

Abstract
The transcription factor NPAS2 is one of nine human core circadian genes that influence a variety of biological processes by regulating the 24-h circadian rhythm. Recently, it has been shown that NPAS2 is a risk biomarker in human cancers and plays a role in tumorigenesis by affecting cancer-related gene expression, and relevant biological pathways. However, it is difficult to study the biological involvement of NPAS2 in cancer development, as little is known about its direct transcriptional targets. The aim of the current study is to create a transcriptional profile of genes regulated by NPAS2, using a human binding ChIP-on-chip analysis of NPAS2 in MCF-7 cells. This genome-wide mapping approach identified 26 genes that contain potential NPAS2 binding regions. Subsequent real-time PCR assays confirmed 16 of these targets, and 9 of these genes (ARHGAP29, CDC25A, CDKN2AIP, CX3CL1, ELF4, GNAL, KDELR1, POU4F2, and THRA) have a known role in tumorigenesis. In addition, a networking analysis of these validated NPAS2 targets revealed that all nine genes, together with REN, are involved in a “Cancer, Cell cycle, Neurological Disease” network. These results report the first list of direct transcriptional targets of NPAS2 and will shed light on the role of circadian genes in tumorigenesis.
Keywords: NPAS2, ChIP-on-chip, Transcriptional targets
1. Introduction
Neuronal PAS domain protein 2 (NPAS2) is the largest of the circadian genes (176.68 kb) and maps on chromosome 2 at 2q11.2. NPAS2 encodes for a member of the basic helix-loop-helix-PAS class of transcription factors and is expressed in the mammalian forebrain and several peripheral tissues. NPAS2 forms heterodimers with BMAL1 (brain and muscle ARNT-like protein 1, also known as aryl hydrocarbon receptor nuclear translocator-like (ARNTL)), and transcriptionally activates expression of the circadian genes PER and CRY, which are required for maintaining biological rhythms in many organisms [1,2]. In animal studies, altered circadian patterns of sleep and behavioral adaptability have been observed in NPAS2-deficient mice [3]. Previous evidence has also suggested that deficiency in NPAS2 leads to decreased oscillation of other circadian genes such as PER1, suggesting a negative impact on the periodicity present in normally functioning molecular clock mechanisms [4].
In addition to its involvement in circadian regulation, emerging data have demonstrated a novel role of NPAS2 as a risk biomarker in human cancers, and a substantial impact of NPAS2 on tumor related biological pathways such as cell cycle checkpoint and DNA repair. For example, our previous genetic epidemiological studies have demonstrated significant associations between a missense polymorphism (Ala394Thr) in NPAS2 and risk of breast cancer [5], prostate cancer [6] and non-Hodgkin’s lymphoma [7]. In addition, the NPAS2/BMAL1 heterodimer has been shown to mediate the binding of the oncogene c-myc and suppress its transcription [8]. Moreover, our recent in vitro functional analyses have provided new evidence that NPAS2 is important for DNA damage response, and functions as a potential tumor suppressor [9].
Despite this growing body of evidence suggesting that NPAS2 may be associated with cancer risk, the underlying mechanisms of NPAS2’s role in tumorigenesis are far from complete, mainly because very few of its direct transcriptional targets have been identified. It has been shown that expression of the circadian genes PER1, PER2, and CRY1 are positively regulated by NPAS2/BMAL1 heterodimers in the circadian feedback loop [4]. However, it is still not clear whether NPAS2 binds directly to the binding sequences of these genes. The aim of this study was to identify direct transcriptional target genes of the circadian gene NPAS2. To this end, we performed a chromatin immunoprecipitation followed by microarray (ChIP-on-chip) assay, the gold standard for determining DNA binding targets for transcription factors. Targets identified by ChIP-on-chip were validated by real-time PCR and siRNA analyses. A networking analysis was also performed using identified target genes to gain more insight into the role of NPAS2 in cancer development.
2. Materials and methods
2.1. Cell culture
The breast cancer cell line MCF-7 and an immortal human epithelial cell line MCF-10A were obtained from American Type Culture Collections (Manassas, VA). MCF-7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 0.01 mg/ml insulin, and 1% penicillin/streptomycin (Sigma–Aldrich, St. Louis, MO). MCF-10A cells were maintained in RPMI modified medium (Invitrogen) supplemented with 5% horse serum (Invitrogen), 13 mg/ml bovine pituitary extract, 10 μg/ml human epidermal growth factor, 5 mg/ml insulin, and 0.5 mg/ml hydrocortisone (Lonza, Walkersville, MD). Cells were incubated in a humidified incubator at 37 °C and 5% CO2.
2.2. Western blotting
Cell lysate was prepared using the standard protocol, and protein concentration was determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA) with BSA as standards. Proteins were resolved on 4–12% Novex Bis-Tris gradient denaturing polyacrylamide gels (Invitrogen), transferred to a polyvinylpyrrolidine difluoride membrane, and blotted with Rabbit polyclonal anti-NPAS2 (sc-28708, Santa Cruz) at 4 °C overnight. The membrane was then washed with fresh blotto three times for 10 min, and incubated with alkaline phosphatase-conjugated secondary antibody for an hour at room temperature. Enhanced chemifluorescence reagent (Amersham Life Science Ltd., Buckinghamshire, United Kingdom) was applied to the membrane according to the manufacturer’s instructions, and the chemiluminescent signal was visualized using a Kodak BIOMAX Light Film.
2.3. Chromatin immunoprecipitation
The ChIP assay was performed using the ChIP Assay Kit (Millipore, Billerica, MA) according to the manufacturer’s protocol. Briefly, cells were grown in 100 mm cell culture plates to 80% confluence. Cross-linking of protein and DNA was performed using cell culture medium containing 1% formaldehyde at room temperature for 10 min. Sonication was performed using an Omni Ruptor 250 Ultrasonic Homogenizer (Omni International, Marietta, GA) to generate input material. This input sample was then incubated with primary antibody (anti-NPAS2) and negative control (Rabbit IgG) for immunoprecipitation (IP). Agarose beads were added to the reactions to bind protein-conjugated antibody. The antibody–protein–DNA complex was then eluted from the agarose beads with freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3) and the cross-linking was reversed by adding 5 M NaCl for 4 h at 65 °C followed by proteinase K digestion at 45 °C for 1 h. The final DNA, representing NPAS2 target sequences, was purified using the GENECLEAN Turbo kit (Qbiogene, Carlsbad, CA) according to the manufacturer’s instructions. Ligation-mediated PCR (LM-PCR) (Agilent Technologies, Foster City, CA) was used to amplify DNA according to the manufacturer’s protocol. Two ChIP assays were performed independently and both amplified DNA samples were used in the subsequent human binding microarray analysis.
2.4. Binding microarray analysis
We worked with MOgene Inc. (St. Louis, MO) to perform the Human Binding Array (Agilent) using the amplified DNA obtained from the ChIP experiment. Labeling, hybridization, and image analyses were all performed at MOgene, an Agilent certified service provider for ChIP-on-chip analysis. The binding array for human promoter includes a total of 488 k probes that target ~17,000 of the best defined human transcripts covering 5.5 kb upstream to 2.5 kb downstream from the transcriptional start sites. A whole-chip error model [10,11] was used for array data analysis and to call a bound gene (target gene). The neighborhood error model takes into account neighboring probes and calls a region as bound, not just a single probe. Genes associated with significant regions are therefore considered target genes. The binding array analysis was performed for both duplicate samples from the ChIP assay. The complete microarray data were uploaded to the Gene Expression Omnibus (GEO) database (<http://www.ncbi.nlm.nih.gov/projects/geo/>): accession #GSE15052.
2.5. Validation of bound genes
Genes identified as potential NPAS2 targets by ChIP-on-chip were further validated by real-time PCR. For each bound region within a target gene we chose the probe with the lowest p-value and designed primers around this 60-mer binding probe. Simultaneously, we chose an internal probe in the same gene and designed an additional set of primers around that internal 60-mer to serve as a control. Therefore, for each gene, two sets of primer pairs were designed to amplify both the NPAS2 binding region and a non-specific control region. The Primer3 program (<http://frodo.wi.mit.edu/>) was used for primer design (Supplemental Table 1), and standard duplicated real-time PCR assays were performed using Power SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA). PCR products from the binding primers were normalized to products from internal primers for both chip-enriched DNA and input material. Fold changes of enrichment were quantified according to the 2−ΔΔCt method.
2.6. NPAS2 knockdown and bound gene expression
siRNA oligos targeting NPAS2 (Duplex Sense: CCCAGGGUCCAAAGCCAAUGAGA-AG; Antisense: CUUCUCAUUGGCUUUGGACCCUGGGUU) and a scrambled-sequence negative control siRNA with no known homology to the human genome to control for the effects of transfection, were chemically synthesized by Integrated DNA Technologies (IDT, Coralville, IA). The oligos were transfected into MCF-7 using the Lipofectamine RNAi Max transfection reagent (Invitrogen) according to the manufacturer’s instructions. Cells were harvested 48 h post-transfection and RNA was isolated using the RNA Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Primers for NPAS2 and all identified target genes from ChIP-on-chip analysis were designed in-house and chemically synthesized by IDT (Supplemental Table 2). Quantitative, real-time reverse transcription PCR (qRT-PCR) conditions were prepared using the Quantitect one-step kit (Qiagen) according to the manufacturer’s protocol. Standard duplicated qRT-PCRs with disassociation curves were performed on a Stratagene MX3000P instrument (Stratagene, La Jolla, CA). RNA quantity was normalized to HPRT, and changes in RNA expression were quantified according to the 2−ΔΔCt method.
2.7. Networking analysis of identified NPAS2 transcriptional targets
To further explore whether identified NPAS2 transcriptional targets are involved in any cancer-related pathways, we built a network of the validated target genes using the Ingenuity Pathway Analysis (IPA) program (Ingenuity Systems Inc., www.ingenuity.com). IPA is a comprehensive pathway analysis software tool that builds up biological pathways from high throughput data such as co-expressed genes identified by expression arrays. It can establish networks of functional relationships (transcriptional regulation, cell signaling, metabolism, etc.) directly among sets of genes or through interactions with other genes, utilizing findings in previous scientific literature. In our study, we input all confirmed NPAS2 binding genes into the program to construct hypothetical dynamic models of the functions of NPAS2 target genes.
3. Results
3.1. NPAS2 expression in breast cancer cell line MCF-7
It is now clear that molecular clocks reside not only in a central pacemaker, but also in peripheral tissues, even in immortalized cells [12,13]. Previous evidence suggests that circadian genes, including NPAS2, show detectable circadian mRNA expression in all tissues except testis [14]. Before beginning our study we confirmed the presence of measurable levels of NPAS2 expression in MCF-7 cells using quantitative reverse transcription PCR (qRT-PCR), as well as the presence of NPAS2 protein in MCF-7 cells by Western blotting. The results from our qRT-PCR assays demonstrate that NPAS2 was expressed at measurable mRNA levels in MCF-7 cells (Fig. 1A). The Western blotting results indicated a strong band corresponding to NPAS2 protein at about 90 kD (Fig. 1B1), and β-actin at 42 kD was used as control (Fig. 1B2).
Fig. 1.
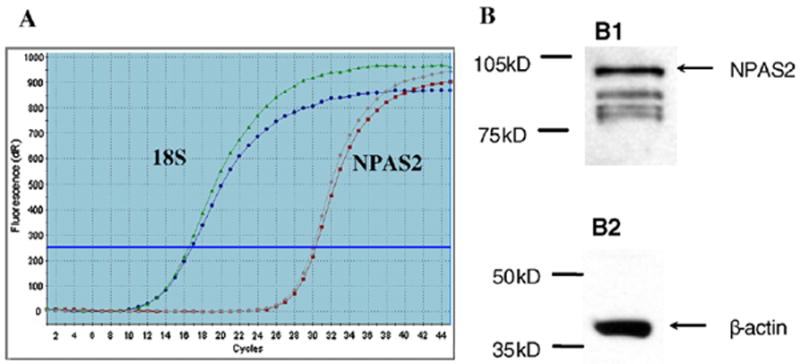
Expression of NPAS2 in MCF-7 cells. A: PCR amplification plot demonstrating measurable mRNA expression of NPAS2 in MCF-7 cells. Curves on the left correspond to 18s rRNA, while those on the right are for NPAS2. Each curve represents the average of two replicates from each of two separately isolated RNA samples. B: NPAS2 protein expression in MCF-7 cells. B1: Membrane blotted with rabbit polyclonal anti-NPAS2, and a strong band corresponding to NPAS2 at about 90 kD. B2: Membrane stripped and blotted with mouse monoclonal anti-β-actin as a loading control at about 42 kD.
3.2. Chromatin immunoprecipitation of NPAS2
In order to obtain accurate results in the ChIP assay, it is critical to have DNA fragments between 100–1000 bp. Therefore, we first tested the sonication conditions using an Omni Ruptor 250 Ultrasonic Homogenizer. We found that 30% power output and 50% pulse generated DNA fragments within the appropriate range, with the majority of the fragments at around 500 bp. Equal amounts of anti-NPAS2 or rabbit IgG control were used in the ChIP assay. To verify the ChIP results, protein was eluted and resolved using a 4–12% Bis-Tris gel (Invitrogen), and blotting was performed using anti-NPAS2. A strong band at about 90 kD was detected in the ChIP with anti-NPAS2 (Fig. 2, lane 1), but not in the ChIP with rabbit IgG (Fig. 2, lane 2). These results showed that the anti-NPAS2 we used in the study is sensitive and specific to bind with NPAS2 protein in the ChIP assay.
Fig. 2.
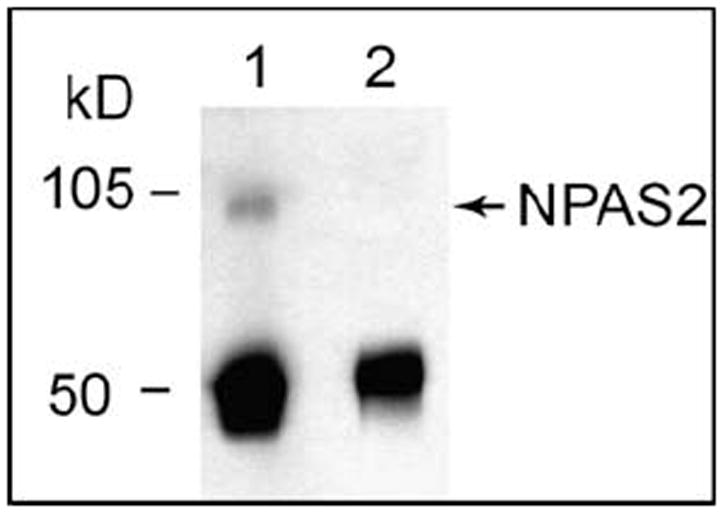
Chromatin immunoprecipitation with anti-NPAS2 antibody. Protein samples from IP with anti-NPAS2 (lane 1) or an equal amount of Rabbit IgG (lane2) were resolved on a 4–12% Bis-Tris gel and blotted with anti-NPAS2. There is a strong band of NPAS2 in the NPAS2 IP sample, but not in the negative control IgG IP sample. The MWs of NPAS2 and IgG are 90 kD and 50 kD, respectively.
3.3. Transcriptional targets of NPAS identified in ChIP-on-chip assay
The ChIP-on-chip assay was repeated twice. There were over 100 NPAS2 binding sequences identified in each experiment, and 64 over-lapping bound genes were detected in both experiments. After removal of repetitive genes and suspected false positives (e.g. genes located on the Y chromosome), 26 replicated target genes were identified for subsequent validation by quantitative real-time PCR. Of this initial 26, 16 targets were confirmed by qPCR to be significantly enriched by ChIP-on-chip analysis of NPAS2. These 16 confirmed genes were: ARHGAP29, BC033172, BRDG1, Clorf188, CADPS2, CDC25A, CDKN2AIP, CX3CL1, DNAH2, ELF4, GNAL, KDELR1, POU4F2, RD3, REN, and THRA. Detailed information regarding these genes, including gene symbol, chromosomal location, brief functionality and fold change of enrichment are shown in Table 1. The gene function description was summarized from information obtained from the Entrez Gene Summary and Ingenuity gene summary databases, which indicate that 9 out of the 16 identified genes are related to tumorigenesis.
Table 1.
Identified transcriptional binding targets of the circadian gene NPAS2.
| Gene symbol | Gene name | NPAS2 binding region | Position to the gene TSSb | Brief functiona | Enrichment fold changes |
|---|---|---|---|---|---|
| ARHGAP29 | Rho GTPase activating protein 29 | chr1:094477485–094477544 | −(1590–1649) | Signal transduction, candidate tumor suppressor gene in MCL | 4.35 |
| BC033172 | Hypothetical protein | chr4:183299087–183299133 | 3’ end +(664–710) | Function unknown | 1.66 |
| BRDG1 | Signal transducing adaptor family member 1 | chr4:068107287–068107346 | +(246–305) | Downstream of Tec tyrosine kinase in B cell antigen receptor signaling | 149.01 |
| C1orf188 | Ring finger protein 207 | chr1:006191778–006191822 | +(3002–3046) | Function unknown | 1.28 |
| CADPS2 | Ca2+-dependent activator protein for secretion 2 | chr7:122313589–122313633 | +(157–201) | Calcium ion binding, exocytosis protein transport. It is related to hyperactive behavior and autism | 1.29 |
| CDC25A | Cell division cycle 25A | chr3:48205294–48205338 | −(489–533) | CDC25A is required for G1/S and G2/M transition. It is an oncogene | 3.26 |
| CDKN2AIP | CDKN2A (p16) interacting protein | chr4:184602132–184602177 | −(651–606) | Binds to p16, p53, and HDM2, and involved in cell cycle, senescence, and DNA damage response | 2.88 |
| CX3CL1 | Chemokine (C-X3-C motif) ligand 1 | chr16:55964583–55964627 | +(668–712) | Mediates antitumor immune response | 1.97 |
| DNAH2 | Dynein, axonemal, heavy chain 2 | chr17:7556327–7556371 | +(7437–7393) | Belongs to dyneins protein family, components of multisubunit motor ATPase complexes | 88.65 |
| ELF4 | E74-like factor 4 | chrX:129072521–129072565 | −(368–412) | Transcription factor. Candidate tumor suppressor gene, regulates cell cycle entry | 14.38 |
| GNAL | Guanine nucleotide binding protein, alpha activating | chr18:11738236–11738288 | +(58972–59025) | Cellular transformation and tumor development | 1.92 |
| KDELR1 | KDEL (Lys-Asp-Glu-Leu) endoplasmic reticulum protein retention receptor 1 | chr19:53589176–53589220 | −(2554–2598) | Intracellular signal transduction, deleted in gliomas | 2.10 |
| POU4F2 | POU class 4 homeobox 2 | chr4:147779300–147779344 | −(151–195) | Transcription factor expressed in human retina ganglion cells. Regulates CDK4, CCND1, and it facilitates breast cancer progression and invasion | 1.61 |
| RD3 | Retinal degeneration 3 | chr1:209731637–209731681 | +(1201–1245) | Gene transcription and splicing | 1.13 |
| REN | Renin | chr1:202402110–202402165 | −(22–77) | Involved in angiotensin maturation, regulation of blood pressure | 1.35 |
| THRA | Thyroid hormone receptor, alpha | chr17:35473541–35473585 | +(952–996) | Mediates activities of thyroid hormone, expression level altered in thyroid and breast cancer | 17.63 |
3.4. Effect of NPAS2 silencing on target gene expression
We performed a loss-of-function experiment using small interfering oligos (siRNA), in order to determine the impact of NPAS2 reduction on the expression of target genes using cells derived from tumor and normal tissue. MCF-7 and MCF-10A cells were transfected with an NPAS2-targeting siRNA oligo, or a non-targeting negative control oligo. Percent reduction in target gene expression (relative to negative control and normalized to HPRT content) was then calculated for each target gene using the following formula: [1 – 2−ΔΔCt] × 100% (Fig. 3). Interestingly, in the MCF-7 cells, 6 of the 16 genes were reduced by 50% or more (ARHGAP29, 61.5%; BRDG1, 58.0%; CX3CL1, 90.2%; GNAL, 99.9%; POU4F2, 74.4%; and RD3, 50.7%). By contrast, 14 of the 16 target genes were significantly reduced in MCF-10A cells (ARHGAP29, 99.8%; BC033172, 91.9%; C1orf188, 71.6%; CAPDS2, 99.2%; CDC25A, 87.3%; CDKN2AIP, 61.7%; CX3CL1, 93.2%; DNAH2, 87.3%; ELF4, 79.3%; POU4F2, 54.6%; RD3, 76.4%; REN, 81.4%; and THRA, 89.0%). The remaining two genes, BRDG1, and GNAL, were reduced by 47.4% and 39.4%, respectively.
Fig. 3.
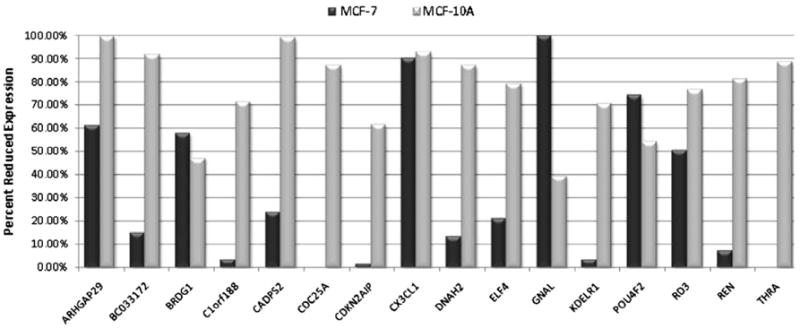
Reduced expression of target genes in MCF-7 and MCF-10A cells following NPAS2 silencing. Expression differences were calculated using the 2−ΔΔCt method comparing expression levels in cells treated with an NPAS2-targeting siRNA oligo to cells treated with a negative control siRNA. Expression of the HPRT gene was used for normalization. Percent reduction was calculated for each gene as: ([1–2−ΔΔCt] × 100%). CDC25A and THRA were moderately up-regulated in MCF-7 cells after NPAS2 knockdown, and are thus shown as 0% reduced. All other genes were down-regulated.
3.5. Cancer related network built from NPAS2 transcriptional targets
All of the 16 confirmed NPAS2 targets were input in the IPA pathway analysis software to determine whether they were functionally related. Ten of these genes were identified in a cancer- and cell cycle-related network connected via transcriptional regulation and protein–protein interactions (Fig. 4). Interestingly, this network contained all nine of the cancer-related target genes (ARHGAP29, CDC25A, CDKN2AIP, CX3CL1, ELF4, GNAL, KDELR1, POU4F2, and THRA). Each of these genes had previously established roles in tumor development, growth, and invasion in breast cancer or other human cancer types. The central molecules of the network were CDKN2A (p16, cyclin-dependent kinase inhibitor 2A), ERK protein, INFG (interferon gamma), and retinoic acid. The major function of the network identified by the IPA tool was “Cancer, Cell cycle, Neurological Disease”. Both CDC25A and CDKN2AIP directly interact with CDKN2A, a well known tumor suppressor that controls cell cycle progression. CX3CL1 and POU4F2 interact with IFNG, an important immune response mediator that has been shown to have a potential therapeutic effect in breast cancer [15]. ELF4 and ARHGAP29 are regulated by retinoic acid. The symbol, name, intracellular location, and gene family of all 35 genes contained in the network are listed in Supplemental Table 3.
Fig. 4.
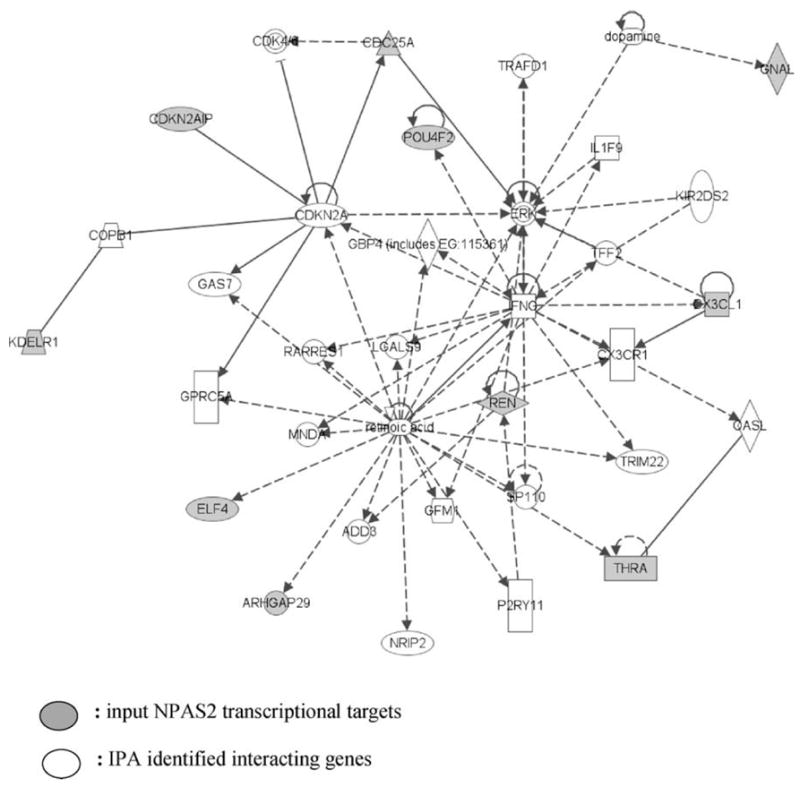
Network of confirmed NPAS2 ChIP enriched genes. Ten of the 16 confirmed genes belong to a network classified as “Cancer, Cell Cycle, Neurological Disease”. The network was generated using the Ingenuity program by inputting the 16 confirmed NPAS2 target genes. All the genes are functionally connected to each other by transcriptional regulation or by protein–protein interactions. The highlighted genes are NPAS2 transcriptional targets revealed by the ChIP-on-chip assay.
4. Discussion
Although little is known about NPAS2’s downstream transcriptional targets, it has been recently shown that NPAS2 is a risk biomarker in human cancers, and plays a role in tumorigenesis by affecting cancer-related genes and relevant biological pathways [5-7,9]. The current genome wide ChIP-on-chip analysis has identified 16 confirmed transcriptional targets of NPAS2, and nine of these genes have a known role in cancer development. Specifically, two NPAS2 targets, CDKN2AIP (also known as p16 interact protein) and CDC25A, are known cell cycle regulators. CDKN2AIP interacts with p16, p53, and MDM2 and is a critical regulator of the p16–p53–p21 pathway [16,17]. CDC25A is a member of the CDC25 family of phosphatases and is required for the G1/S and G2/M transition [18]. Three other target genes, ARHGAP29, POU4F2, and GNAL, have been shown to promote tumorigenesis and progression in human cancers. ARHGAP29, which negatively regulates Rho signaling, is a potential tumor suppressor in mantle cell lymphoma (MCL), since its expression is reduced by several mechanisms such as deletion and promoter methylation in MCL cell lines and primary MCL cases [19,20]. POU4F2 is a member of the POU domain family of transcription factors, which play important roles in the control of cell identity. It has been shown that POU4F2 increases breast cancer proliferation and invasion by transcriptional regulation, such as through augmenting the expression of cyclin D1 and CDK4 [21,22]. GNAL (G protein alpha) increases cell transformation [23], and promotes in vitro cell invasion and survival [24]. CX3CL1 is the sole CX3C chemokine and may mediate immune response against tumor cells [25,26]. KDELR1 is involved in intracellular signal transduction and is frequently lost in gliomas [27]. THRA may mediate biological activities of thyroid hormone and its expression level is altered in thyroid and breast cancer [28,29]. ELF4 is also a transcription factor and potential tumor suppressor gene that regulates cell cycle entry and is [30,31], underexpressed in micrometastatic breast cancer [32]. Overall, the cancer-related involvement of these transcriptional targets support recent findings suggesting NPAS2 is a potential tumor suppressor, in addition to its role in circadian regulation.
Moreover, transcriptional regulation and protein–protein interactions among these nine identified NPAS2 targets form a network which is biologically relevant for processes related to cell cycle and cancer. The central molecules of the network are p16, ERK, Interferon γ, and retinoic acid. They are important signaling molecules involved in multiple cellular processes such as cell cycle control, proliferation, and differentiation. These results suggest a dynamic model in which NPAS2 and its target genes are integral in cancer-related biological pathways such as cell cycle regulation and cell fate. Because the IPA software used in the study established potential gene interaction networks based solely on empirical knowledge extracted from existing published literature, it is possible that NPAS2 affects other cancer-related pathways as well.
Over 60% of the target genes identified in the ChIP-on-chip assay were confirmed by quantitative real-time PCR in our study, which is consistent with the 67–69% confirmation rate reported in a previous study correlating microarray expression data and real-time PCR results using an oligonucleotide based microarray [33]. It has been shown that genes with moderate expression levels are more likely to have high correlation between expression results and real-time PCR quantification, since extreme expression levels (too low or too high) are more likely to cause high false-positive rates in microarray based assays [34]. Indeed, this is what we observed in our ChIP-on-chip array, where the hybridization signals of the majority of the 16 verified genes were in the middle range, whereas most of the unconfirmed genes had signals in the two extreme ends. Specifically, among the 26 targets from the ChIP-on-chip analysis, nine genes have extreme intensity values (>2000 or <50), and only three of them were later verified by real-time PCR. In addition, there are 12 genes located on the Y chromosome that are clearly false positive targets, as MCF-7 cells are derived from a female patient. As expected, all of the 12 genes on the Y chromosome had extremely low intensities on the array (<50). Moreover, it cannot be ruled out that the statistical models used to identify positive binding sequences might generate false positive genes, or miss real targets [35].
Knockdown of NPAS2 by siRNA resulted in greater than 50% reduced expression in six of the 16 identified transcriptional targets in MCF-7 cells. However, the expression of 14 of the 16 genes was significantly affected by NPAS2 silencing in an immortalized “normal” human mammary epithelial cell line MCF-10A, which is non-tumorigenic in athymic mice. The fact that NPAS2 knockdown had a greater impact in normal cells relative to tumor cells underscores the fact that complex transcriptional regulatory processes, including those involving the circadian system, are often dysregulated in tumor cells. Since MCF-10A cells are more likely to mimic the conditions present in normal breast epithelium, it is of note that NPAS2 silencing had the potential to influence a number of cancer-related genes in these cells, as this provides further evidence suggesting that NPAS2 may operate as a tumor suppressor, and loss of NPAS2 function may be an early event in tumor development.
Within the circadian feedback loop, PER1, PER2, and CRY1 are regulated by NPAS2/BMAL1 heterodimers [4]. NPAS2/BMAL1 heterodimers have also been shown to directly bind to binding sites in PER1 and c-MYC [8]. However, neither of these genes were identified as direct NPAS2 transcriptional targets in our study. This might be due to a variety of reasons, including the stringent criteria used to identify true targets, or the tissue specific functions of the NPAS2 gene. It has previously been suggested that expression and regulation of circadian genes might be organ specific [36]. Previous NPAS2 studies were carried out in neuronal tissues or cultured neuron cell lines, whereas epithelial breast cancer cells were used in our ChIP-on-chip assay. It is possible that NPAS2 may regulate different transcriptional targets in different tissue types. Also, some genes have high binding affinity of NPAS2 by ChIP-PCR results (such as DNAH2), but did not show much change in the siRNA experiment. This might be caused by the complex nature of transcription since the binding of a TF to its binding sequence does not guarantee an active transcription as the process can be controlled by many other factors such as association with a co-activator or co-repressor [37].
It is of note that the number of transcriptional targets identified is quite low in ChIP-on chip analysis. One reason might be that the binding array for human promoter used in the study includes probes that target human transcripts covering 5.5 kb upstream to 2.5 kb downstream from the transcriptional start sites. As such, potential binding sites outside these regions can not be detected. Another reason could be that many human genes are not included in the analysis since the current array only covers about 17,000 of the best defined human transcripts. Thus, it is possible that there are potentially more NPAS2 transcriptional targets that have not been identified in the current analysis.
In summary, our results report the first list of direct transcriptional targets of NPAS2 identified from a genome-wide ChIP-on-chip array. Interestingly, over half of these target genes are cancer-related. There are still several questions which remain unanswered, including the identification of consensus binding sequences for NPAS2. E-box sequences (CANNTG) were found in 80% of our confirmed targets, but they were present in 81% of unconfirmed genes as well. As such, it is still not clear whether NPAS2 binds to E-box sequences, or other unknown motifs. Subsequent studies should also aim to address whether NPAS2 has tissue specific binding targets, and whether NPAS2 targets may change following an external challenge, such as exposure to mutagenic agents. Given the increasing evidence supporting a role of NPAS2 in cancer development, identification of its transcriptional targets is an important step towards a better understanding of NPAS2 biology, and will facilitate future mechanistic investigations of NPAS2 as a potential tumor suppressor.
Acknowledgments
This work was supported by the US National Institutes of Health (Grants CA122676, CA110937, and CA108369).
Footnotes
Conflict of interest There is no conflict of interest declared.
Appendix A. Supplementary material Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.canlet.2009.04.017.
References
- 1.Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–725. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley CA, Erbel-Sieler C, Estill SJ, Reick M, Franken P, Pitts S, McKnight SL. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science. 2003;301:379–383. doi: 10.1126/science.1082795. [DOI] [PubMed] [Google Scholar]
- 4.Reick M, Garcia JA, Dudley C, McKnight SL. NPAS2: an analog of clock operative in the mammalian forebrain. Science. 2001;293:506–509. doi: 10.1126/science.1060699. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y, Stevens RG, Leaderer D, Hoffman A, Holford T, Zhang Y, Brown HN, Zheng T. Non-synonymous polymorphisms in the circadian gene NPAS2 and breast cancer risk. Breast Cancer Res Treat. 2008;107:421–425. doi: 10.1007/s10549-007-9565-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu LW, Zhu Y, Yu K, Zheng T, Yu H, Zhang Y, Sesterhenn I, Chokkalingam AP, Danforth KN, Shen MC, Stanczyk FZ, Gao YT, Hsing AW. Variants in circadian genes and prostate cancer risk: a population-based study in China. Prostate Cancer Prostatic Dis. 2007 doi: 10.1038/sj.pcan.4501024. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Y, Leaderer D, Guss C, Brown HN, Zhang Y, Boyle P, Stevens RG, Hoffman A, Qin Q, Han X, Zheng T. Ala394Thr polymorphism in the clock gene NPAS2: a circadian modifier for the risk of non-Hodgkin’s lymphoma. Int J Cancer. 2007;120:432–435. doi: 10.1002/ijc.22321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50. doi: 10.1016/s0092-8674(02)00961-3. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman AE, Zheng T, Ba Y, Zhu Y. The circadian gene NPAS2 a putative tumor suppressor is involved in DNA damage response. Mol Cancer Res. 2008;6:1461–1468. doi: 10.1158/1541-7786.MCR-07-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- 11.Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 12.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 13.Schibler U, Sassone-Corsi P. A web of circadian pacemakers. Cell. 2002;111:919–922. doi: 10.1016/s0092-8674(02)01225-4. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto T, Nakahata Y, Soma H, Akashi M, Mamine T, Takumi T. Transcriptional oscillation of canonical clock genes in mouse peripheral tissues. BMC Mol Biol. 2004;5:18. doi: 10.1186/1471-2199-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Tunon I, Ricote M, Ruiz AA, Fraile B, Paniagua R, Royuela M. Influence of IFN-gamma and its receptors in human breast cancer. BMC Cancer. 2007;7:158. doi: 10.1186/1471-2407-7-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaul SC, Hasan K, Wadhwa R. CARF regulates p19ARF-p53-p21WAF1 senescence pathway by multiple checkpoints. Ann NY Acad Sci. 2006;1067:217–219. doi: 10.1196/annals.1354.026. [DOI] [PubMed] [Google Scholar]
- 17.Kamrul HM, Wadhwa R, Kaul SC. CARF binds to three members (ARF, p53, and HDM2) of the p53 tumor-suppressor pathway. Ann NY Acad Sci. 2007;1100:312–315. doi: 10.1196/annals.1395.033. [DOI] [PubMed] [Google Scholar]
- 18.Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, Okayama H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994;13:1549–1556. doi: 10.1002/j.1460-2075.1994.tb06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saras J, Franzen P, Aspenstrom P, Hellman U, Gonez LJ, Heldin CH. A novel GTPase-activating protein for Rho interacts with a PDZ domain of the protein–tyrosine phosphatase PTPL1. J Biol Chem. 1997;272:24333–24338. doi: 10.1074/jbc.272.39.24333. [DOI] [PubMed] [Google Scholar]
- 20.Ripperger T, von Neuhoff N, Kamphues K, Emura M, Lehmann U, Tauscher M, Schraders M, Groenen P, Skawran B, Rudolph C, Callet-Bauchu E, van Krieken JH, Schlegelberger B, Steinemann D. Promoter methylation of PARG1, a novel candidate tumor suppressor gene in mantle-cell lymphomas. Haematologica. 2007;92:460–468. doi: 10.3324/haematol.10337. [DOI] [PubMed] [Google Scholar]
- 21.Samady L, Dennis J, Budhram-Mahadeo V, Latchman DS. Activation of CDK4 gene expression in human breast cancer cells by the Brn-3b POU family transcription factor. Cancer Biol Ther. 2004;3:317–323. [PubMed] [Google Scholar]
- 22.Budhram-Mahadeo VS, Irshad S, Bowen S, Lee SA, Samady L, Tonini GP, Latchman DS. Proliferation-associated Brn-3b transcription factor can activate cyclin D1 expression in neuroblastoma and breast cancer cells. Oncogene. 2008;27:145–154. doi: 10.1038/sj.onc.1210621. [DOI] [PubMed] [Google Scholar]
- 23.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 24.Regnauld K, Nguyen QD, Vakaet L, Bruyneel E, Launay JM, Endo T, Mareel M, Gespach C, Emami S. G-protein alpha(olf) subunit promotes cellular invasion, survival, and neuroendocrine differentiation in digestive and urogenital epithelial cells. Oncogene. 2002;21:4020–4031. doi: 10.1038/sj.onc.1205498. [DOI] [PubMed] [Google Scholar]
- 25.Yu YR, Fong AM, Combadiere C, Gao JL, Murphy PM, Patel DD. Defective antitumor responses in CX3CR1-deficient mice. Int J Cancer. 2007;121:316–322. doi: 10.1002/ijc.22660. [DOI] [PubMed] [Google Scholar]
- 26.Tang L, Hu HD, Hu P, Lan YH, Peng ML, Chen M, Ren H. Gene therapy with CX3CL1/Fractalkine induces antitumor immunity to regress effectively mouse hepatocellular carcinoma. Gene Ther. 2007;14:1226–1234. doi: 10.1038/sj.gt.3302959. [DOI] [PubMed] [Google Scholar]
- 27.Smith JS, Tachibana I, Pohl U, Lee HK, Thanarajasingam U, Portier BP, Ueki K, Ramaswamy S, Billings SJ, Mohrenweiser HW, Louis DN, Jenkins RB. A transcript map of the chromosome 19q-arm glioma tumor suppressor region. Genomics. 2000;64:44–50. doi: 10.1006/geno.1999.6101. [DOI] [PubMed] [Google Scholar]
- 28.Onda M, Li D, Suzuki S, Nakamura I, Takenoshita S, Brogren CH, Stampanoni S, Rampino N. Expansion of microsatellite in the thyroid hormone receptor-alpha1 gene linked to increased receptor expression and less aggressive thyroid cancer. Clin Cancer Res. 2002;8:2870–2874. [PubMed] [Google Scholar]
- 29.Silva JM, Dominguez G, Gonzalez-Sancho JM, Garcia JM, Silva J, Garcia-Andrade C, Navarro A, Munoz A, Bonilla F. Expression of thyroid hormone receptor/erbA genes is altered in human breast cancer. Oncogene. 2002;21:4307–4316. doi: 10.1038/sj.onc.1205534. [DOI] [PubMed] [Google Scholar]
- 30.Seki Y, Suico MA, Uto A, Hisatsune A, Shuto T, Isohama Y, Kai H. The ETS transcription factor MEF is a candidate tumor suppressor gene on the X chromosome. Cancer Res. 2002;62:6579–6586. [PubMed] [Google Scholar]
- 31.Liu Y, Hedvat CV, Mao S, Zhu XH, Yao J, Nguyen H, Koff A, Nimer SD. The ETS protein MEF is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCFSkp2. Mol Cell Biol. 2006;26:3114–3123. doi: 10.1128/MCB.26.8.3114-3123.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woelfle U, Cloos J, Sauter G, Riethdorf L, Janicke F, van Diest P, Brakenhoff R, Pantel K. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 2003;63:5679–5684. [PubMed] [Google Scholar]
- 33.Dallas PB, Gottardo NG, Firth MJ, Beesley AH, Hoffmann K, Terry PA, Freitas JR, Boag JM, Cummings AJ, Kees UR. Gene expression levels assessed by oligonucleotide microarray analysis and quantitative real-time RT-PCR – how well do they correlate? BMC Genom. 2005;6:59. doi: 10.1186/1471-2164-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Etienne W, Meyer MH, Peppers J, Meyer RA., Jr Comparison of mRNA gene expression by RT-PCR and DNA microarray. Biotechniques. 2004;36:618–626. doi: 10.2144/04364ST02. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Quigg RJ. An integrated strategy for the optimization of microarray data interpretation. Gene Exp. 2005;12:223–230. doi: 10.3727/000000005783991990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002;417:78–83. doi: 10.1038/nature744. [DOI] [PubMed] [Google Scholar]
- 37.Pham TH, Clemente JC, Satou K, Ho TB. Computational discovery of transcriptional regulatory rules. Bioinformatics. 2005;21(2):ii101–ii107. doi: 10.1093/bioinformatics/bti1117. [DOI] [PubMed] [Google Scholar]