

Abstract
Functionally selective lanthanide-based ion mobility shift reagents are presented as a method to elucidate protein or peptide structural information as well as relative quantitation of protein expression profiles. Sequence information and site localization of primary amines (n-terminus and lysine), phosphorylation sites, and cysteine residues can be obtained in a data dependent manner using ion mobility-mass spectrometry (IM-MS). The high mass of the incorporated lanthanide ensures a significant shift of where the signal occurs in IM-MS conformation space. Peptide sequence information provided by the use of IM-MS shift reagents allows for both a more confident identification of peptides from complex mixtures and site localization following tandem MS experiments. Stable isotopes of the lanthanide series may also be used as relative quantitation labels since several lanthanides can be utilized in differential sample analyses.
Keywords: Mass spectrometry, ion mobility, ion mobility-mass spectrometry, proteomics, phosphorylation, phosphoproteomics multiplex labeling
1. Introduction
The rapid rise of mass spectrometry-based proteomics over the past two decades has brought with it many new challenges to detect and characterize lower abundance peptides and proteins. Mass spectrometry (MS) is well suited to address several of the challenges of proteomic analyses since only a minute amount of sample is required and it is readily adaptable to pre-ionization separation techniques (e.g. liquid chromatography, 2D gels, etc.) that reduce sample complexity [1-3]. While pre-ionization fractionation methods have the advantage of simplifying the analysis of complex samples, these methods also suffer from long analysis times as well as the need for a greater amount of sample. Recent MS-based proteomics research has focused on the development and use of post-ionization separations for the analysis of complex biological mixtures.
Post-ionization separation techniques, such as ion mobility spectrometry (IMS), offer the advantage of reducing spectral complexity and greater peak capacity [4]. When combined with MS, as ion mobility-mass spectrometry (IM-MS), it becomes a rapid method for separation and mass identification of the components of complex mixtures. Briefly, ions drift through an IMS drift cell under the direction of a weak electrostatic field in the presence of a background buffer gas at ca. 4-10 Torr. As ions elute from the drift cell they are pulsed orthogonally into a time-off-light MS (TOFMS). Ion mobility separates complex mixtures on the basis of ion structure, thus ions of small collision cross section elute from the drift cell faster than comparatively larger ions that encounter more collisions with the background buffer gas.
IM-MS separations partition complex mixtures with respect to the gas phase packing efficiency of each class of molecule present. For example, an analysis of ca. 600 singly-charged peptide signals indicates that > 99% of these signals occupy a narrow band of 5-7% deviation from an ion mobility versus m/z correlation function [5]. Other classes of biomolecules also provide distinct correlations based on their preferred gas-phase packing efficiencies (i.e. lipids < peptides < carbohydrates < oligonucleotides). IM-MS shift reagents utilize these correlations by shifting specific analytes to areas outside of the correlation band where signals are not predicted to occur.
Ion mobility shift reagents function by shifting signals containing specific chemical functionality to an area of conformation space in which signals for that particular biomolecular class are not predicted to occur in the absence of labeling. For a particular molecular class of given density, ion mobility scales as length squared, while mass scales as length cubed. Therefore, two scenarios can be envisionsed, ion mobility shift reagents of either lower [6] or higher density [7], which shift labeled signals to an area above or below the correlation band, respectively. Lanthanide metal chelating agents [8-9] can function as high density IM-MS shift reagents since the lanthanide metal imparts a large increase in mass to the labeled peptide without a concurrent large increase in collision cross section (Figure 1). This report describes the use of such shift reagents for the selective characterization of primary amines (n-terminus or Lysine), cysteine residues, or sites of phosphorylation.
Figure 1.
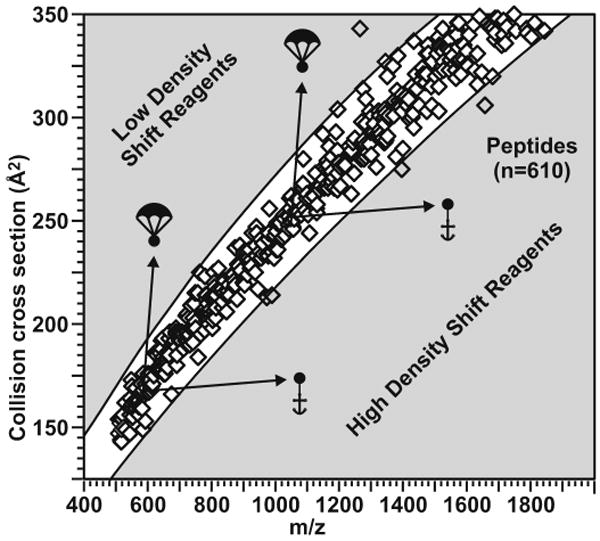
A representation of the peptide correlation band in IM-MS conformation space illustrating the collision cross section of ca. 600 singly-charged peptides. Two shift reagent strategies are illustrated as balloons or anchors. The present manuscript details high density shift reagents illustrated as anchors.
2. Materials and methods
2.1 Reagents
All peptides were purchased from American Peptide Company (Sunnyvale, CA) and proteins, as well as MALDI matrices, were purchased from Sigma Aldrich (St. Louis, MO). Lanthanide metals (as LnCl3 hydrates) were purchased from Strem Chemicals for Research (Newburyport, MA) and Sigma Aldrich. Functionally specific lanthanide-chelating labels were purchased from Macrocyclics (Dallas, TX). All common solvents were also purchased from Sigma Aldrich. Deionized water (18 MΩ cm) was obtained from a Millipore (Bedford, MA) water purification system. All purchased chemicals were used without further purification.
2.2 Primary amine specific shift reagent
Peptides with differing numbers of primary amine sites (e.g. n-terminal primary amine and lysine residues) were selectively labeled by an IM-MS shift reagent consisting of a lanthanide-chelation moiety, a short linker region, and an N-hydroxysuccinimide moiety for the specific modification of primary amine sites. Labeling reactions were performed as illustrated in Scheme 1 whereby primary amine sites were modified by adding a ten-fold molar excess of amine specific label using a borate buffer at a pH of 10. Subsequent to amine modification, a lanthanide (Ln(III)) metal atom is chelated within the macrocycle by adding a ten-fold molar excess of Ln(III) and heating to 80°C in an acetate buffered solution at a pH of 5.6 [10].
Scheme 1.

An illustration of labeling reactions for primary amine specific (R1 = N-hydroxysuccinimide) and cysteine specific (R2 = maleimide) shift reagents. The incorporation of the lanthanide metal (Ln III) provides the necessary properties of high mass and relatively small increased collision cross section required for high density shift reagents.
All labeled peptides were desalted using ziptips (Millipore) prior to analysis. MS analysis was performed on an Applied Biosystems (Carlsbad, CA) Voyager DE MALDI-TOFMS. Ion mobility-mass spectrometry analysis was performed using a Synapt G2 HDMS (Waters Inc., Manchester, England). Labeled peptide samples were spotted onto a MALDI target plate using the dried droplet method with 2,5-dihydroxybenzoic acid (DHB) as the matrix in a matrix:analyte ratio of approximately 100:1.
2.3 Cysteine specific shift reagent
Model cysteine-containing peptides were labeled using a shift reagent consisting of a lanthanide-chelation moiety, a short linker region, and a maleimide group for the selective modification of the thiol sidechain of cysteine residues. Labeling reactions were performed as illustrated in Scheme 1. Briefly, a Ln(III) metal was chelated with the macrocycle by heating a ten-fold excess of metal and label to 80°C in an acetate buffered solution at a pH of 5.6. Subsequent to metal chelation, cysteine-containing peptides were modified by adding a ten-fold excess of lanthanide-shift reagent complex to the peptides [10]. All labeled peptides were desalted using ziptips (Millipore) prior to MS analysis as detailed above.
2.4 Phosphopeptide specific shift reagent
IM-MS separations selective for phosphorylation residues were demonstrated using bovine beta-casein in a proof-of-concept experiment. The protein was dissolved in ammonium bicarbonate buffer (pH 8.0) and thermally denatured at 90°C for 20 minutes and quenched at -20°C [11]. Reduction and alkylation was not performed as there are no native cystine bonds. The protein was subsequently digested with trypsin in a 1:40 weight to weight ratio for 16-20 hours at ∼37°C and purified by C-18 spin columns (Pierce, Rockford, IL) prior to derivatization. Since beta-elimination of glycans is a possible side reaction, enrichment of phosphopeptides using TiO2 or immobilized metal affinity chromatography (IMAC) may be required prior to phosphopeptide labeling.
Labeling reactions were performed as illustrated in Scheme 2, whereby tryptic peptides were subjected to a one-pot beta-elimination (Scheme 2(i)) followed by Michael addition [12] (Scheme 2(ii)), whereby the site of phosphorylation loses phosphoric acid followed by addition of an ethanedithiol linker. This reaction mixture consisted of 2.5 mM EDTA, 0.2 M ethanedithiol, 0.5 M NaOH, 1.5 M acetonitrile, 1.5 M ethanol, 5 M DMSO, and water. The reaction was allowed to progress for 1-2 hrs under nitrogen at 55°C in a manner similar to reaction conditions described previously [13-15]. The samples were then neutralized and purified by gel filtration (Sephadex G-10, Sigma). Subsequently, the thiolated peptides were labeled with a 10-fold excess of maleimido-DOTA (Scheme 2(iii)) in a mixture containing acetate buffer (pH 5.5) and DMSO in 1:1 ratio (v/v), resulting in a covalent bond between the free sulfhydryl group and the maleimide portion of the lanthanide-based tag. Finally, selected lanthanide metals were chelated to the maleimide portion of the tag by adding a 100-fold molar excess of metal to peptide and heating to 80°C for 45 minutes (Scheme 1(iv)) [10].
Scheme 2.
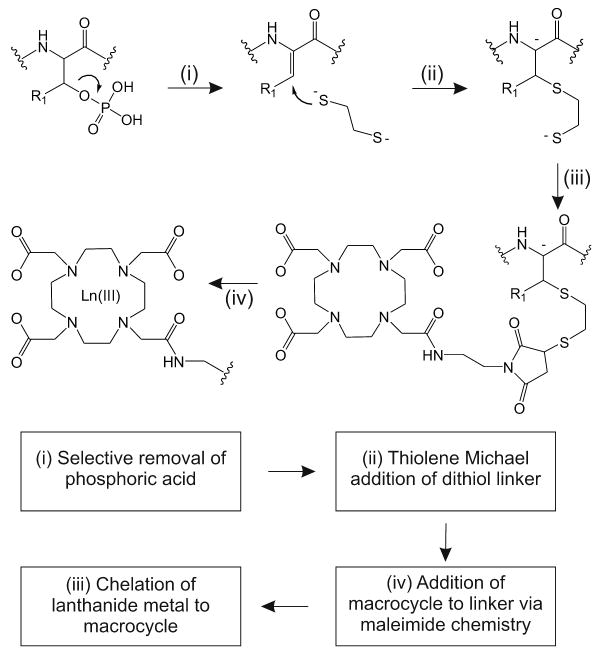
A schematic diagram of the labeling reactions utilized for selective labeling at sites of phosphorylation. See text for a description of the specific reaction conditions.
Differentially labeled samples were then combined and desalted by C-18 spin columns (according to Pierce protocol) and analyzed by MALDI-IM-MS. Peaks exhibiting a negative deviation from the peptide correlation line were selected for IM-MS/MS fragmentation in the transfer portion of the IM-MS.
2.5 Protein digestion protocol
Proteins used for simulation of complex mixtures were digested according to previously published methods [11]. Briefly, proteins were reduced (if required) and heat denatured at 90°C. Ammonium bicarbonate buffer (pH ∼8) was added followed by trypsin in a ratio of 1:50 w:w. Digestion was allowed to proceed overnight (approximately 18-24 hours) at ∼37°C. Upon completion, the resulting peptides were desalted using C18 spin columns. Labeled peptides were finally desalted using ziptips prior to analysis.
2.6 Multiplexed labeling
Multiplexed labeling was accomplished by labeling primary amines first, followed by the modification of cysteine residues. Briefly, primary amine sites were modified followed immediately by incorporation of a lanthanide metal. In a separate reaction, a different lanthanide metal was chelated with the cysteine specific label. The lanthanide-cysteine label complex was then used to modify cysteine residues on the labeled peptide. Different metals can be used to optimize the overall mass shift of the multiply labeled peptide. Samples subjected to multiplex labeling were desalted using Ziptips (Millipore) prior to MS and IM-MS analysis as described above.
3. Results and discussion
3.1 Utility of ion mobility shift reagents
High density shift reagents function by shifting specific peptide signals to an area in IM-MS conformation space where the parent signal is not predicted to occur in the absence of labeling. For peptides this corresponds to a drift time shorter than 5-7% of the deviation about the ion mobility versus m/z correlation at a given mass. The large mass of the lanthanide and the minimal branching of the label ensures that labeled peptide signals are shifted to an area below the peptide correlation band as illustrated in (Figure 2). Qualitatively, shift reagents allow for rapid, confident identification of peptides since the selected reagent is selective for reaction at a specific chemical functionality.
Figure 2.
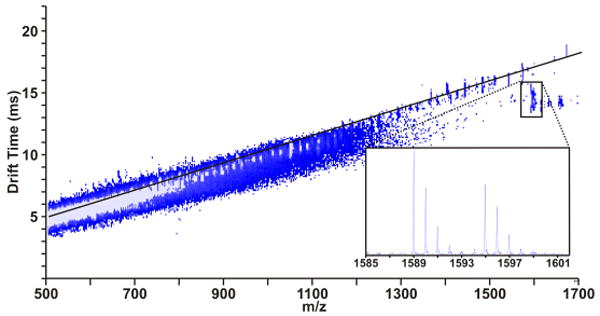
A representation of IM-MS relative quantitation analysis of a angiotensin II (DRVYIHPF) labeled with the primary amine specific label (Tb:Ho) in a 2:1 ratio. The extracted mass spectrum of the labeled pair illustrating relative quantitation is shown in the inset. Analysis was carried out using MALDI-IM-MS.
Labeling primary amines ensures that peptides from a complex mixture (e.g. a tryptic digest) each are labeled at the n-terminus. Peptides that contain a lysine residue are labeled twice (i.e. the n-terminus and lysine side chain). While the correlation band is shifted since each peptide is labeled, the addition of subsequent labels shift the signal even further from the unlabeled peptide correlation band. Through using different lanthanides to chelate the label, relative quantitation between different samples can be performed [20]. Furthermore, lanthanide-based shift reagents are useful in the site localization of specific functionality since the labels remain intact during typical tandem MS fragmentation conditions as discussed below.
3.2 Phosphopeptide labelling
Protein phosphorylation constitutes a large portion of biologically important post-translational modifications [16-18], and elucidating sites of phosphorylation presently remains challenging [19]. Challenges in phosphoproteomics continue to be lability of the phosphate group and the substoichiometric amounts of phosphorylation. By selective labeling of this PTM with a high density shift reagent followed by IM-MS analysis, phosphorylated peptides and proteins are shifted to regions of IM-MS conformation space where signal is not predicted to occur. In this manner, phosphorylated peptides may be identified for fragmentation and further analysis (i.e., site identification) in a manner more rapid and more efficient than traditional data-dependent and data-independent tandem MS methodologies.
To demonstrate our phosphorylation labeling strategy, bovine beta-casein, a model phosphorylated protein, was selectively labeled with a Ho-chelated mobility-shift label to visually identify phosphorylated peptides in 2D IM-MS conformation space prior to fragmentation. The resulting underivatized tryptic peaks that establish the peptide correlation line and the derivatized phosphorylated peptides are shown in Figure 3(a). Upon visualization of a negative deviation from the peptide correlation line, mobility shifted phosphorylated peptides were selected for fragmentation to elucidate the site of phosphorylation. One mobility-shifted peak was identified as a Ho-labeled tryptic peptide FQpSEEQQQTEDELQDK. The tandem spectrum for this selected peptide is shown in Figure 3b. The expected b/y series ions were observed, including b ion coverage from the b3 to the b15 ion, labeled y14 and y15 ions, y9, y11, y13, and several labeled water loss ions with little evidence for fragmentation of the label. The stability of this label enables predictable mass shifts of anticipated b and y ions which gives an indication of the site of modification previously phosphorylated [7]. Furthermore, because this tag is not labile and does not show phosphorylation site rearrangement, it has an added advantage of more confident phosphorylation site assignment when rearrangement reactions are possible [19].
Figure 3.
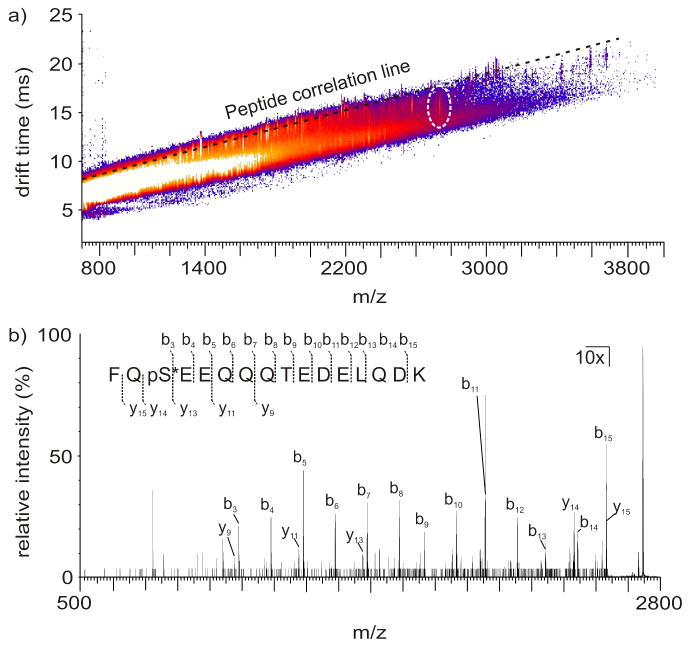
(a) 2D IM-MS plot of derivatized tryptic beta-casein. The signal corresponding to derivatized FQpSEEQQQTEDELQDK (indicated in a white dashed circle) exhibits a negative deviation from the peptide correlation line (indicated by a black dashed line), facilitating visual identification prior to fragmentation. (b) Fragmentation spectrum of a Ho-labeled tryptic beta-casein phosphorylated peptide having the sequence FQpSEEQQQTEDELQDK. Spectral peaks from 500 m/z to 2730 m/z were enhanced 10x to increase visibility of b and y spectral assignments, as the [M+H]+ ion dominated the spectrum. Observed fragmentation peaks are indicated on the peptide sequence above the spectrum and indicated in the spectrum. Fragmentation coverage of 86.7% and 33.3% of the labeled peptide was observed for b and y ions, respectively. One additional y ion was located, but were not reported due to inadequate S/N. Importantly, all of the anticipated ions corresponding to labeled positions are observed, demonstrating the utility of this label for phosphopeptide site identification as well as relative quantitation.
3.3 Multiplexed labeling
Multiplexed labeling, or labeling multiple functionalities on a single peptide, is also demonstrated using lanthanide-based shift reagents (Figure 4). For example, labeling of primary amine and cysteine sites are accomplished using sequential labeling reactions. Using a different lanthanide metal for each label affords the ability to quantify the relative ratio of multiple functionalities on a single peptide, for example using inductively coupled plasma-based instruments. The relative ratio of functionalities would allow for higher confidence in assigning peptide identification. Since the labels remain intact through collision induced dissociation, tandem MS experiments for peptide identification and functionality site localization are possible.
Figure 4.
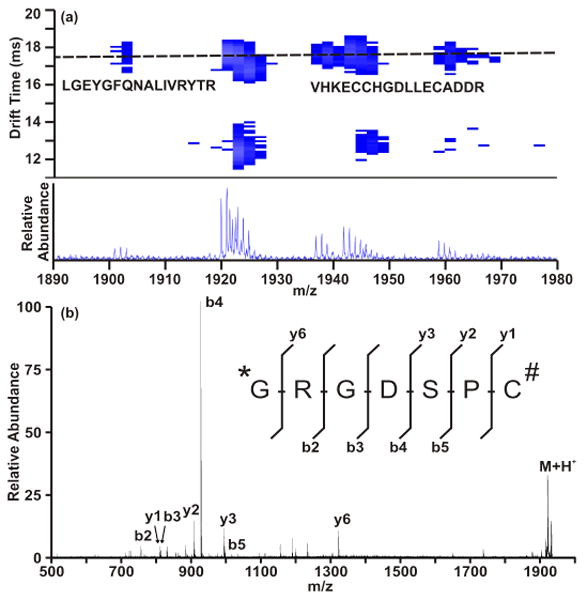
(A) representation of IM-MS analysis of multiplexed labeling. Briefly, a peptide (sequence shown in part (B)) was labeled at both the N-terminal primary amine (Tb labeled) and a cysteine residue (Ho labeled) according to scheme 1. Panel (A) illustrates the peptide correlation band (illustrated as a dashed line) showing a tryptic digest of bovine serum albumin. The lower peaks are due to the multiply labeled peptide shown in (B). The labeled peptide was subjected to MS/MS to ensure confident identification and label stability.
4. Conclusion
IM-MS shift reagents can be used to gain several types of information from samples. These reagents can be used to identify several relevant chemical functionalities, including primary amine sites, cysteine sites, and phosphorylation sites. In addition, the use of different metals for different samples affords the utility of relative quantitation experiments. Primary amine sites work well as targets for relative quantitation labels since nearly all tryptic peptides have at least one primary amine site (n-terminus). Cysteine labeling can be used to locate cysteine residues within peptides, thus increasing the confidence of peptide identification. Furthermore, phosphorylation sites may be elucidated in a manner more rapid and with greater confidence than traditional data-dependent tandem MS methods. Multiplexed labeling can also be used to rapidly identify specific peptides that contain both primary amine sites (e.g. lys residues) and cysteine residues for enhanced characterization capabilities over contemporary proteomic approaches.
Acknowledgments
One of the authors (JAM) had the distinct pleasure and opportunity to perform research with the Jülich mass spectrometry group under the direction of Dr. Hans-Joahim Dietze and Dr. J. Sabine Becker. These opportunities provided extraordinary experiences and insight into technological developments in mass spectrometric approaches. On the occasion of Dr. Dietze's 75th birthday I would like to express my deepest gratitude to him for his willingness to share his deep scientific insights, intellectual curiosity, and constant cheerfulness with young scientists at formative stages of their careers and colleagues at all stages. His personal and scientific contributions are profound at all levels. Financial support for this work was provided by the NIH (1R01GM092218-01 and RC2DA028981), the U.S. Defense Threat Reduction Agency (HDTRA-09-1-0013), and the Vanderbilt University College of Arts and Sciences, the Vanderbilt Institute of Chemical Biology (pilot project grant), the Vanderbilt Institute for Integrative Biosystems Research and Education, and the American Society for Mass Spectrometry (Research Award to JAM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Unwin RD, Evans CA, Wheton AD. Relative quantification in proteomics: new approaches for biochemistry. Trends in Biochem Sci. 2006;31:473–484. doi: 10.1016/j.tibs.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Chelius D, Bondarenko PV. Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J of Proteome Research. 2002;1:317–323. doi: 10.1021/pr025517j. [DOI] [PubMed] [Google Scholar]
- 4.McLean JA, Ruotolo BT, Gillig KJ, Russell DH. Ion mobility-mass spectrometry: a new paradigm for proteomics. Int J of Mass Spectrometry. 2005;240:301–315. [Google Scholar]
- 5.Tao L, McLean JR, McLean JA, Russell DH. A collision cross-section database of singly-charged peptide ions. J Am Soc Mass Spectrom. 2007;18:1232–1238. doi: 10.1016/j.jasms.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Hilderbrand AE, Myung S, Clemmer DE. Exploring crown ethers as shift reagents for ion mobility spectrometry. Anal Chemistry. 2006;78:6792–6800. doi: 10.1021/ac060439v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gant-Branum RL, Kerr TJ, McLean JA. Labeling strategies in mass spectrometry-based protein quantitation. Analyst. 2009;134:1525–1530. doi: 10.1039/b904643g. [DOI] [PubMed] [Google Scholar]
- 8.Whetstone PA, Butlin NG, Corneille TM, Meares CF. Element-coded affinity tags for peptides and proteins. Bioconjugate chemistry. 2004;15:3–6. doi: 10.1021/bc034150l. [DOI] [PubMed] [Google Scholar]
- 9.Ahrends R, Pieper S, Kuhn A, Weisshoff H, Hamester M, Lindemann T, Scheler C, Lehmann K, Taubner K, Linscheid MW. A metal-coded affinity tag approach to quantitative proteomics. Moll and Cell Proteomics. 2007;6:1907–1916. doi: 10.1074/mcp.M700152-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Zhu X, Lever SZ. Formation kinetics and stability studies on the lanthanide complexes of 1,4,7,10- tetraazacyclododecane-N,N′,N″,N‴ -tetraacetic acid by capillary electrophoresis. Electrophoresis. 2002;23:1348–1356. doi: 10.1002/1522-2683(200205)23:9<1348::AID-ELPS1348>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 11.Park ZY, Russell DH. Thermal denaturation: a useful technique in peptide mass mapping. Anal Chem. 2000;72:2667–2670. doi: 10.1021/ac991444k. [DOI] [PubMed] [Google Scholar]
- 12.Hoyle CE, Bowman CN. Thiol-ene click chemistry. Angewandte Chemie Int Ed. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 13.Meyer HE, Hoffmann-Posorske E, Korte H, Heilmeyer LMG., Jr Sequence analysis of phosphoserine-containing peptides: modification for picomolar sensitivity. FEBS letters. 1986;204:61–66. doi: 10.1016/0014-5793(86)81388-6. [DOI] [PubMed] [Google Scholar]
- 14.Mega T, Hamazume Y, Nong YM, Ikenaka T. Studies on the methods for the determination of phosphorylation sites in highly phosphorylated peptides or proteins: phosphorylation sites of hen egg white riboflavin binding protein. J Biochem. 1986;100:1109–1116. doi: 10.1093/oxfordjournals.jbchem.a121814. [DOI] [PubMed] [Google Scholar]
- 15.Fadden P, Haystead TAJ. Quantitative and selective fluorophore labeling of phosphoserine on peptides and proteins: characterization at the attomole level by capillary electrophoresis and laser-induced fluorescence. Anal Biochem. 1995;225:81–88. doi: 10.1006/abio.1995.1111. [DOI] [PubMed] [Google Scholar]
- 16.Mann M, Ong SE, Grønborg M, Steen H, Jensen ON, Pandey A. Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends in Biotech. 2002;20:261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 17.Schroeder MJ, Webb DJ, Shabanowitz J, Horwitz AF, Hunt DF. Methods for the detection of paxillin post-translational modifications and interacting proteins by mass spectrometry. 2005;4:1832–1841. doi: 10.1021/pr0502020. [DOI] [PubMed] [Google Scholar]
- 18.Garcia BA, Shabanowitz J, Hunt DF. Analysis of protein phosphorylation by mass spectrometry. Methods. 2005;35:256–264. doi: 10.1016/j.ymeth.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Palumbo AM, Reid GE. Evaluation of gas-phase rearrangement and competing fragmentation reactions on protein phosphorylation site assignment using collision induced dissociation-ms/ms and ms3. Anal Chem. 2008;80:9735–9747. doi: 10.1021/ac801768s. [DOI] [PubMed] [Google Scholar]
- 20.Kerr TJ, McLean JA. Peptide quantitation using primary amine selective metal chelation labels for mass spectrometry. Chem Comm. 2010;46:5479–5481. doi: 10.1039/b926290c. [DOI] [PubMed] [Google Scholar]