

Abstract
A phytochemical investigation of the leaves of Vitex quinata (Lour.) F.N. Williams (Verbenaceae), guided by the MCF-7 human breast cancer cell line, led to the isolation of a new δ-truxinate derivative (1) and a new phytonoic acid derivative (2), together with 12 known compounds. The structures of the new compounds were determined by spectroscopic methods as dimethyl 3,4,3′,4′-tetrahydroxy-δ-truxinate (1) and methyl 10R-methoxy-12-oxo-9(13),16E-phytodienoate (2), respectively. In a cytotoxicity assay, (S)-5-hydroxy-7,4′-dimethoxyflavanone (3) was found to be the sole active principle, with ED50 values of 1.1-6.7 μM, respectively, when tested against a panel of three human cancer cells. Methyl 3,4,5-O-tricaffeoyl quinate (4) showed activity in an enzyme-based ELISA NF-κB p65 assay, with an ED50 value of 10.3 μM.
Keywords: Cytotoxicity; (S)-5-Hydroxy-7,4′-dimethoxyflavanone; Methyl 3,4,5-O-tricaffeoyl quinate; Methyl 10R-methoxy-12-oxo-9(13); 16E-phytodienoate; NF-κB assay; 3,4,3′4′-Tetrahydroxy-δ-truxinate; Vitex quinata
1. Introduction
Vitex is one of the largest genera in the family Verbenaceae, with about 250 species distributed mainly in tropical regions of the world (Chen and Gilbert, 1994). Some species of this genus have been used in folk medicine to treat various diseases. Vitex agnus-castus, commonly known as “vitex”, “chaste tree”, “chasteberry”, or “monk's pepper”, is used currently as a botanical dietary supplement in the U.S., and has purported actions in the alleviation of premenstrual symtoms (Low Dog, 2009). Phytochemical investigations on about 30 species from the genus Vitex have revealed that the major secondary metabolites of these plants are diterpenoids, ecdysteroids, flavonoids and iridoid glycosides. Other components, such as lignans, phenylpropanoids, sesquiterpenoids, and triterpenoids have been found in some Vitex plants, but with less frequency. Within this genus, the species V agnus-castus, V. negundo, V rotundifolia, and V trifolia are the most thoroughly investigated phytochemically. Reviews on the chemical constituents of Vitex species and their biological effects have been published in recent years (Ganapaty and Vidyadhar, 2005; Li et al., 2005; Filho et al, 2008).
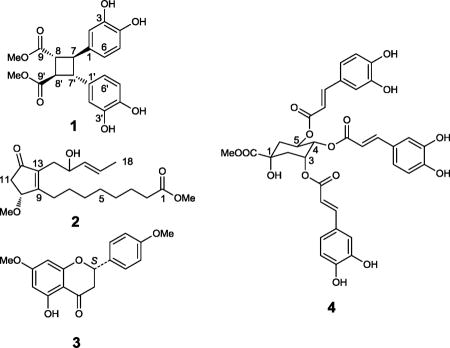
Vitex quinata (Lour.) F. N. Williams is a 4-12 m tall evergreen tree found mainly in temperate and tropical Asia (Chen and Gilbert, 1994). A previous study on Vitex quinata led to the isolation of five compounds, namely, daucosterol, 3,5-O-dicaffeoyl quinic acid, 20-hydroxyecdysone 20,22-monoacetonide, β-sitosterol, and vitexin, but with no biological data reported (Cheng et al, 2007). As part of a collaborative, multi-disciplinary approach toward the discovery of new naturally occurring anticancer agents (Kinghorn et al., 2009), the leaves of V quinata collected from West Java, Indonesia, were selected for further study, when a chloroform extract was found to be active in an initial cytotoxicity screening procedure using the MCF-7 human breast cancer cell line, with an ED50 value of 3.1 μg/mL. The chloroform-soluble extract was active also against two other human cancer cell lines: LNCaP (hormone-dependent prostate) and Lu1 (lung), with ED50 values of 3.1 and 2.5 μg/mL, respectively. A phytochemical investigation of the chloroform and ethyl acetate-soluble extracts of the leaves of V quinata led to the isolation of a new δ-truxinate derivative, dimethyl 3,4,3′,4′-tetrahydroxy-δ-truxinate (1), and a new phytonoic acid derivative, methyl 10R-methoxy-12-oxo-9(13),16E-phytodienoate (2), together with 12 known compounds. The known compounds were identified by spectroscopic data comparison with reported data, as (S)-5-hydroxy-7,4′-dimethoxyflavanone (3) (Kamperdick, 2002), (S)-isosakuranetin (Wagner et al., 1976), 2′-hydroxy-4,4′,6′-trimethoxychalcone (Aponte et al., 2008), 2′,6′-dihydroxy-4,4′-dimethoxychalcone (Zhang et al., 2006; Tu et al., 2007), 3,5-dihydroxy-7,4′-dimethoxyflavonone (Rossi et al., 1997), rhamnocitrin (Zhang et al., 2006; Tu et al., 2007), (-)-loliolide (Valdes, 1986; Chavez et al., 1997), methyl 3,4-O-dicaffeoylquinate (Ma et al., 2009), methyl 3,5-O-dicaffeoylquinate (Choi et al., 2004), methyl 4,5-O-dicaffeoyl quinate (Jiang et al., 2002), methyl 3,4,5-O-tricaffeoyl quinate (4) (Merfort, 1992), and β-sitosterol (Kovganko et al., 1999).
2. Results and discussion
Compound 1 (3.0 mg) was isolated from the ethyl acetate-soluble extract of Vitex quinata leaves as a light yellow gum. Its molecular formula, C20H20O8, was determined by the observation of a sodiated molecular ion peak at m/z 411.1061 (calcd for C20H20O8Na, 411.1057) in the HRESIMS, with a calculated unsaturation number of 11. However, its 13C NMR spectrum showed only ten resonance signals, which suggested that the molecule is composed of two identical units. These carbon resonance signals were assignable to two such units. Each unit was observed to contain an ester functional group [δC 173.6 (C-9 and 9′) and 52.3 (C-10 and 10′)], an aromatic ring with substitution by two hydroxy groups [δC 145.9 (C-3 and 3′), 145.0 (C-4 and 4′), 134.2 (C-4 and 4′), 119.1 (C-6 and 6′), 116.1 (C-5 and 5′), 114.8 (C-2 and 2′)], and two methine carbons [δC 48.5 (C-7 and 7′) and 45.6 (C-8 and 8′)]. It was evident from the unsaturation number that a cyclobutane ring was present also in the molecule of 1. Further analysis of the 1H NMR spectrum of 1 suggested the molecule to be a modified lignan with two phenylpropenoid units coupled at the C-7 (C-7′) and C-8 (C-8′) positions, from the observation of an AA′B′B system of methine signals at δH 3.46 (2H, dd like, H-7 and H-7′) and 3.30 ppm (2H, dd like, H-8 and H-8′). Other resonance signals observed in the 1H NMR spectrum were assignable to two aromatic rings at δH 6.80 (2H, d, J = 2.1 Hz, H-2 and H-2′), 6.77 (2H, d, J = 8.1 Hz, H-5 and H-5′), and 6.65 (2H, dd, J = 2.1, 8.1 Hz, H-6 and H-6′), as well as two carboxymethyl groups both resonating at δH 3.68 (6H, s, H-10 and 10′). The shape of the AA′B′B system of methine signals at δH 3.46 and 3.30 ppm implied a chemically equivalent but magnetically non-equivalent environment for the cyclobutanoid proton set, and the relative configuration of the cyclobutyl ring was determined to be the same as that of δ-truxinic acid, based on a comparison of the 1H NMR chemical shifts of compound 1 with reported data of various truxillic and truxinic acid derivatives (Montaudo and Caccamese, 1973; Ben-Efraim and Green, 1974). This conclusion was supported by a further comparison of the NMR data of compound 1 with those of several δ-truxinic acid derivatives reported more recently (Ito et al, 2003; Saito et al, 2004). The assignments of the 1H and 13C NMR signals of compound 1 were finalized with DEPT, 1H-1H COSY, HSQC, HMBC, and NOESY experiments. Therefore, the structure of the new compound 1 was determined as dimethyl 3,3′,4,4′-tetrahydroxy-δ-truxinate.
Compound 2 (6.0 mg) was obtained from the chloroform partition as a colorless amorphous powder and exhibited a sodiated molecular ion peak at m/z 375.2136 (calculated for C20H32O5Na, 375.2142) in the HRESIMS. An unusual double bond carbon signal observed in the downfield region at δC 174.0 (C-9) of the 13C NMR spectrum, together with signals at δC 207.0 (C-12), 139.2 (C-13), 78.0 (C-10), and 40.5 (C-11), suggested the presence of a conjugated cyclopentenone structure, which was supported by comparison with reported NMR data of a similar ring system (Challener, 1993). The carbonyl carbon signal at δC 174.1 (C-1) and seven methylene signals at δC 34.0 (C-2), 29.7 (C-6), 29.0 (C-4 and 5), 28.1 (C-7), 27.4 (C-8), and 24.9 (C-3), were assignable to a fatty acid chain moiety. Another chain containing a terminal methyl (δC 17.6, C-18), a double bond (δC 133.3 and 126.6, C-16 and C-17), an oxymethine (δC 71.2, C-15), and a methylene (δC 32.1, C-14) functionality was also inferred from the 13C NMR spectrum. The 1H NMR spectrum of 2 showed signals at δH 4.46 (1H, br d, J = 5.2, Hz, H-10), 2.64 (1H, dd, J = 18.2, 5.8 Hz, H-11), and 2.32 (1H, dd, J = 18.2, 1.9 Hz, H-11), assignable to the above-mentioned cyclopentenone ring. Resonance signals belonging to the fatty acid chain moiety were also observed in the range δH 1.34 to 2.33. The planar structure of another five-membered chain was determined by analyzing chemical shifts and coupling constants of the remaining signals in the 1H NMR spectrum of 2. From the terminus of this chain, the connectivity was established in the following sequence: a methyl group (δH 1.66, 3H, d, J = 6.5 Hz, H-18), a trans-double bond [δH 5.65 (1H, dqd, J = 15.2, 6.4, 0.4 Hz, H-17) and 5.43 (1H, ddd, J = 15.2, 6.8, 1.5 Hz, H-16)], an oxymethine functionality (δH 4.22, 1H, q like, J = 5.8 Hz, H-15), and a methylene group (δH 2.44, 2H, d, J = 6.3 Hz, H-14). The linkage of this short chain to the cyclopentanone ring at C-13 position was established by the correlation of H-15 to C-13 observed in the HMBC spectrum. DEPT and 2D NMR techniques were used to finalize the planar structure of 2.
The absolute configuration at the C-10 position in compound 2 was assigned according to a circular dichroism (CD) spectroscopic measurement. Analysis of the conformation of the unsaturated cyclopentanone ring present in the molecule suggested that a pseudoaxial orientation of the C-10 methoxy group was favored over a pseudoequatorial orientation, because of reduced repulsive interactions with the H-11 protons, as depicted in Figure 1 (a and b) (Bifulco et al., 2007). Further investigation of the doublet 1H NMR resonance signal of H-10 revealed that this proton only coupled with one of the two H-11 protons, with a coupling constant of 5.2 Hz. This value corresponds to a 30-40° dihedral angle between these two coupled protons, and an 80-90° dihedral angle between H-10 and the uncoupled H-11 proton, consistent with the aforementioned conformational analysis. Therefore, projection diagrams for the two possible configurations of C-10 could be drawn from the application of the octant rule, as shown in Figure 1 (c and d). The CD spectrum of compound 2 (Figure 1, e) exhibited a strong negative Cotton effect around 315 nm, resulting from the n→π* transition in the cyclopentanone ketone double bond. In the case of unsaturated cyclopentanones, it has been suggested that an “inverse” octant rule may be applied to determine absolute configuration (Lavie et al., 1971). Accordingly, the observed negative n→π* Cotton effect is related to a perturber located in the positive region in the projection diagram (Figure 1, c). Thus, the absolute configuration of C-10 could be determined as R, and the structure of the new compound 2 was proposed as methyl 10R-methoxy-12-oxo-9(13),16E-phytodienoate. The absolute configuration of the C-15 hydroxy group remains unresolved.
Figure 1.

Conformation analysis (a and b), octant rule analysis (c and d), and circular dichroism (CD) spectrum (e) of methyl 10R-methoxy-12-oxo-9,16E-phytodienoate (2)
All the isolated compounds isolated from V. quinata were evaluated for their cytotoxicity against LNCaP hormone-dependent prostate, Lu1 human lung, and MCF-7 human breast cancer cells. Of these isolates, only (S)-5-hydroxy-7,4′-dimethoxyflavanone (3) was found to be an active principle, with ED50 values of 6.7, 4.7, and 1.1 μM, respectively, against these three cell lines.
Compounds 2′-hydroxy-4,4′,6′-trimethoxychalcone, 3,5-dihydroxy-7,4′-dimethoxyflavonone, rhamnocitrin, (-)-loliolide, methyl 3,4-O-dicaffeoyl quinate, methyl 3,5-O-dicaffeoyl quinate, methyl 4,5-O-dicaffeoyl quinate, methyl 3,4,5-O-tricaffeoyl quinate (4) were tested in an enzyme-based ELISA NF-κB p65 assay to evaluate their potential in inhibiting the binding of NF-κB to DNA. Among these compounds, 4 showed weak activity, with an ED50 value of 10.3 μM.
3. Experimental
3.1. General experimental procedures
Optical rotations were obtained on a Perkin-Elmer 343 automatic polarimeter. UV spectra were measured with a Perkin-Elmer Lambda 10 UV/vis spectrometer. CD spectra were run on a JASCO J-810 spectrometer. IR spectra were run on a Thermo Scientific Nicolet 6700 FT-IR spectrometer. NMR spectroscopic data were recorded at room temperature on a Bruker Avance DPX-300 or Bruker Avance DRX-400 spectrometer. Column chromatography was performed with 65-250 or 230-400 mesh silica gel (Sorbent Technologies, Atlanta, GA). Analytical thin-layer chromatography was conducted on 250 μm thickness Partisil® silica gel 60 F254 glass plates (Whatman, Clifton, NJ). Analytical and semi-preparative HPLC were carried out on a Waters system composed of a 600 controller, a 717 plus autosampler, and a 2487 dual wavelength absorbance detector, with Waters Sunfire analytical (4.6 × 150 mm) and preparative (19 × 150 mm) C18 columns. All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
3.2. Plant material
The leaf sample of V. quinata (Lour.) F. N. Williams was collected from a secondary forest at Gunung Kancana (Kancana Mountain) in Cianjur, West Java, Indonesia, at geographic coordinates -8°17′58.62″ S and +116°24′31.32″ E, at an altitude of 750 m, on August 13, 2003, by Soedarsano Riswan and colleagues. The plant is known locally under the common name of “Kibangbara”. The voucher herbarium specimen (SR-CJR 96) was identified by one of the authors (Soedarsano Riswan) and has been has been deposited at the John G Searle Herbarium of the Field Museum of Natural History, Chicago, Illinois, under accession number #2286401.
3.3. Extraction and isolation of compounds
The dried, powdered leaves of V. quinata (830 g) were extracted with methanol at room temperature three times for two days each time. The methanol extract (125 g) was suspended in a mixture of methanol-water (9:1) and then partitioned between hexanes, chloroform, and ethyl acetate, successively. The chloroform extract (3.2 g) was chromatographed over a silica gel column (65-250 mesh, 2.2 × 47 cm), eluted with a gradient solvent system of increasing polarity (chloroform-methanol, 50:1 to 5:1, then pure methanol) to give nine fractions (D2F1-D2F9).
A white solid precipitate that formed from fraction D2F2 was washed with hexane to give β-sitosterol. The remainder of fraction D2F2 (100 mg) was subjected to silica gel chromatography (230-400 mesh, 1 × 40 cm), using chloroform-acetone (10:1) for elution, to afford 15 subfractions (D3F2F1-D3F2F15). Further purification of subfraction D3F2F11, using a silica gel column (230-400 mesh, 1 × 30 cm) eluted with hexane-ethyl acetate (3:1), led to the isolation of the new compound, methyl 10R-methoxy-12-oxo-9(13),16E-phytodienoate (2, 6 mg). Subfraction D3F2F14 was purified on a silica gel column (230-400 mesh, 1 × 30 cm), using hexane-ethyl acetate (3:1) as eluting solvent, to give 2′-hydroxy-4,4′,6′-trimethoxychalcone (2 mg).
Fraction D2F3 (180 mg) was subjected to silica gel column chromatography (230-400 mesh, 1 × 40 cm), eluted with a gradient solvent system of increasing polarity (hexanes-acetone, 5:1 to 3:1), to give eight subfractions (D2F3F1-D2F3F8). Purification of subfraction D2F3F3 (40 mg) using silica gel column chromatography (230-400 mesh, 1 × 20 cm), eluted with hexane-ethyl acetate (5:1), led to the isolation of (-)-loliolide (15 mg). Subfraction D2F3F5 (33 mg) was separated using a silica gel column (230-400 mesh, 1 × 35 cm) to give (S)-5-hydroxy-7,4′-dimethoxyflavanone (3, 3 mg). Subfraction D2F3F8 (20 mg) was chromatographed over a silica gel column (230-400 mesh, 1 × 20 cm), using hexane-ethyl acetate (5:1) as eluting solvent, to afford 2′,6′-dihydroxy-4,4′-dimethoxychalcone (2 mg).
Fraction D2F5 (80 mg) was chromatographed over a Sephadex LH-20 column (4 × 50 cm), using pure methanol as eluting solvent, to give five subfractions (D2F5F1-D2F5F5). Sufraction D2F5F4 (36 mg) was separated by preparative thin-layer chromatography (PTLC), using chloroform-methanol (10:1) as developing solvent, to give 3,5-dihydroxy-7,4′-dimethoxyflavonone (3 mg, Rf= 0.45).
The ethyl acetate extract (16 g) was fractionated over a silica gel column (63-200 mesh, 8.5 × 19 cm), eluted with a gradient of increasing polarity containing chloroform-methanol (from 20:1 to 2:1), and was finally washed with pure methanol. Altogether, six pooled fractions (D3F1-D3F6) were collected. Fraction D3F2 (270 mg) was chromatographed over a Sephadex LH-20 column (4 × 50 cm), using pure methanol as eluting solvent, to give four subfractions (D3F2F1-D3F2F4). Subfraction D3F2F2 (40 mg) was subjected to passage over a silica gel column (230-400 mesh, 1 × 40 cm), eluted with chloroform-methanol (30:1), to give rhamnocitrin (2 mg).
Fraction D3F3 (1.6 g) was subjected to passage over a Sephadex LH-20 column (4 × 50 cm), eluted with pure methanol, to give six subfractions (D3F3F1-D3F3F6). Subfraction D3F4F2 (290 mg) was separated using a silica gel column (230-400 mesh, 20 × 1.5 cm) to afford methyl 3,4-dicaffeoyl quinic acid ester (30 mg) and methyl 4,5-dicaffeoyl quinic acid ester (4 mg). Subfraction D3F3F3 (80 mg) was further fractionated by silica gel column chromatography, using chloroform-methanol (15:1), to give a mixture containing dimethyl 3,3′,4,4′-tetrahydroxy-δ-truxinate. The mixture was further purified by semi-preparative HPLC, using acetonitrile-0.5% formic acid water solution (30:70, 8 mL/min) as eluting solvent, to give the new compound dimethyl 3,4,3′,4′-tetrahydroxy-δ-truxinate (1, 4 mg, t R = 11.4 min). Another subfraction, D3F3F5 (230 mg), was subjected to passage over a silica gel column (1.0 × 40 cm), eluted with chloroform-acetone (8:1), to furnish compound (S)-isosakuranetin (8 mg).
Fraction D3F4 (2.5 g) was chromatographed over a Sephadex LH-20 column (4 × 50 cm), eluted with pure methanol, to afford five subfractions (D3F4F1-D3F4F5). Subfraction D3F4F2 (1 g) was purified over a silica gel column (230-400 mesh, 1.8 × 35 cm), using chloroform-acetone (6:1) as solvent, to give methyl 3,4,5-O-tricaffeoyl quinate (4, 500 mg). A mixture obtained from subfraction D3F4F4 was further purified using semi-preparative HPLC, eluted with methanol-water (45:55, 8 mL/min), to afford methyl 3,5-dicaffeoyl quinic acid ester (65 mg, tR = 13 min).
3.4. Characterization of dimethyl 3,4,3′,4′-tetrahydroxy-δ-truxinate (1)
Light yellow gum; [α]20D +5.4 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 215 (4.07), 345 (3.88) nm; IR (film) vmax 3350 (br), 3100, 2956, 1710, 1624, 1514, 1440, 1165, 825 cm-1; 1H NMR (400 MHz, acetone-d6) δH 6.80 (2H, d, J = 2.1 Hz, H-2, -2′), 6.77 (2H, d, J = 8.1 Hz, H-5, -5′), 6.65 (2H, dd, J = 8.1, 2.1 Hz, H-6, -6′), 3.68 (6H, s, H-10, -10′), 3.46 (2H, dd like, H-7, -7′), 3.30 (2H, dd like, H-8, -8′); 13C NMR (100 MHz, acetone-d6) δC 173.6 (C-9, -9′), 145.9 (C-3, -3′), 145.0 (C-4, -4′), 134.2 (C-4, -4′), 119.1 (C-6, -6′), 116.1 (C5, -5′), 114.8 (C-2, -2′), 52.3 (C-10, -10′), 48.5 (C-7, -7′), 45.6 (C-8, -8′); HRESIMS m/z 411.1061 [M + Na]+ (calcd for C20H20O8Na, 411.1057).
3.5. Characterization of methyl 10R-methoxy-12-oxo-9(13),16E-phytodienoate (2)
Colorless gum; [α]20D -19.8 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 208 (3.87), 310 (3.72) nm; IR (film) vmax 3300, 2944, 2925, 1725, 1672, 1543, 1432, 1375, 1244 cm-1; 1H NMR (400 MHz, CDCl3) δH 5.65 (1H, dqd, J = 15.2, 6.4, 0.4 Hz, H-17), 5.43 (1H, ddd, J = 15.2, 6.8, 1.5 Hz, H-16), 4.46 (1H, br d, J = 5.2, Hz, H-10), 4.22 (1H, q like, J = 5.8 Hz, H-15), 3.67 (3H, s, OCH3-1), 3.38 (3H, s, OCH3-10), 2.64 (1H, dd, J = 18.2, 5.8 Hz, H-11), 2.44 (2H, d, J = 6.3 Hz, H-14), 2.42 (2H, m, H-8), 2.32 (1H, dd, J = 18.2, 1.9 Hz, H-11), 2.31 (2H, t, J = 7.5 Hz, H-2), 1.66 (3H, d, J = 6.5 Hz, H-18), 1.63 (2H, m, H-3), 1.60 (1H, m, H-7), 1.47 (1H, m, H-7), 1.34 (2H, m, H-6), 1.33 (4H, m, H-4 and 5); 13C NMR (100 MHz, CDCl3) δC 207.0 (C-12), (C-15), 174.3 (C-1), 174.0 (C-9), 139.2 (C-13), 133.3 (C-16), 126.6 (C-17), 78.0 (C-10), 71.2 (C-15), 57.1 (OCH3-1), 51.5 (OCH3-10), 40.5 (C-11), 34.0 (C-2), 32.1 (C-14), 29.7 (C-6), 29.0 (C-4 and 5), 28.1 (C-7), 27.4 (C-8), 24.9 (C-3), 17.6 (C-18); HRESIMS m/z 375.2136 [M + Na]+ (calcd for C20H32O5Na, 375.2142).
3.6 Cytotoxicity Assay
Cytotoxicity assays on extracts, chromatographic fractions, and isolated compounds from V. quinata were tested against three different human cancer cell lines, including MCF-7, HT-29, and Lu1, using an established protocol (Seo et al., 2001).
3.7. Enzyme-based ELISA NF-κB Assay
An established protocol was used to carry out the enzyme-based NF-κB assay (Renard et al, 2001; Salim et al., 2007). A nuclear extract was prepared from a culture of Hela cell line obtained from the American Type Culture Collection. An EZ-Detect™ Transcription Factor Assay System ELISA kit (Pierce Biotechnology, Rockford, IL) was used to evaluate the specific binding ability of activated NF-κB to the biotinylated-consensus sequence in the presence of tested isolates. The activity of the p65 subunit of NF-κB was measured by detecting the chemiluminescent signal in a Fluostar Optima plate reader (BMG Labtech Inc., Durham, NC). Rocaglamide was used as a positive control with an ED50 value of 0.075 μM in this assay.
Acknowledgments
This study was supported by grants U19 CA52956 and P01 CA125066 awarded to Prof. A. Douglas Kinghorn, from the National Cancer Institute, National Institutes of Health (NCI, NIH), Bethesda, MD, USA. We thank John W. Fowble, College of Pharmacy, The Ohio State University, for facilitating the acquisition of the 300 and 400 MHz NMR spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aponte JC, Verastegui M, Malaga E, Zimic M, Quiliano M, Vaisberg AJ, Gilman RH, Hammond GB. Synthesis, cytotoxicity, and anti-Trypanosoma cruzi activity of new chalcones. J Med Chem. 2008;51:6230–6234. doi: 10.1021/jm800812k. [DOI] [PubMed] [Google Scholar]
- Ben-Efraim DA, Green BS. The use of mid-points or average NMR chemical shifts in stereochemical assignments. Tetrahedron. 1974;30:2357–2364. [Google Scholar]
- Bifulco G, Dambruoso P, Gomez-Paloma L, Riccio R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem Rev. 2007;107:3744–3779. doi: 10.1021/cr030733c. [DOI] [PubMed] [Google Scholar]
- Challener CA, Wulff WD, Anderson BA, Chamberlin S, Faron KL, Kim OK, Murray CK, Xu YC, Yang DC, Darling SD. Cyclopentenone formation via hydrogen activation in the reactions of chromium carbene complexes with alkynes. J Am Chem Soc. 1993;115:1359–1376. [Google Scholar]
- Chavez JP, dos Santos ID, Cruz FG, David JM, Yang SW, Cordell GA. A quinoline alkaloid from Acanthosyris paulo-alvinii. Phytochemistry. 1997;46:967–968. [Google Scholar]
- Chen SL, Gilbert MG. Flora of China. Vol. 17. Science Press; Beijing: 1994. [Google Scholar]
- Cheng WX, Chen HY, Zhang YP, Qin XL, Gu K. Chemical constituents of Vitex quinata. Nat Prod Res Dev. 2007;19:244–246. [Google Scholar]
- Choi SZ, Choi SU, Lee KR. Phytochemical constituents of the aerial parts from Solidago virga-aurea var. gigantea. Arch Pharm Res. 2004;27:164–168. doi: 10.1007/BF02980100. [DOI] [PubMed] [Google Scholar]
- Low Dog T. Chaste tree extract in women's health. Altern Complement Ther. 2009;15:119–125. [Google Scholar]
- Filho JGS, Duringer J, Maia GLA, Tavares JF, Xavier HS, Sobral da Silva M, da-Cunha EVL, Barbosa-Filho JM. Ecdysteroids from Vitex species: distribution and compilation of their 13C-NMR spectral data. Chem Biodivers. 2008;5:707–713. doi: 10.1002/cbdv.200890067. [DOI] [PubMed] [Google Scholar]
- Ganapaty S, Vidyadhar KN. Phytoconstituents and biological activites of Vitex – a review. J Nat Remed. 2005;5:75–79. [Google Scholar]
- Ito Y, Tetsuya K, Horiguchi M. Coerced photodimerization reaction in the solid state through amine salt formation. Tetrahedron. 2003;59:7323–7329. [Google Scholar]
- Jiang HL, Xu LZ, Yang XD, Zhang D, Yang SL, Zou ZM. Quinic acid esters from herba of Siphonostegia chinensis. Chin J Chin Mat Med. 2002;27:923–926. [PubMed] [Google Scholar]
- Kamperdick C, Van NH, Sung TV. Constituents from Miliusa balansae (Annonaceae) Phytochemistry. 2002;61:991–994. doi: 10.1016/s0031-9422(02)00374-6. [DOI] [PubMed] [Google Scholar]
- Kinghorn AD, Carcache de Blanco EJ, Chai HB, Orjala J, Farnsworth NR, Soejarto DD, Oberlies NH, Wani MC, Kroll DJ, Pearce CJ, Swanson SM, Kramer RA, Rose WC, Fairchild CR, Vite GD, Emanuel S, Jarjoura D, Cope FO. Discovery of anticancer agents of diverse natural origin. Pure Appl Chem. 2009;81:1051–1063. doi: 10.1351/PAC-CON-08-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovganko NV, Kashkan ZN, Borisov EV, Batura EV. 13C NMR spectra of β-sitosterol derivatives with oxidized rings A and B. Chem Nat Comp. 1999;35:646–649. [Google Scholar]
- Lavie D, Levy EC, Jain MK. Limonoids of biogenetic interest from Melia azadirachta L. Tetrahedron. 1971;27:3927–3939. [Google Scholar]
- Li CZ, Su YF, Jin XJ. Advances in studies on chemical constituents from plants of Vitex and their bioactivities. Chin Trad Herb Drugs. 2005;36:930–938. [Google Scholar]
- Ma JL, Li N, Li X. Caffeoylquinic acid derivatives from leaves of Lonicera japonica. Chin J Chin Mat Med. 2009;34:10–12. [PubMed] [Google Scholar]
- Merfort I. Caffeoylquinic acids from flowers of Arnica montana and Arnica chamissonis. Phytochemistry. 1992;31:2111–2113. [Google Scholar]
- Montaudo G, Caccamese S. Structure and conformation of chalcone photodimers and related compounds. J Org Chem. 1973;38:710–716. [Google Scholar]
- Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J. Development of a sensitive multi-well colorimetric assay for active NFκB. Nucleic Acids Res. 2001;29:e21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi MH, Yoshida M, Maia JGS. Neolignans, styrylpyrones and flavonoids from Aniba species. Phytochemistry. 1997;45:1263–1269. [Google Scholar]
- Saito H, Mori T, Wada T, Inoue Y. Diastereoselective [2+2] photocycloaddition of stibene to chiral fumarate, direct versus charger-transfer excitation. J Am Chem Soc. 2004;126:1900–1906. doi: 10.1021/ja0370140. [DOI] [PubMed] [Google Scholar]
- Salim AA, Pawlus AD, Chai HB, Farnsworth NR, Kinghorn AD, Carcache-Blanco EJ. Ponapensin, a cyclopenta[bc]benzopyran with potent NF-κB inhibitory activity from Aglaia ponapensis. Bioorg Med Chem Lett. 2007;17:109–112. doi: 10.1016/j.bmcl.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo EK, Kim NC, Mi Q, Chai H, Wall ME, Wani MC, Navarro HA, Burgess JP, Graham JG, Cabieses F, Tan GT, Farnsworth NR, Pezzuto JM, Kinghorn AD. Macharistol, a new cytotoxic cinnamylphenol from the stems of Machaerium aristulatum. J Nat Prod. 2001;64:1483–1485. doi: 10.1021/np0103158. [DOI] [PubMed] [Google Scholar]
- Tu YC, Lian TW, Yen JH, Chen ZT, Wu MJJ. Antiatherogenic effects of kaempferol and rhamnocitrin. J Agric Food Chem. 2007;55:9969–9976. doi: 10.1021/jf0717788. [DOI] [PubMed] [Google Scholar]
- Valdes LJ. Loliolide from Salvia divinorum. J Nat Prod. 1986;49:171. doi: 10.1021/np50043a031. [DOI] [PubMed] [Google Scholar]
- Wagner H, Chari VM, Sonnenbichler J. Carbon-13 NMR spectra of naturally occurring flavonoids. Tetrahedron Lett. 1976;21:1799–1802. [Google Scholar]
- Zhang XF, Hung TM, Phuong PT, Ngoc TM, Min BS, Song KS, Seong YH, Bae KH. Anti-inflammatory activity of flavonoids from Populus davidiana. Arch Pharm Res. 2006;29:1102–1108. doi: 10.1007/BF02969299. [DOI] [PubMed] [Google Scholar]