PTX sensitive, and we also provide evidence for the existence of ERα-mediated G-protein- and β-arrestin-dependent mechanisms in neurons, which regulate some of E2-mediated functions.
Many E2-dependent pathways are coupled to G-protein effector systems (Gu and Moss, 1996; Wyckoff et al., 2001; Simoncini et al., 2006). In neurons and other cell types, E2 has been observed to enhance the expression and activity of Gαi and Gβγ proteins and to modulate direct interactions between G-protein subunits and ERα receptors (Wyckoff et al., 2001; Thompson and Certain, 2005). The localization of ERs and the link between specific G-proteins and E2-mediated pathways has remained widely unknown, partially attributable to the ambiguity of the ERs and G proteins involved. Despite this, it has been shown that ERα-like receptors coupled to G-protein subunits were responsible for the activation of the ERK pathway and other signaling cascades (Moss and Gu, 1999; D'Souza et al., 2004; Belcher et al., 2005). In fact, a recent study has found that ERα exhibits Gαi- and Gβγ-binding domains (251–260 and 271–585) and that during stimulation, G-protein subunits are released from ERα; this interaction was found to be involved in ERK activation and disrupted by PTX (Kumar et al., 2007).
Studies have shown that acute E2 stimulation resulted in interactions between ERα and Gαi; however, the role of such interactions is unclear, although it has been suggested that ERα stimulation of Gβγ leads to activation of the ERK pathway (Wyckoff et al., 2001; Razandi et al., 2003; Lu et al., 2004). Activation of G-protein-induced ERK activation is a major regulatory mechanism of GPCR-mediated effects (Navarro et al., 2003). Free Gβγ subunits have been reported to activate c-Src and GRK2, resulting in phosphorylation of GPCRs and β-arrestin-1 and formation of a GPCRβ-arrestin-1 receptor signaling complex (Luttrell et al., 1996; Lopez-Ilasaca, 1998; Pitcher et al., 1999). The formation of the receptor complex initiates the activation of ERK pathway components. Formation of the GPCRβ-arrestin-1/ERK complex is required for clathrin-mediated endocytosis, where receptors are sorted to be recycled or sent for degradation (Luttrell et al., 1999; Eichmann et al., 2003). These pathways are involved in GPCR desensitization and downregulation.
Rapid and transient activation of ERK by E2 has been repeatedly shown in different cellular systems. The signal is initiated within minutes (∼5 min), reaching a maximum after 30 min of hormone stimulation, and returning toward basal levels after 60–120 min; this action of E2 is reported to involve receptor tyrosine kinases (Migliaccio et al., 1996; Singer et al., 1999; Singh et al., 1999; Miller et al., 2000). This could also be explained by activation of GPCR-signaling systems (Filardo et al., 2007). The mechanism linking E2 and ERK activation has remained widely unknown, although available evidence indicates that ERs and Src kinase form a complex along with adaptor proteins such as Shc/Grb2, modulator of nongenomic actions of the estrogen receptor (MNAR), and striatin (Singh et al., 1999; Honda et al., 2000; Razandi et al., 2003; Song et al., 2005). In contrast, not much is known regarding the links between mechanisms regulating E2-mediated GPCR signaling and G-protein- and β-arrestin-coupled pathways.
Our data indicate that E2-mediated neuroprotection is PTX sensitive and that E2 stimulation rapidly induces GRK2 and β-arrestin-1 activation. Decreased β-arrestin-1 gene expression prevented E2-mediated ERK activation, whereas E2 treatment resulted in increased ERα association with β-arrestin-1 and c-Src. These findings support the idea that E2-induced ERK activation and possibly neuroprotection require G-protein- and β-arrestin-mediated mechanisms. Long-term E2 treatment has been shown to enhance GRK2 function in cerebral cortex of female rats, whereas overexpression of GRK2 in vascular epithelial and in immortalized cell lines prevented E2-induced ERK pathway activation (Ansonoff and Etgen, 2001; Razandi et al., 2003; Kumar et al., 2007); no study has yet addressed the involvement of β-arrestins in membrane-initiated E2 signaling. Cell transfection with β-arrestin siRNAs has previously been used to block β-arrestin-dependent ERK pathway activation by stimulation of membrane localized GPCRs (Ahn et al., 2004; Shenoy et al., 2006). Although there are multiple pathways involved in E2-induced ERK activation (Bulayeva et al., 2004; Boulware et al., 2005), studies have shown that G-protein-dependent ERK activation is sensitive to knockdown of endogenous β-arrestin-1 (Shenoy et al., 2006). Our results indicated that even partial depletion of β-arrestin-1 levels in cortical neuronal cultures prevented E2 stimulation of the ERK pathway, suggesting that this adaptor protein is involved in activation of the pathway and may be involved in regulating other effects of E2 in neurons.
GRK2 and β-arrestin-1 activation has previously been shown to be involved in the regulation of ERK activation, as GRK2 activation negatively regulates the ERK pathway (Elorza et al., 2000; Iacovelli et al., 2004). Interestingly, the time course for recruitment of β-arrestin-1 to ERα in our study was closely related to that of maximum (30 min) E2-induced ERK activation (Migliaccio et al., 1996; Singer et al., 1999; Singh et al., 1999; Dominguez et al., 2007). Moreover, E2-induced GRK2 maximal activation coincided in time with the termination of E2-induced ERK activation (60 min). Thus, in light of our findings, we propose the following hypothesis for the links between E2 receptor activation and downstream events (Fig. 10): after E2 activation of membrane-bound ERα, G-protein activation results in GRK2 activation leading to β-arrestin-1 recruitment to ERα receptors and then to Src and ERK activation. It still remains unclear whether GRK2 and β-arrestin-1 have a direct role in E2-mediated neuroprotection (Waters et al., 2004) or whether transactivation of GPCRs is responsible (Micevych and Mermelstein, 2008).
Figure 10.
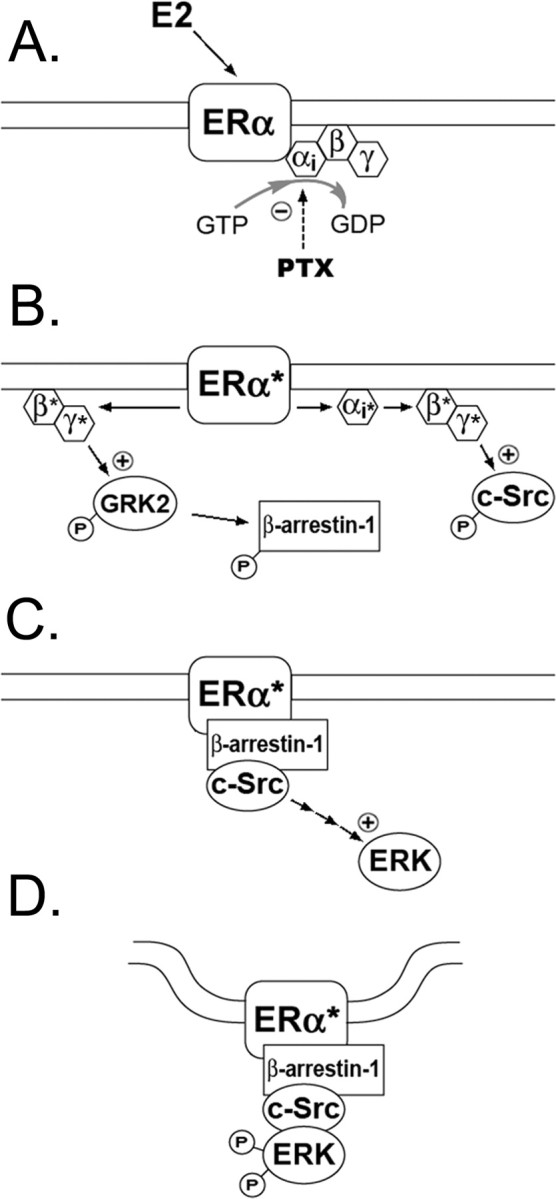
A proposed mechanism for ERα signaling. A, Many of estrogen's rapid actions are mediated by PTX-sensitive G proteins. B, Ligand-activated ERα receptors cause dissociation of Gαi and Gβγ subunits from the receptor and activation of their downstream effector systems. E2-induced Gβγ protein activation increases the phosphorylation of GRK2 and β-arrestin-1. C, Activation of ERα initiates the recruitment of β-arrestin-1 and activation of ERK pathway components. D, Formation of an ERαβ-arrestin-1 receptor/signaling complex leads to internalization of the receptor.
In addition to GRK2 and β-arrestin-1 activation, E2 also stimulated the internalization of ER receptors in cortical neurons. In cortical synaptoneurosomes, E2 treatment resulted in translocation of ERα receptors from the mitochondrial to the postmitochondrial tissue fractions. In goat uterine membranes preparation, it has been reported that acute E2 stimulation of plasma membranes resulted in the internalization of a membrane-localized ER, whereas in embryonic hypothalamic neurons, E2 increased the amount of surface biotinylated ERα (Karthikeyan and Thampan, 1996; Gorosito et al., 2008). Moreover, in hypothalamic neurons, E2 application rapidly (∼1 min) increased the appearance of endocytotic and exocytotic pits at the plasma membrane, an event associated with clathrin-mediated endocytosis and receptor trafficking (Garcia-Segura et al., 1987, 1988; Karthikeyan and Thampan, 1996; Sreeja and Thampan, 2004). These findings suggest that membrane ERs are regulated similarly to other membrane receptors and in particular that prolonged activation of the receptors results in desensitization and downregulation mediated through phosphorylation and internalization (Sinchak and Micevych, 2003; Micevych and Mermelstein, 2008).
In addition to these findings, Western immunoblots of cortical synaptoneurosomes revealed the presence of novel ER immunoreactive bands. Similar findings have been described in protein extracts derived from microsomes and purified membranes from septal and hippocampal and cell lines expressing endogenous ERs (Marin et al., 2006). Western immunoblots identified ERα immunoreactive bands at 67 kDa as well as at 55, 62, and 80 kDa using several antibodies directed against different domain regions of the receptor. Surface biotinylation experiments (Gorosito et al., 2008) identified an ERα immunoreactive protein at 55 kDa, whereas in purified membrane caveolar preparation from postnatal rat cortex, a 63 kDa immunoreactive protein has been identified called ER-X (Toran-Allerand, 2005). In our synaptoneurosome preparation, we immunologically identified a 55 kDa and a 63 kDa ER. It is unknown whether these proteins represent classical ERα or canonical ERs or are products of epitope variation attributable to experimental treatments or proteasomal degradation. We also identified higher molecular weight ERs at ∼80 kDa and ∼110–120 kDa, but like lower molecular weight proteins, it is unclear whether these represent functional ERs. It has been suggested that higher molecular weight ER-like proteins are similar to those described in rodent cortical and liver tissue (Asaithambi et al., 1997; Rao, 1998) and may be products of post-transcriptional modifications (Marin et al., 2006).
Additional evidence supporting ERα internalization is provided by reports that ERα receptors can selectively partition into plasma membrane lipid raft microdomains and bind to caveolin-1, a protein involved in endocytosis (Schlegel et al., 2001; Gilad et al., 2005; Marin et al., 2007). Verification of ER receptor sequestration was evident by the internalization of E-6-BSA-FITC and E-6-biotin in cortical neurons. BSA-conjugated E2 compounds have previously been shown to be internalized within 5 min as well as found to be bound to subcellular organelle membranes (e.g., mitochondria, lysosomes, microsomes) in different cell types (Karthikeyan and Thampan, 1996; Moats and Ramirez, 1998, 2000; Marin et al., 2006). The internalization of E-6-125I-BSA has been reported to become apparent after 30 min, and after 60 min, 75% of labeled organelles were mitochondria and lysosomes (Moats and Ramirez, 1998, 2000). We also observed that E-6-BSA-FITC binding was colocalized with lipid rafts and was present in intracellular compartments of neurons, suggesting it was internalized by a similar mechanism as GM1. These findings further support the notion that E2-mediated stimulation of ERα results in receptor internalization, possibly through β-arrestin-dependent endocytosis. In contrast, other studies have reported that both ERα and ERβ are trafficked toward the plasma membrane after E2 stimulation in neurons (Hart et al., 2007; Sheldahl et al., 2008).
In conclusion, our studies provide evidence that E2-induced ERK activation is mediated through a ERα/Gβγ-coupled mechanism and that E2 stimulation induces the formation of a ERαβ-arrestin-1 receptor complex. The results also underline a new framework to explain some of membrane ERα's GPCR-like characteristics and link to ERK activation. A better understanding of these E2-mediated actions may help to elucidate some of the signaling events involved in E2 actions in the brain and, in particular, its involvement in neuroprotection.
Footnotes
This research was supported by National Institute on Aging Grant AG14751 (awarded to Dr. C. E. Finch). Special thanks to Dr. Hussam Jourdi for editing and insight during the preparation of this manuscript.
References
- Acconcia F, Ascenzi P, Bocedi A, Spisni E, Tomasi V, Trentalance A, Visca P, Marino M. Palmitoylation-dependent estrogen receptor alpha membrane localization: regulation by 17beta-estradiol. Mol Biol Cell. 2005;16:231–237. doi: 10.1091/mbc.E04-07-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Alexaki VI, Charalampopoulos I, Kampa M, Nifli AP, Hatzoglou A, Gravanis A, Castanas E. Activation of membrane estrogen receptors induce pro-survival kinases. J Steroid Biochem Mol Biol. 2006;98:97–110. doi: 10.1016/j.jsbmb.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Ansonoff MA, Etgen AM. Estrogen increases G protein coupled receptor kinase 2 in the cortex of female rats. Brain Res. 2001;898:186–189. doi: 10.1016/s0006-8993(01)02161-8. [DOI] [PubMed] [Google Scholar]
- Asaithambi A, Mukherjee S, Thakur MK. Expression of 112-kDa estrogen receptor in mouse brain cortex and its autoregulation with age. Biochem Biophys Res Commun. 1997;231:683–685. doi: 10.1006/bbrc.1997.6173. [DOI] [PubMed] [Google Scholar]
- Bains M, Cousins JC, Roberts JL. Neuroprotection by estrogen against MPP+-induced dopamine neuron death is mediated by ERalpha in primary cultures of mouse mesencephalon. Exp Neurol. 2007;204:767–776. doi: 10.1016/j.expneurol.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher SM, Le HH, Spurling L, Wong JK. Rapid estrogenic regulation of extracellular signal- regulated kinase 1/2 signaling in cerebellar granule cells involves a G protein- and protein kinase A-dependent mechanism and intracellular activation of protein phosphatase 2A. Endocrinology. 2005;146:5397–5406. doi: 10.1210/en.2005-0564. [DOI] [PubMed] [Google Scholar]
- Benten WP, Stephan C, Lieberherr M, Wunderlich F. Estradiol signaling via sequestrable surface receptors. Endocrinology. 2001;142:1669–1677. doi: 10.1210/endo.142.4.8094. [DOI] [PubMed] [Google Scholar]
- Bi R, Broutman G, Foy MR, Thompson RF, Baudry M. The tyrosine kinase and mitogen-activated protein kinase pathways mediate multiple effects of estrogen in hippocampus. Proc Natl Acad Sci U S A. 2000;97:3602–3607. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi R, Foy MR, Thompson RF, Baudry M. Effects of estrogen, age, and calpain on MAP kinase and NMDA receptors in female rat brain. Neurobiol Aging. 2003;24:977–983. doi: 10.1016/s0197-4580(03)00012-5. [DOI] [PubMed] [Google Scholar]
- Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, Mermelstein PG. Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J Neurosci. 2005;25:5066–5078. doi: 10.1523/JNEUROSCI.1427-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulware MI, Kordasiewicz H, Mermelstein PG. Caveolin proteins are essential for distinct effects of membrane estrogen receptors in neurons. J Neurosci. 2007;27:9941–9950. doi: 10.1523/JNEUROSCI.1647-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Bruce AJ, Sakhi S, Schreiber SS, Baudry M. Development of kainic acid and N-methyl-D-aspartic acid toxicity in organotypic hippocampal cultures. Exp Neurol. 1995;132:209–219. doi: 10.1016/0014-4886(95)90026-8. [DOI] [PubMed] [Google Scholar]
- Bryant DN, Sheldahl LC, Marriott LK, Shapiro RA, Dorsa DM. Multiple pathways transmit neuroprotective effects of gonadal steroids. Endocrine. 2006;29:199–207. doi: 10.1385/ENDO:29:2:199. [DOI] [PubMed] [Google Scholar]
- Bulayeva NN, Gametchu B, Watson CS. Quantitative measurement of estrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Steroids. 2004;69:181–192. doi: 10.1016/j.steroids.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Insights into G protein structure, function, and regulation. Endocr Rev. 2003;24:765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- Chinnapen DJ, Chinnapen H, Saslowsky D, Lencer WI. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol Lett. 2007;266:129–137. doi: 10.1111/j.1574-6968.2006.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke CH, Norfleet AM, Clarke MS, Watson CS, Cunningham KA, Thomas ML. Perimembrane localization of the estrogen receptor alpha protein in neuronal processes of cultured hippocampal neurons. Neuroendocrinology. 2000;71:34–42. doi: 10.1159/000054518. [DOI] [PubMed] [Google Scholar]
- Cordey M, Pike CJ. Neuroprotective properties of selective estrogen receptor agonists in cultured neurons. Brain Res. 2005;1045:217–223. doi: 10.1016/j.brainres.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Dewing P, Boulware MI, Sinchak K, Christensen A, Mermelstein PG, Micevych P. Membrane estrogen receptor-alpha interactions with metabotropic glutamate receptor 1a modulate female sexual receptivity in rats. J Neurosci. 2007;27:9294–9300. doi: 10.1523/JNEUROSCI.0592-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez R, Liu R, Baudry M. 17-Beta-estradiol-mediated activation of extracellular-signal regulated kinase, phosphatidylinositol 3-kinase/protein kinase B-Akt and N-methyl-D-aspartate receptor phosphorylation in cortical synaptoneurosomes. J Neurochem. 2007;101:232–240. doi: 10.1111/j.1471-4159.2006.04360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza DN, Zhang Y, Damjanoska KJ, Carrasco GA, Sullivan NR, Garcia F, Battaglia G, Doncarlos LL, Muma NA, Van de Kar LD. Estrogen reduces serotonin-1A receptor-mediated oxytocin release and Galpha(i/o/z) proteins in the hypothalamus of ovariectomized rats. Neuroendocrinology. 2004;80:31–41. doi: 10.1159/000080795. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichmann T, Lorenz K, Hoffmann M, Brockmann J, Krasel C, Lohse MJ, Quitterer U. The amino-terminal domain of G-protein-coupled receptor kinase 2 is a regulatory Gbeta gamma binding site. J Biol Chem. 2003;278:8052–8057. doi: 10.1074/jbc.M204795200. [DOI] [PubMed] [Google Scholar]
- Elorza A, Sarnago S, Mayor F., Jr Agonist-dependent modulation of G protein-coupled receptor kinase 2 by mitogen-activated protein kinases. Mol Pharmacol. 2000;57:778–783. doi: 10.1124/mol.57.4.778. [DOI] [PubMed] [Google Scholar]
- Evinger AJ, 3rd, Levin ER. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005;70:361–363. doi: 10.1016/j.steroids.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Filardo E, Quinn J, Pang Y, Graeber C, Shaw S, Dong J, Thomas P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology. 2007;148:3236–3245. doi: 10.1210/en.2006-1605. [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Olmos G, Tranque P, Naftolin F. Rapid effects of gonadal steroids upon hypothalamic neuronal membrane ultrastructure. J Steroid Biochem. 1987;27:615–623. doi: 10.1016/0022-4731(87)90361-x. [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Hernandez P, Olmos G, Tranque PA, Naftolin F. Neuronal membrane remodelling during the oestrus cycle: a freeze-fracture study in the arcuate nucleus of the rat hypothalamus. J Neurocytol. 1988;17:377–383. doi: 10.1007/BF01187859. [DOI] [PubMed] [Google Scholar]
- Gilad LA, Bresler T, Gnainsky J, Smirnoff P, Schwartz B. Regulation of vitamin D receptor expression via estrogen-induced activation of the ERK 1/2 signaling pathway in colon and breast cancer cells. J Endocrinol. 2005;185:577–592. doi: 10.1677/joe.1.05770. [DOI] [PubMed] [Google Scholar]
- Gorosito SV, Lorenzo AG, Cambiasso MJ. Estrogen receptor alpha is expressed on the cell-surface of embryonic hypothalamic neurons. Neuroscience. 2008;154:1173–1177. doi: 10.1016/j.neuroscience.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Gu Q, Moss RL. 17β-Estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J Neurosci. 1996;16:3620–3629. doi: 10.1523/JNEUROSCI.16-11-03620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart SA, Snyder MA, Smejkalova T, Woolley CS. Estrogen mobilizes a subset of estrogen receptor-α-immunoreactive vesicles in inhibitory presynaptic boutons in hippocampal CA1. J Neurosci. 2007;27:2102–2111. doi: 10.1523/JNEUROSCI.5436-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth EB, McNeal ET, Burton JL, Williams RJ, Daly JW, Creveling CR. Biochemical characterization of a filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3′:5′-monophosphate-generating systems, receptors, and enzymes. J Neurosci. 1985;5:2240–2253. doi: 10.1523/JNEUROSCI.05-08-02240.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Sawada H, Kihara T, Urushitani M, Nakamizo T, Akaike A, Shimohama S. Phosphatidylinositol 3-kinase mediates neuroprotection by estrogen in cultured cortical neurons. J Neurosci Res. 2000;60:321–327. doi: 10.1002/(SICI)1097-4547(20000501)60:3<321::AID-JNR6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Capobianco L, Iula M, Di Giorgi Gerevini V, Picascia A, Blahos J, Melchiorri D, Nicoletti F, De Blasi A. Regulation of mGlu4 metabotropic glutamate receptor signaling by type-2 G-protein coupled receptor kinase (GRK2) Mol Pharmacol. 2004;65:1103–1110. doi: 10.1124/mol.65.5.1103. [DOI] [PubMed] [Google Scholar]
- Karthikeyan N, Thampan RV. Plasma membrane is the primary site of localization of the nonactivated estrogen receptor in the goat uterus: hormone binding causes receptor internalization. Arch Biochem Biophys. 1996;325:47–57. doi: 10.1006/abbi.1996.0006. [DOI] [PubMed] [Google Scholar]
- Kumar P, Wu Q, Chambliss KL, Yuhanna IS, Mumby SM, Mineo C, Tall GG, Shaul PW. Direct interactions with G alpha i and G betagamma mediate nongenomic signaling by estrogen receptor alpha. Mol Endocrinol. 2007;21:1370–1380. doi: 10.1210/me.2006-0360. [DOI] [PubMed] [Google Scholar]
- Kuo J, Hariri OR, Bondar G, Ogi J, Micevych P. Membrane estrogen receptor-alpha interacts with metabotropic glutamate receptor 1a to mobilize intracellular calcium in hypothalamic astrocytes. Endocrinology. 2009;150:1369–1376. doi: 10.1210/en.2008-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannigan DA. Estrogen receptor phosphorylation. Steroids. 2003;68:1–9. doi: 10.1016/s0039-128x(02)00110-1. [DOI] [PubMed] [Google Scholar]
- Lopez-Ilasaca M. Signaling from G-protein-coupled receptors to mitogen-activated protein (MAP)-kinase cascades. Biochem Pharmacol. 1998;56:269–277. doi: 10.1016/s0006-2952(98)00059-8. [DOI] [PubMed] [Google Scholar]
- Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor alpha. Proc Natl Acad Sci U S A. 2004;101:17126–17131. doi: 10.1073/pnas.0407492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem. 1996;271:19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- Marin R, Guerra B, Morales A, Diaz M, Alonso R. An ICI 182,780-sensitive, membrane-related estrogen receptor contributes to estrogenic neuroprotective actions against amyloid-beta toxicity. Ann N Y Acad Sci. 2003;1007:108–116. doi: 10.1196/annals.1286.011. [DOI] [PubMed] [Google Scholar]
- Marin R, Ramírez CM, González M, Alonso R, Díaz M. Alternative estrogen receptors homologous to classical receptor alpha in murine neural tissues. Neurosci Lett. 2006;395:7–11. doi: 10.1016/j.neulet.2005.10.047. [DOI] [PubMed] [Google Scholar]
- Marin R, Ramírez CM, González M, González-Muñoz E, Zorzano A, Camps M, Alonso R, Díaz M. Voltage-dependent anion channel (VDAC) participates in amyloid beta-induced toxicity and interacts with plasma membrane estrogen receptor alpha in septal and hippocampal neurons. Mol Membr Biol. 2007;24:148–160. doi: 10.1080/09687860601055559. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I, Dellovade TL, Shughrue PJ. Neuroprotection by estrogen in animal models of global and focal ischemia. Ann N Y Acad Sci. 2003;1007:89–100. doi: 10.1196/annals.1286.009. [DOI] [PubMed] [Google Scholar]
- Micevych PE, Mermelstein PG. Membrane estrogen receptors acting through metabotropic glutamate receptors: an emerging mechanism of estrogen action in brain. Mol Neurobiol. 2008;38:66–77. doi: 10.1007/s12035-008-8034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- Miller WE, Maudsley S, Ahn S, Khan KD, Luttrell LM, Lefkowitz RJ. beta-arrestin1 interacts with the catalytic domain of the tyrosine kinase c-SRC. Role of beta-arrestin1-dependent targeting of c-SRC in receptor endocytosis. J Biol Chem. 2000;275:11312–11319. doi: 10.1074/jbc.275.15.11312. [DOI] [PubMed] [Google Scholar]
- Moats RK, 2nd, Ramirez VD. Rapid uptake and binding of estradiol-17beta-6-(O-carboxymethyl)oxime:125I-labeled BSA by female rat liver. Biol Reprod. 1998;58:531–538. doi: 10.1095/biolreprod58.2.531. [DOI] [PubMed] [Google Scholar]
- Moats RK, 2nd, Ramirez VD. Electron microscopic visualization of membrane-mediated uptake and translocation of estrogen-BSA:colloidal gold by hep G2 cells. J Endocrinol. 2000;166:631–647. doi: 10.1677/joe.0.1660631. [DOI] [PubMed] [Google Scholar]
- Moss RL, Gu Q. Estrogen: mechanisms for a rapid action in CA1 hippocampal neurons. Steroids. 1999;64:14–21. doi: 10.1016/s0039-128x(98)00092-0. [DOI] [PubMed] [Google Scholar]
- Navarro CE, Abdul Saeed S, Murdock C, Martinez-Fuentes AJ, Arora KK, Krsmanovic LZ, Catt KJ. Regulation of cyclic adenosine 3′,5′-monophosphate signaling and pulsatile neurosecretion by Gi-coupled plasma membrane estrogen receptors in immortalized gonadotropin-releasing hormone neurons. Mol Endocrinol. 2003;17:1792–1804. doi: 10.1210/me.2003-0040. [DOI] [PubMed] [Google Scholar]
- Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 1995;9:404–410. doi: 10.1096/fasebj.9.5.7896011. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;20:1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007;282:22278–22288. doi: 10.1074/jbc.M611877200. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Tesmer JJ, Freeman JL, Capel WD, Stone WC, Lefkowitz RJ. Feedback inhibition of G protein-coupled receptor kinase 2 (GRK2) activity by extracellular signal-regulated kinases. J Biol Chem. 1999;274:34531–34534. doi: 10.1074/jbc.274.49.34531. [DOI] [PubMed] [Google Scholar]
- Rao BR. Isolation and characterization of an estrogen binding protein which may integrate the plethora of estrogenic actions in non-reproductive organs. J Steroid Biochem Mol Biol. 1998;65:3–41. doi: 10.1016/s0960-0760(98)00019-3. [DOI] [PubMed] [Google Scholar]
- Raval AP, Bramlett H, Perez-Pinzon MA. Estrogen preconditioning protects the hippocampal CA1 against ischemia. Neuroscience. 2006;141:1721–1730. doi: 10.1016/j.neuroscience.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16:140–153. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Park ST, Levin ER. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. 2003;278:2701–2712. doi: 10.1074/jbc.M205692200. [DOI] [PubMed] [Google Scholar]
- Sato M, Suzuki K, Nakanishi S. NMDA receptor stimulation and brain-derived neurotrophic factor upregulate homer 1a mRNA via the mitogen-activated protein kinase cascade in cultured cerebellar granule cells. J Neurosci. 2001;21:3797–3805. doi: 10.1523/JNEUROSCI.21-11-03797.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel A, Wang C, Pestell RG, Lisanti MP. Ligand-independent activation of oestrogen receptor alpha by caveolin-1. Biochem J. 2001;359:203–210. doi: 10.1042/0264-6021:3590203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldahl LC, Shapiro RA, Bryant DN, Koerner IP, Dorsa DM. Estrogen induces rapid translocation of estrogen receptor beta, but not estrogen receptor alpha, to the neuronal plasma membrane. Neuroscience. 2008;153:751–761. doi: 10.1016/j.neuroscience.2008.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Scorticati C, Mannella P, Fadiel A, Giretti MS, Fu XD, Baldacci C, Garibaldi S, Caruso A, Fornari L, Naftolin F, Genazzani AR. Estrogen receptor alpha interacts with Galpha13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol Endocrinol. 2006;20:1756–1771. doi: 10.1210/me.2005-0259. [DOI] [PubMed] [Google Scholar]
- Sinchak K, Micevych P. Visualizing activation of opioid circuits by internalization of G protein-coupled receptors. Mol Neurobiol. 2003;27:197–222. doi: 10.1385/MN:27:2:197. [DOI] [PubMed] [Google Scholar]
- Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Sétáló G, Jr, Guan X, Warren M, Toran-Allerand CD. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci. 1999;19:1179–1188. doi: 10.1523/JNEUROSCI.19-04-01179.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab. 2005;16:347–353. doi: 10.1016/j.tem.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Sreeja S, Thampan RV. Estradiol-mediated internalisation of the non-activated estrogen receptor from the goat uterine plasma membrane: identification of the proteins involved. Mol Cell Biochem. 2004;259:131–140. doi: 10.1023/b:mcbi.0000021358.79359.c9. [DOI] [PubMed] [Google Scholar]
- Taguchi Y, Koslowski M, Bodenner DL. Binding of estrogen receptor with estrogen conjugated to bovine serum albumin (BSA) Nucl Recept. 2004;2:5. doi: 10.1186/1478-1336-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson TL, Certain ME. Estrogen mediated inhibition of dopamine transport in the striatum: regulation by G alpha i/o. Eur J Pharmacol. 2005;511:121–126. doi: 10.1016/j.ejphar.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD. Minireview: a plethora of estrogen receptors in the brain: where will it end? Endocrinology. 2004;145:1069–1074. doi: 10.1210/en.2003-1462. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD. Estrogen and the brain: beyond ER-alpha, ER-beta, and 17beta-estradiol. Ann N Y Acad Sci. 2005;1052:136–144. doi: 10.1196/annals.1347.009. [DOI] [PubMed] [Google Scholar]
- Waters C, Pyne S, Pyne NJ. The role of G-protein coupled receptors and associated proteins in receptor tyrosine kinase signal transduction. Semin Cell Dev Biol. 2004;15:309–323. doi: 10.1016/j.semcdb.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Weaver CE, Jr, Park-Chung M, Gibbs TT, Farb DH. 17beta-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res. 1997;761:338–341. doi: 10.1016/s0006-8993(97)00449-6. [DOI] [PubMed] [Google Scholar]
- Wu TW, Wang JM, Chen S, Brinton RD. 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, Shaul PW. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i) J Biol Chem. 2001;276:27071–27076. doi: 10.1074/jbc.M100312200. [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhang W, Klaus J, Young J, Koerner I, Sheldahl LC, Hurn PD, Martínez-Murillo F, Alkayed NJ. Role of cocaine- and amphetamine-regulated transcript in estradiol-mediated neuroprotection. Proc Natl Acad Sci U S A. 2006;103:14489–14494. doi: 10.1073/pnas.0602932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wu TW, Brinton RD. Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Res. 2004;1010:22–34. doi: 10.1016/j.brainres.2004.02.066. [DOI] [PubMed] [Google Scholar]