

Abstract
The mechanism by which Hepatocyte Growth Factor (HGF) induces tight junction disassembly prior to cell scattering is largely unknown. Here, we show that HGF-stimulates rapid loss of the TJ assembly protein Par6 from the TJ in an Erk-dependent manner. Erk activation by HGF is found to mediate the interaction of Par6 with GTP-loaded Cdc42. The Cdc42 GTPase activating protein cdGAP is shown to interact with Pkcζ at baseline and prevent Par6-Cdc42 association. Erk, by phosphorylating cdGAP at threonine776, can inhibit the GAP activity, thereby increasing Par6-Cdc42 association and TJ disassembly. Our findings reveal a novel pathway for regulating HGF signaling to the Par proteins through Erk-cdGAP, resulting in TJ disassembly and cell scattering.
Introduction
Epithelial cells spontaneously organize into small islands when cultured under subconfluent conditions. Membrane-associated proteins that make up the adherens junctions (E-cadherin and β-catenin e.g.) and tight junctions (Zo-1 and occludin e.g.) accumulate at the sites of cell-cell contact. Stimulation of sub-confluent monolayers with Hepatocyte Growth Factor (HGF) has been shown to induce disassociation of cell-cell contacts, resulting in cell scattering [1]. Paradoxically, HGF stimulation of confluent cell monolayers leads to increased transepithelial resistance (suggesting the formation of more complex cell-cell contacts [2]) and cells stimulated with HGF during culture in a three-dimensional matrix organize into complex branching tubules in which cell-cell junctions are maintained [1; 3; 4]. These observations suggest that HGF can regulate both junctional assembly and disassembly, depending on the cell context.
In tubular epithelial cells, cell-cell contacts are comprised primarily of laterally positioned adherens junctions (AJ) and apico-laterally positioned tight junctions (TJ). During the process of cell scattering, HGF-stimulated adherens junction disassembly is believed to be mediated by Src-dependent phosphorylation of β-catenin, leading to disruption of the E-cadherin/β-catenin complex that is necessary for AJ stabilization [5]. However, little is known at present regarding the mechanism by which HGF induces TJ disassembly.
TJ assembly and stabilization are believed to be regulated by signaling pathways that involve the interaction of integral TJ proteins such as occludin, claudins and Junctional Adhesion Molecule (JAM) with several binding partners, including partitioning-defective3 (Par3), Par6 and atypical protein kinases C (aPkc; including Pkcζ,λ, and/ι) [6; 7]. Par6, aPkc and Par3 associate to form a ternary complex that was originally identified as a key factor in the establishment of anterior-posterior polarity in Caenorhabditis elegans embryos. In both Drosophila and mammalian epithelial cells, this Par protein complex has since been found to have an indispensable role in the formation of TJs and the establishment of apical-basal polarity [8; 9; 10; 11]. Interestingly, Par proteins have also been demonstrated to regulate cell migration in astrocytes and MDCK cells, suggesting roles outside of the TJ [12; 13; 14; 15].
In the present study, we focused on the signaling pathways that regulate TJ disassembly following HGF stimulation. In MDCK cells, HGF stimulation was found to result in early Par6 re-localization away from cell-cell contacts, preceding the re-localization of Zo-1 and TJ disassembly. Inhibition of HGF-stimulated Erk activation diminished the Cdc42-Par6 association, partially prevented Par6 re-localization, and markedly inhibited TJ disassembly and cell scattering. Furthermore, cdGAP, a GTPase activating protein (GAP) for Cdc42 and Rac1, associated with aPKC and regulated HGF-stimulated Par6-Cdc42 interaction and Par6 re-localization in an Erk dependent manner.
MATERIALS AND METHODS
Cell Culture and Reagents
Madin-Darby canine kidney (MDCK) type II cells were provided by Michael Caplan (Yale University). Rabbit anti-Par6 and rabbit anti-Par3 (which recognizes the 180, 150 and 100kDa splice variants of Par3) antibodies were raised against canine Par6 or Par3 synthetic peptides, respectively [16]. Anti-Pals-1 antibody was a generous gift of Dr. Ben Margolis (University of Michigan Medical School). The following commercially available antibodies were used: rabbit anti-Par3 (recognizes the 180kDa form of Par3 with higher efficiency than the pan-isoform antibody) and rabbit anti-Zo-1 (Invitrogen); rabbit anti-phospho Erk (Cell Signaling, Beverly, MA); rabbit anti-Pkcζ, (C20, Santa Cruz Biotechnology, Santa Cruz, CA); rat anti-HA (Roche, Mannheim, Germany); Ez view Red Anti-HA Affinity Gel (SIGMA, St. Louis. MO); mouse anti-Cdc42 (BD Bioscience, Palo Alto, CA); mouse anti-Zo-1 antibody and mouse anti-Flag antibody (Sigma); mouse anti-myc antibody (4A6, Upstate/Millipore, Billerica, MA); Alexa Fluor 488/594 (Molecular Probe/Invitrogen).
Bisindolylmaleimide was purchased from Calbiochem. U0126 and LY294002 were purchased from Promega (Madison, WI). Recombinant HGF (H1404) was purchased from Sigma. HA-tagged Par6A, B, C and myc-tagged Par3b were kindly provided by Dr. Ian Macara (University of Virginia School of Medicine). Flag-tagged Pkcζ was kindly provided by Dr. Alex Toker (Beth Israel Deaconess Medical Center, Harvard Medical School). Myc-tagged cdGAP was kindly provided by Dr. Nathalie Lamarche-Vane (McGill University).
Transient transfection, immunoprecipitation and Western Analysis
Transient transfections were carried out using Lipofectamin 2000 according to the manufacture’s instruction (Invitrogen). After 4 hours incubation, the DNA-Lipofectamin complex containing medium was replaced with 0.1% FBS containing medium and subsequently incubated for 16–20 h before cell harvest. Cells were then stimulated with HGF (40ng/ml) for the indicated times and lysed in ice cold lysis buffer (150mM NaCl, 20 mM Tris-Cl pH 7.4, 2 mM EDTA, 1 mM, 1% Triton X-100, 10% glycerol, 1 mM sodium fluoride, 1 mM sodium vanadate, 1 mM phenylmethylsulfonayl fluoride, and 1μg/ml pepstatin).
Predesigned human cdGAP siRNA (Ambion) was used for cdGAP knockdown. Transient transfection of cdGAP siRNA into 293T cells was carried out using RNAiMax (Invitrogen) according to the manufactures instruction. 24 hours after transfection, mRNA was extracted using the RNAeasy kit (Qiagen) and cDNAs were generated using Superscript II kit (Invitrogen). cdGAP knockdown efficiency was confirmed by real-time PCR using Rotor-Gene3000 with QuantiTect SYBR Green PCR Kit (Qiagen).
For co-immunoprecipitation experiments, 500–1000 μg of cell lysates were immunoprecipitated with the indicated antibody, collected by adding protein G-Sepharose (Sigma), and washed 3 times with lysis buffer. Associated proteins were analyzed by immunoblotting. Quantification of co-immunoprecipitating proteins was performed using the NIH Image 1.61 software (http://rsb.info.nih.gov/nih-image/).
Immuofluorescence and Image Quantification
8-chamber glass slides (BD Falcon/BD Bioscience, Bedford, MA) were coated as previously described [17] with slight modification. Collagen type I (Upstate/Millipore) was diluted to 3ug/ml in 0.2N acetic acid. Coating was done by incubation for 2h at 37C°. Coated surfaces were washed three times with phosphate buffer saline (PBS) and incubated with 10% FBS containing culture medium for 1h. In one well, 3×103 MDCKII cells were seeded and cultured for 2 days. The medium was replaced to 0.1% FBS containing medium and incubated for at least 16h. Cells were treated ± HGF (40ng/ml) for the indicated time, washed, fixed and permeabilized with cold methanol: acetone (1:1) for 4–8min at −20C°, and processed for immunofluorescence. Slides were incubated with primary antibody (over-night at 4C°) and secondary antibody. Slides were washed and mounted using VECTASHIELD/DAPI (Vector Laboratories, Inc. CA) and sealed before viewing. 0.4um filter (Transwell) were also used for immnofluorescence studies without collagen coating.
For purifying cdGAP mutant expressing cells, myc-cdGAPT776A and EGFP-C1 plasmids were co-transfected to MDCK cells. 24 hour after transfection, GFP-positive cells were isolated by FACS. Cells were then seeded on 0.4um filter (Transwell), cultured for 48 hours and used for immunofluorescence study. For quantification, digital images of 5 to 10 isolated cell islands (15–55 cells/island) were obtained using a TE200 Nikon microscope and a SPOT RT CCD camera (Diagnostic Instruments, Sterling Heights, MI) or FV1000 confocal laser scanning microscope (Olympus). Cells were scored as positive for TJ localization of either Par6, Par3 or Zo-1 if protein expression was detected at all cell-cell contacts, and the percentage of positive cells was compared at the indicated time points and conditions (Supplemental Figure 1). At least 140 cells from 5 to 10 islands were quantified for each condition and three independent experiments were evaluated for statistical analysis.
For real-time imaging of cell scattering, MDCK cells were serum starved for 12h and stimulated with HGF (20ng/ml) while maintained in a stage top incubation system (LiveCell, Neue Bioscience, Inc.) or Nikon Biostation (Nikon, Japan). Video images were captured every 5 min for 10–14h.
Cdc42 activation assay
Detection of GTP-loaded Cdc42 was performed using a GST-linked PAK binding domain pull-down assay (Upstate/Invitrogen). 500–700mg of lysates from MDCK cells stimulated ± HGF were incubated with 10μg of the agarose conjugated PAK-1 PBD for 45 min at 4°C, washed, and immunoblotted with anti-Cdc42 antibody. In some experiments cells were pretreated with the MEK inhibitor U0126 (10 μM for 20 min).
Statistics
Data are presented as mean± SEM. P values were calculated using the one-tailed Student’s T test.
RESULTS
HGF stimulates Par6 disappearance from the TJ
To study the mechanism of HGF-stimulated TJ disassembly, we utilized the in vitro model of cell scattering. Immunolocalization of Par6 and Zo-1 in serum starved isolated MDCK colonies revealed these proteins were localized to cell-cell junctions as previously reported [18; 19; 20; 21] (Figure 1A). HGF stimulation induced rapid flattening and spreading of cells at the center of the colonies, resulting in an increase in colony area prior to any loss of cell-cell junctions. Coincident with spreading, Par6 immunostaining at cell-cell junctions in these centrally located cells detectably decreased within 2–4 hours of HGF treatment, while cells at the rim of the colony demonstrated partial Par6 re-localization to the free edge of the cell (Figure 1B, arrow, Supplemental Figure 2, arrow). There was nearly complete loss of detectable Par6 at cell-cell junctions in all colony cells following 4–8 hours of HGF stimulation (Figure 1D arrow, and quantitated in Figure 1B; Supplemental Figure 2, arrowheads), with a concomitant increase in the cytosolic Par6 signal. In contrast, Par3 and Pals1, another binding partner of Par6, relocalization from cell-cell contacts were slower, with detectable Par3 and Pals1 at many cell-cell contacts even 8 hours after HGF stimulation (data not shown). Consistent with previous reports [17], Zo-1 remained at cell-cell contacts until the cells began to physically separate following 8–12 hours of stimulation (Figure 1E–H). Thus, these studies demonstrate that HGF-stimulated TJ disassembly proceeds in a step-wise fashion, with Par6 re-localization preceding Par3 and Zo-1.
Figure 1. HGF induces Par6 relocalization from cell-cell contacts prior to Zo-1.
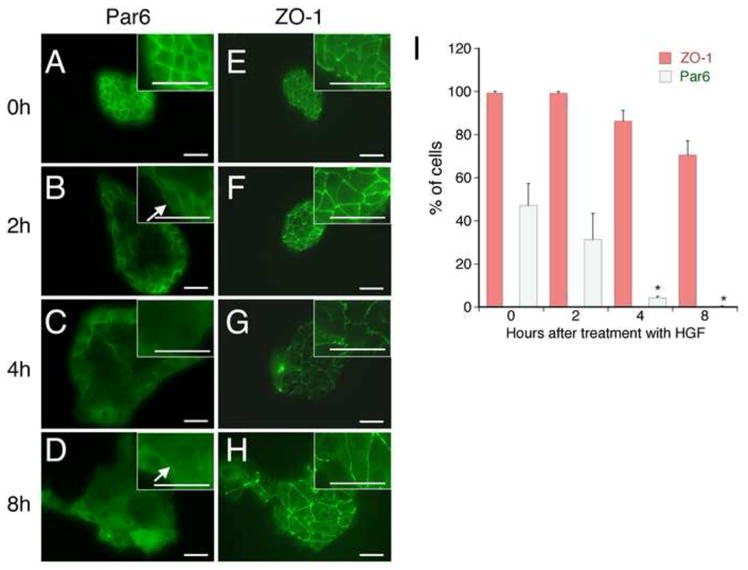
A: MDCK cells were stimulated with HGF for the indicated times. Cells were immunostained for Par6 or the TJ marker protein, Zo-1. Scale bar = 50μm. B: Quantitation of the percentage of the cells with preserved Par6 and Zo-1 expression at all cell-cell contacts. *p<0.02 vs. baseline. n=3 separate experiments with quantitation of 5 colonies/experiment. Scale bar = 50μm.
Activated Erk is required for HGF-stimulated Par6 re-localization
The Erk-MAPK pathway has been shown to play an important role in HGF induced scattering in MDCK cells [22; 23]. Consistent with these studies, cells stimulated with HGF in the presence of the Mek inhibitor U0126 flattened and spread, but do not exhibit disruption of cell-cell junctions and remained associated in discreet colonies for up to 14 hours (Figure 2A). A time course of Erk activation following U0126 treatment confirms that the Mek inhibitor suppressed Erk activation for the entire 14 hour time period (Figure 2B).
Figure 2. Erk activation is required for TJ disassembly.
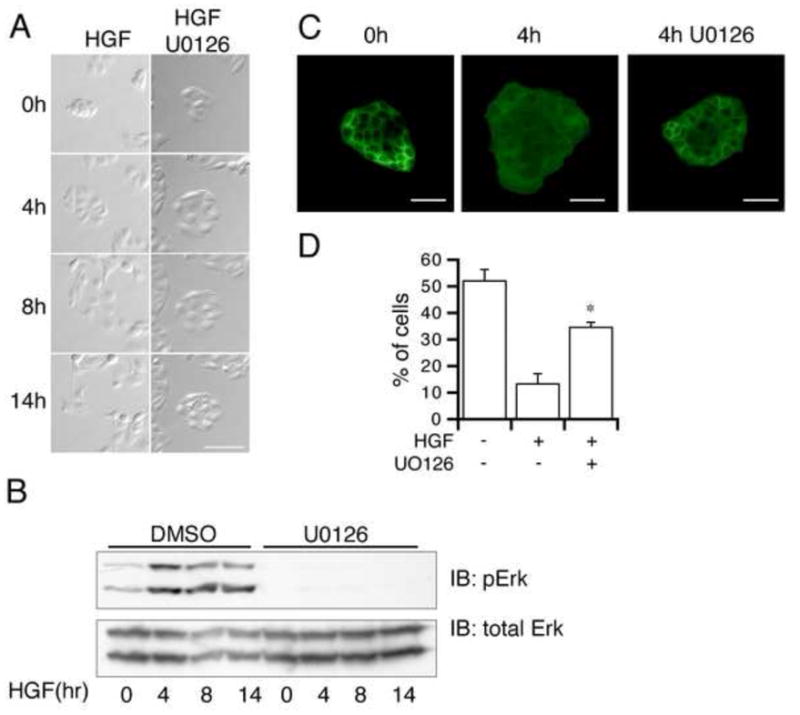
A) MDCK cells were incubated with HGF ± U0126 and images of the same individual colonies acquired at the indicated time points using time-lapse photography. Scale bar = 50μm. B) Cells were treated with HGF + DMSO (control) or HGF + U0126. The lysates from the indicated time points were immunoblotted with anti-pErk and anti-total Erk antibodies. C) Cells were treated with HGF ± U0126 for the indicated times and immunostained for Par6. Scale bar = 50μm. D) Quantitation of the % of cells with Par6 at all cell-cell junctions 4 hours after HGF stimulation ± U0126. n=3 separate experiments with quantitation of 10 colonies/experiment. *p<0.02 vs. HGF alone.
To determine if Erk activation is required for HGF-stimulated junctional protein re-localization, MDCK cells were plated as small islands and stimulated with HGF ± U0126 followed by immunostaining for Par6 and Zo-1. The loss of Par6 from cell-cell contacts following HGF stimulation was partially blocked by U0126 (Figure 2C, quantified in 2D). Thus Erk activation appears to be required for Par6 re-localization and subsequent TJ disassembly.
Erk-activation is required for Par6-Cdc42 interaction during TJ disassembly
Cdc42 is a member of the Rho family of small GTP binding proteins. These proteins have been found to be critical regulators of actin rearrangement, and more recently to associate with, and regulate the activity of the Par protein complex [18; 24; 25]. Immunoprecipitation of HA-Par6c from MDCK cells treated ±HGF revealed that HGF increases the Par6/Cdc42 association. Furthermore, pretreatment with the MEK inhibitor, U0126, markedly inhibited this increase in association of Par6c/Cdc42 (Figure 3A, quantitated in 3B). GTP-loaded Cdc42 has a higher binding affinity for Par6 than does GDP-Cdc42 [26; 27]. Therefore, we hypothesized that Erk might locally activate a Cdc42 guanine nucleotide exchange factor (GEF) or inhibit a Cdc42 GAP, thus increasing the local availability of GTP-Cdc42. cdGAP, a GAP for Rac1 and Cdc42, has been shown to be a target of activated Erk. Erk phosphorylation at threonine 776 decreases its GAP activity [28] and therefore increases GTP-Cdc42 levels. To examine the possible involvement of cdGAP in HGF-stimulated TJ disassembly, HEK293T cells were transiently transfected with myc-cdGAP and Flag-Pkcζ, demonstrating that cdGAP can interact with the Par protein complex via association with Pkcζ (Figure 3C). Consistent with a role for cdGAP in regulating GTP-Cdc42 levels, over-expression of myc-cdGAP was found to inhibit the interaction of Par6C and Cdc42 (Figure 3D).
Figure 3. HGF induces the Erk-dependent association of Cdc42 and Par6.
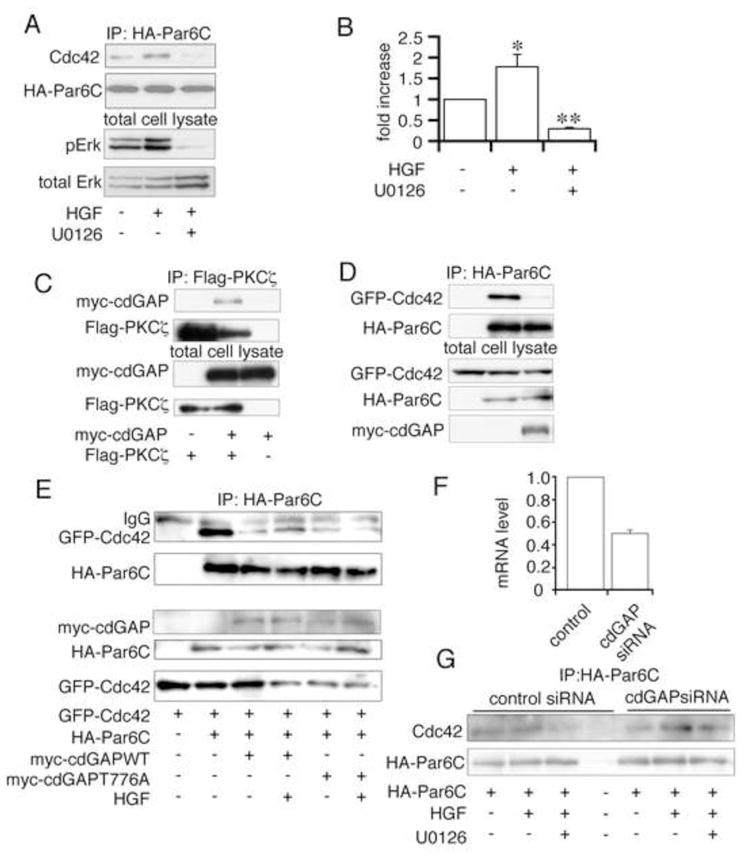
A) HA-Par6c transfected cells were incubated ±U0126 ±HGF for 90 min, lysed, immunoprecipitated with anti-HA, and immunoblotted with anti-Cdc42 (upper panel) and anti-HA antibodies (lower panel). B) Densitometric quantification of 6 experiments performed as in A, normalized to the amount of HA-Par6c immunoprecipitated for each condition. * p<0.05 vs. control, ** P<0.002 vs. HGF alone. cdGAP associates with Pkcζ and regulates Par6-Cdc42 interaction. C)Myc-cdGAP and Flag-Pkcζ transfected HEK293T cells were lysed, immunoprecipitated with anti-Flag, and immunoblotted with anti-myc and anti-Flag antibodies. D) Myc-cdGAP, HA-Par6C and GFP-Cdc42 transfected HEK293T cells were lysed, immunoprecipitated with anti-HA and immunoblotted with anti-myc, anti-HA and anti-GFP antibodies. E) HA-Par6c, GFP-Cdc42 and myc-cdGAP(WT/T776A) transfected MDCK cells were incubated ±HGF, lysed, immunoprecipitated with anti-HA, and immunoblotted with anti-GFP and anti-HA antibodies. F) cdGAP siRNA or scrambled siRNA were transiently transfected in HEK293T cells. mRNA was extracted and quantified by real-time RT-PCR. G) HA-Par6C and cdGAP or scrambled siRNA transfected cells were incubated ±U0126 ±HGF, lysed, immunoprecipitated with anti-HA, and immunoblotted with anti-Cdc42 and anti-HA antibodies.
To determine whether the cdGAP mediated inhibition of Par6-Cdc42 association is regulated by Erk phosphorylation of cdGAP, either myc-cdGAP-WT or a non-phosphorylatable myc-cdGAP-T776A mutant were co-transfected with GFP-Cdc42 and HA-Par6C. WT-cdGAP inhibited Par6C-Cdc42 interaction as noted above, and HGF stimulation partially restores this association (Figure 3E, lane 4 vs. 3). However, in cdGAP-T776A transfected MDCK cells, the inhibition of the Par6-Cdc42 interaction was not reversed by HGF stimulation (Figure 3E, lane 6 vs. 5). Furthermore, we tested whether silencing expression of cdGAP modulates the Par6-Cdc42 association. HEK293T cells were used as the highest efficiency of cdGAP silencing was achieved with the human siRNA. Transfection of cdGAP siRNA resulted in a 50% decrease in the mRNA levels of cdGAP (Figure 3F). cdGAP silencing did not change the basal HA-Par6-Cdc42 (endogenous) interaction (Figure 3G, lane 1 vs. lane 5). But the HGF stimulated association of Par6 and Cdc42 (lane 2 vs. lane 6) was increased by cdGAP silencing, and this interaction was relatively resistant to U0126 treatment (lane 3 vs. lane 7). These data suggest that cdGAP normally acts to prevent the recruitment of Cdc42 to the junctional complex, and that HGF stimulation of Erk leads to inhibition of cdGAP and thus promotes the Cdc42-Par6 association.
To define the importance of cdGAP in Par6 relocalization and TJ disruption, MDCK cells transfected with cdGAP-T776A or empty vector were stimulated with HGF, fixed and stained with anti-Par6 antibody. Similar to wild-type cells (Figure 1A), cells transfected with the empty vector were flattened and Par6 re-localization from cell-cell contacts was observed 4 hours after HGF treatment. However, in cdGAP-T776A transfected cells there was no detectable change in cell morphology following HGF stimulation, and the loss of Par6 from cell-cell contacts was significantly inhibited (Figure 4A, quantitated in 4B). These data suggest that Erk-dependent inhibition of cdGAP activity is required for HGF-stimulated GTP-Cdc42 recruitment to the junctional complex and subsequent Par6 re-localization from cell-cell contacts and eventual cell scattering.
Figure 4. cdGAP inhibits re-localization of Par6 from cell-cell contacts following HGF stimulation.
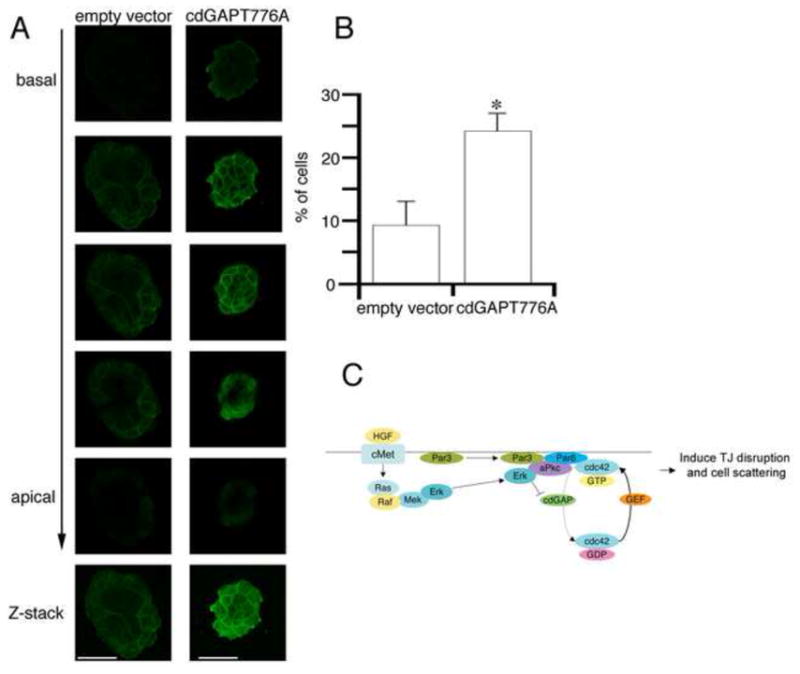
A) MDCK cells co-expressing the empty vector or cdGAPT776A along with EGFP were collected by FACS and seeded on 0.4um filter (Transwell), cultured for 48 hours and stimulated with HGF for 4 hours. B) Quantitation of Par6 localization from 10 colonies/experiment. n=3 separate experiments *p<0.05 vs. empty vector transfected cells. Scale bar = 20μm. C) Model of HGF-stimulated Par protein complex signaling during TJ disruption.
DISCUSSION
In the present study, we demonstrate that stimulation with HGF results in the Erk-dependent recruitment of Cdc42 to the Par3-Par6-Pkcζ junctional complex followed by re-localization of Par6 from cell-cell contacts. In the setting of MAPK inhibition, GTP-Cdc42 does not associate with Par6 and TJ disassembly is markedly slowed, preventing HGF-induced cell scattering. We identify cdGAP as a regulatory protein in this process, with over-expressed cdGAP preventing the HGF-stimulated Par6-Cdc42 interaction and Par6 relocalization. These studies support the idea that tight junction disassembly is a carefully regulated process that requires the highly coordinated recruitment and activation of a complex set of signaling molecules. Based on these results, we propose a model (Figure 4C) in which HGF stimulates Erk activation at the junctional complex, leading to phosphorylation of cdGAP and thus a local increase in GTP-Cdc42. GTP-Cdc42 then associates with Par6 and is required for efficient Par6 relocalization, TJ disruption and cell scattering.
The attractive feature of a local scaffold as the mechanism for Erk activation at tight junctions is that this could help explain the paradoxical observation that HGF-stimulation can result in TJ disassembly in subconfluent epithelial islands (scattering) whereas TJ’s become more complex (resulting in higher trans-epithelial resistance) in confluent monolayers stimulated with HGF. We have previously reported that fully confluent renal epithelial cells still activate the Met receptor normally in response to HGF, but that downstream signaling is altered [31]. In that study it was demonstrated that in confluent monolayers, phosphoinositide 3-kinase (PI 3-K) activation occurred normally at the Met receptor but was downregulated at focal adhesions, inhibiting HGF-stimulated migratory responses. Thus it is conceivable that in fully confluent epithelial cells, MAPK scaffolding and subsequent Erk activation at tight junctions may be downregulated, preventing HGF-stimulated TJ disassembly.
In a previous report by Johansson and coworkers, Par6 and Zo-1 localization were examined in MDCK cells at 4 and 16 hours after HGF stimulation [19]. Although a semi-quantitative analysis was not provided, it appeared that both were equally maintained in cell-cell junctions at 4 hours, with significant loss of both at 16 hours although actually scattering had not yet occurred at this later time point. This is in contrast to our findings in which the cells demonstrate early loss of Par6 localization and are fully scattered by 12 hours. The more rapid loss of Par6 and scattering observed in our study might be due to differences in the MDCK II clones used (different levels of Met receptor expression, e.g.) or the different source of HGF.
Our finding that Erk activation is required for the HGF-stimulated association of Cdc42 with Par6 led us to hypothesize that Erk activation at the TJ may locally activate a Cdc42 GEF or inhibit a Cdc42 GAP, thus increasing the local availability of GTP-Cdc42. CdGAP, a GAP for Rac1 and Cdc42, has been demonstrated to be inhibited by Erk [28] and to localize to focal adhesions and be necessary for adhesion-dependent signaling in U2OS cell [29]. However, the role of cdGAP in TJ regulation is unclear. In the present study, we demonstrate that cdGAP associates with the Par protein complex via interaction with aPKC and can inhibit the Par6-Cdc42 interaction and Par6 re-localization from cell-cell contacts following HGF stimulation.
In early embryos of C. elegans a newly identified RhoGAP, PAC-1, has been found to inactivate Cdc42 at cell-cell contacts and therefore restrict GTP-Cdc42 to contact-free surfaces, where it associates with Par6 re-localized from cell-cell contacts [30]. In a similar manner, the current results suggest that the GAP activity of cdGAP serves to prevent Par6-Cdc42 association and to promote Par6 re-localization from sites of cell-cell contact.
Supplementary Material
Cells were stained with anti-Par6 and DAPI. Number of nuclei was counted by DAPI (right panel). Cells that are completely surrounded by Par6 (*) were counted as positive (center panel). From these images, 10 out of 22 cells were Par6 positive at cell-cell contacts.
Acknowledgments
This work was supported by an NIH grant to LGC (DK065109), an NRSA grant to JS (F32GM072162-03), and an AHA grant to SI (575043N). The authors would like to thank I. Macara for HA-Par6 constructs, B. Margolis for Pals1 antibody, A. Toker for Flag-Pkcζ constructs and N. Lamarche-Vane for myc-cdGAP WT and T776A constructs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stoker M, Gherardi E. Factors affecting epithelial interactions. Ciba Found Symp. 1987;125:217–39. doi: 10.1002/9780470513408.ch13. [DOI] [PubMed] [Google Scholar]
- 2.Pollack AL, Apodaca G, Mostov KE. Hepatocyte growth factor induces MDCK cell morphogenesis without causing loss of tight junction functional integrity. Am J Physiol Cell Physiol. 2004;286:C482–94. doi: 10.1152/ajpcell.00377.2003. [DOI] [PubMed] [Google Scholar]
- 3.Montesano R, Matsumoto K, Nakamura T, Orci L. Identification of a fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell. 1991;67:901–8. doi: 10.1016/0092-8674(91)90363-4. [DOI] [PubMed] [Google Scholar]
- 4.Cantley LG, Barros EJ, Gandhi M, Rauchman M, Nigam SK. Regulation of mitogenesis, motogenesis, and tubulogenesis by hepatocyte growth factor in renal collecting duct cells. Am J Physiol. 1994;267:F271–80. doi: 10.1152/ajprenal.1994.267.2.F271. [DOI] [PubMed] [Google Scholar]
- 5.Cozzolino M, Stagni V, Spinardi L, Campioni N, Fiorentini C, Salvati E, Alema S, Salvatore AM. p120 Catenin is required for growth factor-dependent cell motility and scattering in epithelial cells. Mol Biol Cell. 2003;14:1964–77. doi: 10.1091/mbc.E02-08-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyoshi J, Takai Y. Molecular perspective on tight-junction assembly and epithelial polarity. Adv Drug Deliv Rev. 2005;57:815–55. doi: 10.1016/j.addr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–35. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- 8.Macara IG. Parsing the polarity code. Nat Rev Mol Cell Biol. 2004;5:220–31. doi: 10.1038/nrm1332. [DOI] [PubMed] [Google Scholar]
- 9.Matter K, Balda MS. Signalling to and from tight junctions. Nat Rev Mol Cell Biol. 2003;4:225–36. doi: 10.1038/nrm1055. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–87. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- 11.Ooshio T, Fujita N, Yamada A, Sato T, Kitagawa Y, Okamoto R, Nakata S, Miki A, Irie K, Takai Y. Cooperative roles of Par-3 and afadin in the formation of adherens and tight junctions. J Cell Sci. 2007;120:2352–65. doi: 10.1242/jcs.03470. [DOI] [PubMed] [Google Scholar]
- 12.Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–98. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 13.Mishima A, Suzuki A, Enaka M, Hirose T, Mizuno K, Ohnishi T, Mohri H, Ishigatsubo Y, Ohno S. Over-expression of PAR-3 suppresses contact-mediated inhibition of cell migration in MDCK cells. Genes Cells. 2002;7:581–96. doi: 10.1046/j.1365-2443.2002.00540.x. [DOI] [PubMed] [Google Scholar]
- 14.Shin K, Wang Q, Margolis B. PATJ regulates directional migration of mammalian epithelial cells. EMBO Rep. 2007;8:158–64. doi: 10.1038/sj.embor.7400890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q, Margolis B. Apical junctional complexes and cell polarity. Kidney Int. 2007;72:1448–58. doi: 10.1038/sj.ki.5002579. [DOI] [PubMed] [Google Scholar]
- 16.Sfakianos J, Togawa A, Maday S, Hull M, Pypaert M, Cantley L, Toomre D, Mellman I. Par3 functions in the biogenesis of the primary cilium in polarized epithelial cells. J Cell Biol. 2007;179:1133–40. doi: 10.1083/jcb.200709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Rooij J, Kerstens A, Danuser G, Schwartz MA, Waterman-Storer CM. Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J Cell Biol. 2005;171:153–64. doi: 10.1083/jcb.200506152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, Akimoto K, Izumi Y, Ohnishi T, Ohno S. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J Cell Biol. 2001;152:1183–96. doi: 10.1083/jcb.152.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johansson A, Driessens M, Aspenstrom P. The mammalian homologue of the Caenorhabditis elegans polarity protein PAR-6 is a binding partner for the Rho GTPases Cdc42 and Rac1. J Cell Sci. 2000;113(Pt 18):3267–75. doi: 10.1242/jcs.113.18.3267. [DOI] [PubMed] [Google Scholar]
- 20.Gao L, Joberty G, Macara IG. Assembly of epithelial tight junctions is negatively regulated by Par6. Curr Biol. 2002;12:221–5. doi: 10.1016/s0960-9822(01)00663-7. [DOI] [PubMed] [Google Scholar]
- 21.Gopalakrishnan S, Hallett MA, Atkinson SJ, Marrs JA. aPKC-PAR complex dysfunction and tight junction disassembly in renal epithelial cells during ATP depletion. Am J Physiol Cell Physiol. 2007;292:C1094–102. doi: 10.1152/ajpcell.00099.2006. [DOI] [PubMed] [Google Scholar]
- 22.Royal I, Park M. Hepatocyte growth factor-induced scatter of Madin-Darby canine kidney cells requires phosphatidylinositol 3-kinase. J Biol Chem. 1995;270:27780–7. doi: 10.1074/jbc.270.46.27780. [DOI] [PubMed] [Google Scholar]
- 23.Potempa S, Ridley AJ. Activation of both MAP kinase and phosphatidylinositide 3-kinase by Ras is required for hepatocyte growth factor/scatter factor-induced adherens junction disassembly. Mol Biol Cell. 1998;9:2185–200. doi: 10.1091/mbc.9.8.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2:540–7. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- 25.Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–9. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- 26.Hurd TW, Fan S, Liu CJ, Kweon HK, Hakansson K, Margolis B. Phosphorylation-dependent binding of 14-3-3 to the polarity protein Par3 regulates cell polarity in mammalian epithelia. Curr Biol. 2003;13:2082–90. doi: 10.1016/j.cub.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 27.Gao L, Macara IG. Isoforms of the polarity protein par6 have distinct functions. J Biol Chem. 2004;279:41557–62. doi: 10.1074/jbc.M403723200. [DOI] [PubMed] [Google Scholar]
- 28.Tcherkezian J, Danek EI, Jenna S, Triki I, Lamarche-Vane N. Extracellular signal-regulated kinase 1 interacts with and phosphorylates CdGAP at an important regulatory site. Mol Cell Biol. 2005;25:6314–29. doi: 10.1128/MCB.25.15.6314-6329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LaLonde DP, Grubinger M, Lamarche-Vane N, Turner CE. CdGAP associates with actopaxin to regulate integrin-dependent changes in cell morphology and motility. Curr Biol. 2006;16:1375–85. doi: 10.1016/j.cub.2006.05.057. [DOI] [PubMed] [Google Scholar]
- 30.Anderson DC, Gill JS, Cinalli RM, Nance J. Polarization of the C. elegans embryo by RhoGAP-mediated exclusion of PAR-6 from cell contacts. Science. 2008;320:1771–4. doi: 10.1126/science.1156063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishibe S, Haydu JE, Togawa A, Marlier A, Cantley LG. Cell confluence regulates hepatocyte growth factor-stimulated cell morphogenesis in a beta-catenin-dependent manner. Mol Cell Biol. 2006;26:9232–43. doi: 10.1128/MCB.01312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cells were stained with anti-Par6 and DAPI. Number of nuclei was counted by DAPI (right panel). Cells that are completely surrounded by Par6 (*) were counted as positive (center panel). From these images, 10 out of 22 cells were Par6 positive at cell-cell contacts.