

Abstract
Fast, temporally-precise, and consistent synaptic transmission is required to encode features of acoustic stimuli. Neurons of nucleus magnocellularis (NM) in the auditory brain stem of the chick possess numerous adaptations to optimize the coding of temporal information. One potential problem for the system is the depression of synaptic transmission during a prolonged stimulus. The present studies tested the hypothesis that cannabinoid receptor one (CB1) signaling may limit synaptic depression at the auditory nerve-NM synapse. In situ hybridization was used to confirm that CB1 mRNA is expressed in the cochlear ganglion; immunohistochemistry was used to confirm the presence of CB1 protein in NM. These findings are consistent with the common presynaptic locus of CB1 in the brain. Rate-dependent synaptic depression was then examined in a brain slice preparation before and after administration of WIN 55,212-2 (WIN), a potent CB1 agonist. WIN decreased the amplitude of excitatory postsynaptic currents and also reduced depression across a train of stimuli. The effect was most obvious late in the pulse train and during high rates of stimulation. This CB1-mediated influence could allow for lower, but more consistent activation of NM neurons, which could be of importance for optimizing the coding of prolonged, temporally-locked acoustic stimuli.
Keywords: cochlear nucleus; synaptic depression; nucleus magnocellularis; brain slice; WIN 55,212-2; calyx
Acoustic stimuli drive high rates of neural activity and these stimuli can be relatively long-lasting. In vivo spontaneous firing rates average 100Hz even lacking auditory stimuli (Warchol and Dallos, 1990). Neurons must be able to respond in a consistent manner throughout the entirety of the stimulus to code this information effectively. Additionally, the encoding of important information about acoustic stimuli is not simply a duration code as it requires that action potentials be precisely timed with respect to the acoustic stimulus. Neurons in the brain stem auditory system possess a number of features that optimize their ability to maintain precisely timed and sustained responses.
Important adaptations to the rigorous demands of coding acoustic information have been identified in the cochlear nucleus of the chick, nucleus magnocellularis (NM). This area is key for processing time-locked signals from the auditory nerve (cnVIII) and relaying that information to nucleus laminaris (NL), where the analyses of interaural time differences assist in tasks such as the localization of a sound source (Sullivan and Konishi, 1984, Takahashi et al., 1984, Carr and Konishi, 1990, Warchol and Dallos, 1990, Overholt et al., 1992).
One adaptation towards accomplishing NM's key role of accurately coding timed information can be seen in the nature of its innervation. NM neurons receive large excitatory calyceal input from only 2-3 cnVIII fibers (Carr and Boudreau, 1991). Each of these afferents is capable of producing suprathreshold currents through a large release of glutamate, resulting in a high neurotransmission safety factor (Parks and Rubel, 1978, Hackett et al., 1982, Zhang and Trussell, 1994b, a). NM neurons produce a single, strong excitatory post-synaptic potential (EPSP) mediated primarily via AMPA receptor activation. Little jitter or temporal summation is observed which is aided by low-threshold K+ channels to help ensure a minimal refractory period (Reyes et al., 1994, Zhang and Trussell, 1994b).
While these synaptic adaptations allow NM to fire at very high rates, synaptic depression can become a serious impediment to the coding of ongoing auditory stimuli. This short-term effect is seen as a progressive decline in the size of postsynaptic responses to the same stimulus. AMPA receptor desensitization appears to be a major limitation of sustained responses (Trussell et al., 1993, Otis et al., 1996), but vesicle depletion presents a second limitation to sustained action potential (AP) firing (Brenowitz and Trussell, 2001). In addition, glutamatatergic autoreceptor activation may contribute to this phenomenon (Barnes-Davies and Forsythe, 1995, von Gersdorff et al., 1997). Failing to limit synaptic depression would be detrimental to temporal coding as it would prevent NM from coding throughout the duration of a stimulus.
Strong synaptic depression is seen in both NM (Hackett et al., 1982, Zhang and Trussell, 1994a) and NL (Kuba et al., 2002, Cook et al., 2003) in brain stem slice preparations. It is possible that the procedure itself, i.e. extraction of a brain slice, removes endogenous signals necessary to attenuate depression. For example, descending GABAergic inhibition from the superior olivary nucleus (SON) may play a dual role, working both pre- and post-synaptically at the cnVIII-NM synapse to preserve temporal coding (Carr et al., 1989, Code et al., 1989, Lachica et al., 1994). GABAA receptors influence NM excitability, tightening the window for summation postsynaptically (Hyson et al., 1995, Monsivais et al., 2000) while GABAB receptors help conserve glutamate release during high rates of activity (Brenowitz et al., 1998). In doing so, GABAB activation reduces synaptic depression across a train of stimuli, limiting the progressive decrease in response.
Another potential mechanism for regulating glutamatergic signaling is cannabinoid (CB) signaling, which has already been shown to have a modulatory role in other sensory systems (Straiker et al., 1999a, Straiker et al., 1999b, Czesnik et al., 2007, Deshmukh et al., 2007, Thornton-Jones et al., 2007, Nyilas et al., 2009). Our laboratory recently reported the distribution of the cannabinoid receptor 1 (CB1) mRNA throughout the brain of the chick (Stincic and Hyson, 2008). While little mRNA expression was found in NM or NL, the cnVIII ganglion (vestibular portion) displayed robust CB1 mRNA expression. This finding suggested that CB1 mRNA expression may be found in the cochlear ganglion (CG) as well. CB1 is expressed in the dorsal cochlear nucleus of rats (Mailleux and Vanderhaeghen, 1992) and CBs can modulate synaptic plasticity in auditory nuclei (Penzo and Peña, 2009, Zhao et al., 2009). CB mediated retroinhibition has also been reported in the medial nucleus of the trapezoid body (MNTB) (Kushmerick et al., 2004). This is significant because the MNTB is an auditory nucleus that also receives calyceal input. Taken together, these previous reports suggest that CB signaling in the chick auditory brain stem is likely as well.
CB signaling acts primarily through metabotropic receptors that share some traits with classical neurotransmitter systems, but differs in key aspects. When activated, the receptors trigger 2nd messenger signaling cascades through Gi/o-proteins (Howlett and Fleming, 1984, Howlett, 1985, Howlett et al., 1986). Vesicles, however, are not required for transport since endogenous CBs (eCBs) are lipophillic and capable of crossing the cell membrane either unaided or by facilitated diffusion (Beltramo et al., 1997, Hillard et al., 1997). Once released into the synapse from postsynaptic cells, eCBs activate CB receptors located on the presynaptic cell. Functional CB1 has been localized presynaptically to axonal terminals with almost no exceptions in adult animals (Schlicker and Kathmann, 2001).
A major effect of CB1 activation is inhibition of voltage-gated Ca2+ channels (N-type and P/Q-type) (Caulfield and Brown, 1992, Felder et al., 1993, Mackie et al., 1993), leading to a reduction in the number of vesicles released upon arrival of the action potential to the terminal. In this way, CB1 activation can reduce the amount of neurotransmitter released into the synaptic cleft, subsequently lessening the factors that contribute to synaptic depression.
If CB signaling is an additional adaptation for optimizing coding at the cnVIII-NM synapse, it must be present and effective at modulating synaptic transmission. Our study first establishes the presence of CB1 at the cnVIII-NM synapse using in situ hybridization (ISH) and immunohistochemistry (IHC), and then documents the influence of CB1 activation on synaptic depression.
2. Experimental
Subjects
Subjects were prehatch (E18-19) or posthatch (P10) chicks (Gallus domesticus) incubated at Florida State University from eggs obtained from a local supplier. Subjects of either sex were chosen randomly.
In situ hybridization
The 702 base pair fragment of chick CB1 mRNA was re-subcloned into the pGEM vector (Promega). Both an anti-sense and a corresponding sense-control riboprobe were synthesized using a protocol adapted from previously published work (McCullumsmith et al., 2003, Krause et al., 2006). Alternating tissue sections were hybridized with either sense or anti-sense 35S riboprobe and exposed to film.
For riboprobe synthesis, 8μl of Uridine 5”-(a-thio)triphosphate [35S] (Perkin Elmer, NEG039H) and 4.0μl 5X transcription buffer, 2.0μl of 0.1mM dithiothreitol, 1.5μl adenosine triphosphate, cytosine triphosphate and guanidine triphosphate, 2.0μl linearized plasmid (1μg/ml−1) DNA 1.0μl RNAse inhibitor (40 units μl−1), and 1.5μl T7 RNA polymerase (15 units μl−1) was combined and incubated for 2 hours at 37°C. DNAse (1.0μl: RNAse-free) was added and the mixture was incubated for 15 minutes at room temperature. Radio-labeled probe was purified with microspin chromatography columns (Bio-Rad, Hercules, CA, USA).
Slides were removed from −80°C storage and post-fixed in 4% paraformaldehyde at room temperature for 1 hour. After fixation, the slides were washed in 2X Standard Sodium Citrate, (SSC, where 1X SSC is 300mM NaCl and 30mM sodium citrate), pH 7.2, three times for 5 minutes each. Slides were then washed in deionized water for 1 minute and placed in 0.1mM triethanolamine (pH 8.0)–acetic anhydride, 400: 1 (v/v) on a stir plate, for 10 minutes. The final rinse was in 2X SSC for 5 minutes, followed by dehydration through graded alcohols and air-drying for 30 minutes. A coverslip with 60μl of hybridization buffer (75% formamide,10% dextran sulphate, 3X SSC (pH 7.2), 50mM Na2HPO4 (pH 7.4), 10 mM dithiothreitol, 1X Denhardt's solution (Sigma-Aldrich, St. Louis, MO, USA), 100μg (ml)−1 yeast tRNA) was placed on each slide. Strength of probe in 60μl of hybridization buffer was approximately 1–2 million counts per minute as assessed by a liquid scintillation counter (Beckman Coulter). Slides were placed in a covered tray with filter paper saturated with 75% formamide. After overnight incubation at 55°C, coverslips were removed and slides were placed at room temperature in 2X SSC for 5 minutes, followed by RNAse (200μg (ml)−1 in 10mM Tris-HCl, pH 8.0, 0.5mM NaCl) at 37°C for 30 minutes and then: 2X SSC at room temperature for 10 minutes, 1X SSC for 10 minutes at room temperature; 0.5X SSC at 55°C for 60 minutes; and 0.5X SSC for 10 minutes at room temperature. The slides were dehydrated in graded ethanol solutions, air dried, placed in X-ray cassettes, and apposed to BioMax MR Film for 10 days.
Immunohistochemistry
Antibodies used were CB1 anti-Zebra Finch (Dr. Ken Soderstrom), CB1 anti-rat (Cayman Laboratories, Ann Arbor, MI), and SV2 (University of Iowa, Iowa City, IA). Specificity of the custom zebra finch CB1 antibody was demonstrated through preabsorption with the antigen peptide which completely blocked all labeling. SV2 antibody was developed by Kathleen M. Buckley and was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. This particular SV2 antibody has been used successfully by our laboratory in the past (Wilkinson et al., 2003).
Subjects were deeply anesthetized with pentobarbital and perfused with 0.9% saline followed by ice cold 4% paraformaldehyde. Brainstems were blocked and post-fixed in 4% paraformaldehyde for 1–2 hours followed by overnight cryoprotection at 4°C in phosphate buffered saline (PBS) containing 20% sucrose. The tissue was rapidly frozen in 2-methylbutane on dry ice and embedded in tissue freezing medium for cryosectioning using a Leica CM 1850 cryostat. Sections were cut at 20 µm and were collected into ice cold PBS for later processing and mounting. Every section containing NM was collected.
Sections were then washed 2 × 10 minutes in PBS and endogenous peroxidase activity was quenched by incubating in 0.03% H2O2 in methanol for 10 minutes. Following 3 10-minute rinses in PBS, sections were placed in a blocking solution containing 2% bovine serum albumin (BSA), 2-4% normal goat serum (NGS) (CB1 antibody) or 2% normal horse serum (NHS) (SV2), and 0.1% Triton-X100 in PBS for 1 hour. Sections were transferred to various concentrations of the primary antibodies, CB1 (1:1,000-5,000) and SV2 (1:500), in blocking solution and incubated on a rotor overnight at 4°C.
The following day, the sections were removed from 4°C and allowed to incubate at RT for 2 hours. Sections were washed 3 ×10 minutes in PBS. Sections were then incubated in a 1:200 concentration of goat anti-rabbit or horse anti-mouse secondary antibodies in blocking solution for 1 hour. Following 3 10-minute washes with PBS, sections were incubated in avidin–biotin-peroxidase complex (ABC; Vector Laboratories, Burlingame, CA, USA) for 1 hour. After another round of washes, with PBS, sections were reacted with diaminobenzidine (DAB) tetrahydrochloride and 0.01% H2O2 for visualization. Following a final round of washes, sections were mounted on slides and allowed to dry overnight. The following day, slides were dehydrated in a graded series of ethanol and cleared in xylenes. Slides were coverslipped in DPX mounting medium and allowed to dry overnight.
For sections undergoing double-labeling with both anti-CB1 and anti-SV2, secondary antibodies conjugated to fluorophores, Cy3 (Goat anti-rabbit, Red) and FITC (Horse anti-mouse, Green), were used. The standard IHC protocol was modified by removal of quenching, ABC kit, and DAB staining. Rather, the tissue sections were washed 3 × 10 minutes in PBS in light-tight containers after secondary antibody incubation. Sections were mounted onto glass slides using a gelatin solution. A single drop of Vectashield (Vector Laboratories, Burlingame, CA) was placed over the sections and a coverslip was sealed onto the slide using clear nail polish. Slides were allowed to dry overnight at 4°C in a light-tight container. Fluorescent images were captured using confocal microscopy and photo-merged with LSM software. LSM files were converted to tiff files.
Brain slice preparation
This protocol is adapted from previous work in our laboratory (Hyson et al., 1995). All procedures were approved by the Animal Care and Use Committee at Florida State University. Chicken embryos were anesthetized by cooling and then sacrificed by rapid decapitation. A 4 mm segment of the caudal skull containing the brain stem was removed with a razor blade and quickly submerged in room temperature artificial cerebral spinal fluid (ACSF). ACSF contained the following (in mM): 130 NaCl, 26 NaH2CO3, 3 KCl, 3 CaCl2, 1 MgCl2, 1.25 NaH2PO4, and 10 dextrose. ACSF was constantly gassed with 95% O2-5% CO2 (pH 7.4). A brain stem segment was dissected out of the cranium and transferred to a vibrating blade tissue slicer where it was mounted with cyanoacrylate glue, supported by a gelatin solution (30% gelatin in ACSF), and cut in ACSF. Transverse slices (300μm) containing NM were then transferred to a submersion-type recording chamber and perfused (2-3 ml/minute by gravity) with heated oxygenated ACSF (30°C), using an in-line heater. 30°C was chosen as bath temperature to standardize recordings, but still allow for robust depression as depression is reduced with increases in bath temperature (Brenowitz et al., 1998). All reported physiological measurements were made from heated slices after a 60 minute recovery period. The slice was held in the recording chamber under a piece of nylon mesh. Brain slices typically remained viable for 4-6 hours, but the majority of recordings were made between 1 to 4 hours after dissection. Slices that were administered CB drugs were discarded regardless of experiment completion due to the difficulties in washing out the drugs. After CB drug administration, 10ml of 70% ethanol followed by 40ml of dH20 was run through the perfusion system to clean the lines and bath. Flow rates and temperature were recalibrated 10 minutes after returning to normal ACSF.
Voltage clamp
Standard methods for whole-cell voltage clamp recordings were used. Recordings were made using glass micropipettes. Patch electrodes (3-7 MΩ) were drawn on a Brown-Flaming electrode puller from borosilicate glass micropipets (ID 0.86, OD 1.5mm, WPI Inc). Typical electrode solution consisted of (in nM) 105 K-gluconate, 35 KCl, 5 EGTA, 1 MgCl2, 10 K+-HEPES, 4 ATP-Mg, 0.3 GTP-Na pH adjusted to 7.2 with KOH. Liquid junction potential was 10mV and corrected after data collection. Cells were clamped at -70mV for the duration of the experiment.
The electrode was guided to the cell of interest using an upright fixed-stage microscope (Olympus BX50WI) with a water immersion lens (40 or 60 X) and infrared DIC optics. Slight positive pressure was applied before the electrode entered the bath. Upon contact with a cell negative pressure was applied until a tight seal (>1GΩ) was formed. Additional negative pressure was then applied to rupture the membrane. Cells were given 10 minutes to stabilize before recordings began. Recordings were digitized at 20kHz and filtered at 5kHz. Series resistance was compensated 80-90% and monitored via a 5mV hyperpolarizing command (5ms duration) before each synaptic stimulation protocol. Recordings were rejected if series resistance increased more than 20%. Signals were amplified, digitized, stored and then analyzed by computer (pClamp 9.2, Molecular Devices; Excel 2007, Microsoft; SPSS).
Slices were stimulated unilaterally with a bipolar electrode constructed from a Teflon-coated platinum wire placed on the fibers of cnVIII. Stimulation was provided by a constant current stimulator (Neurodata SIU90) controlled through the Clampex software. Each stimulus was a 20μs monophasic current pulse, generally 0.1-10mA. The placement of the stimulating electrode and the number of undamaged cnVIII fibers lead to variability in the necessary stimulation amplitude. Therefore, the stimulation amplitude for any investigated neuron was adjusted 10 minutes after patch to produce maximal excitatory postsynaptic current (EPSC) amplitude.
Drug application
Where indicated in the text, the following drugs were bath applied: CB agonist (WIN 55,212-2 (WIN) 5-10μm) and GABAA antagonist (Picrotoxin, 100μM). WIN was dissolved in dimethyl sulfoxide (DMSO), but final DMSO concentration was <0.1%.
Synaptic depression
The voltage clamp configuration was used to maintain the membrane potential at -70mV. Whole cell patch recordings of NM EPSCs were taken before and after a 10 minute holding period. Control cells had normal ACSF throughout the entire experiment. Drug groups (5 and 10μM WIN) were treated with the CB agonist during the 10 minute holding period. Post-holding measurements were made with WIN still present in the bath. GABAA antagonist picrotoxin (100μM) was present in the bath during the entire experiment for a subset of each group.
4 different rates of stimulation were chosen to represent activity below that seen in vivo (20Hz), the spontaneous rate of activity in vivo (100Hz), and high rates of stimuli (200 and 250Hz). 10-pulse trains were delivered at each rate. Each stimulation rate was repeated 5 times with a 30 second resting period in between trials. Cells were then given 60 seconds of unstimulated holding before switching to a different stimulation rate. The order of stimulation rates was counterbalanced to prevent sequence effects.
Data analysis
The average peak amplitude for each EPSC (1-10) was measured. To control for differences in EPSC amplitude across cells, the amplitude of EPSC peaks were standardized to the amplitude of the 1st EPSC in each train (EPSCn/EPSC1), giving 9 values (EPSCs 2 through 10). Initial analyses (detailed below) showed no effects of picrotoxin, so groups were subsequently collapsed to form Control, 5μM WIN, and 10μM WIN groups. The collapsed data was analyzed using GLM repeated measures (SPSS). There were 3 within-subjects factors: pre versus post holding (2 levels), stimulation rate (4 levels), and pulse number (9 levels). Drug treatment acted as a between-subjects factor (3 levels). To visualize reduction in synaptic depression, the difference between standardized values for pre-drug and post-drug for EPSCs 2-10 was calculated, with lower numbers representing reduced depression.
3. Results
The anatomical analyses documented that CB1 mRNA is present in CG cells and that CB1 protein is present in NM. The physiological analyses showed that CB1 activation reduces the amplitude of EPSCs, but also reduces the decrease in EPSCs across a train of stimuli.
3.1 CB1 mRNA is expressed in the cochlear ganglion of the chick
Our previous survey of CB1 distribution throughout the chick brain (Stincic and Hyson, 2008) revealed the robust expression of CB1 mRNA in the vestibular ganglion of the chick, but the CG was not preserved during the brain extraction procedure. The present study preserved the periphery during sectioning. The low density of the chick skull allowed cryostat sectioning of fresh frozen P10 chick heads that preserved both the CG cells and the local morphology.
Figure 1 displays a sample autoradiogram from a section containing the CG. The pattern of expression observed with skull-intact sections matched that found in extracted-brain sections (Stincic and Hyson, 2008). For example, the cerebellum displayed strong labeling in the granular layer, the tectum showed the same laminar-restricted labeling, and the brainstem was relatively devoid of any CB1 expression. Some nonspecific labeling could be seen in both the otoliths and surrounding bone, however, the CG was easily identifiable and well above background. Nearly all labeling was abolished when using a sense version of the CB1 riboprobe, suggesting labeling in the CG was specific.
Figure 1.

CB1 mRNA is expressed in the cochlear ganglion. White arrows point to cochlear ganglion. Nissl: Micrograph of skull-intact coronal section stained following in situ hybridization. Arrows point the cochlear ganglia on each side. C = cerebellum, c.d. = cochlear duct, IV = fourth ventricle, n.VIII = auditory nerve, t = tectum, O = otoconia.,CB1: Autoradiograph displays robust CB1 mRNA labeling of the cochlear ganglion. Intense labeling of the cerebellum and tectum is also observed. Sense: Autoradiograph using a sense control probe suggests that the antisense CB1 labeling in the cochlear ganglion and other neural areas is specific. Only the otoconia (O) shows non-specific labeling. Scale bar = 1mm.
3.2 CB1 protein appears to be presynaptic to NM neurons
As can be seen in Figure 2, CB1 antibody visualized with DAB produced staining around NM neurons. The annular pattern of labeling was similar to that produced by the presynaptic marker SV2. SV2 is a synaptic vesicle protein localized to vesicles involved in neurosecretion. The SV2 antibody obtained from the Developmental Studies Hybridoma Bank has been used successfully by our laboratory in the past to determine the pre- or post-synaptic localization of another protein (Wilkinson et al., 2003). SV2 (green) strongly labels vesicles in the calyceal synapse formed on NM neurons with no appreciable background.
Figure 2.
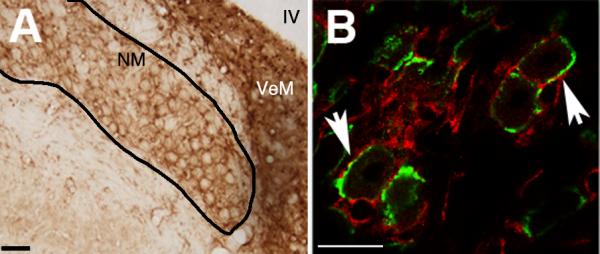
CB1 Immunohistochemistry labels area surrounding NM cell bodies. A: Micrograph of NM following immunolabeling with anti-CB1. There is little staining the soma of NM neurons, but ample staining around the somata. Scale bar = 50μM B: Double-labeling with CB1 (red) and SV2 (green), a presynaptic marker, show similar patterns, supporting the common finding that CB1 is located in the presynaptic neuron. IV = fourth ventricle, NM = nucleus magnocellularis, VeM = medial vestibular nucleus. Scale bar = 25μM.
SV2 labeling was consistent with these previous results and clearly labeled presynaptic vesicles with little to no background (see Figure 2). The CB1 antibody did not demonstrate much cellular labeling. CB1 labeling did appear concentrated in the area surrounding NM cells like SV2, albeit more diffusely and not co-localized. While not overlapping, the patterns of labeling observed with SV2 and CB1 are similar, suggesting that they both reside in the same subcellular compartment. Endbulbs of Held do not possess typical terminal boutons, but rather numerous active zones that form synapses and contain vesicles. The lack of co-localization suggests the majority of CB1 protein is located perisynaptically to SV2.
3.3 Cannabinoids reduce synaptic depression during high rates of stimulation
Control subjects (n =11) showed a clear and consistent pattern of rate–dependent depression across a train of afferent stimuli. Little depression was seen at 20Hz with a stable 25% decrease in EPSC amplitude after the first pulse. 100Hz stimulation resulted in a little over 40% depression by the end of the train. High rates of stimulation, 200 and 250Hz, produced greater depression earlier in the train, ultimately reducing EPSC peak values by 60-70%.
As predicted, application of WIN (n = 9 for each dose) reduced the amplitude of EPSCs, but also reduced the degree of synaptic depression across the train. An example of this effect can be observed in Figure 3. The overlaid traces (each an average of 5 sweeps) are slightly offset in time to allow easier visualization of the two trains of EPSCs. Although the first EPSC of each train is lower following WIN application, there is less decrease in the size of the EPSC across the train. Hence, the degree of activity-dependent synaptic depression was reduced in the presence of the cannabinoid agonist. Picrotoxin was included in a subset of recordings (n = 4, 5 and 4 for control, 5μM and 10μM WIN, respectively) to isolate CB effects on glutamatergic signaling and no differences in the patterns of results were observed in when the GABAA receptors were blocked.
Figure 3.

Stimulation rate and cannabinoid agonist affect synaptic depression. Displayed is an example from an NM neuron before (black) and during (gray) treatment with 5 μM WIN 55,212-2. The traces in the presence of the CB1 agonist are offset in the time axis to allow easier comparison of the EPSCs. As expected, there is greater synaptic depression across the train at higher rates of stimulation. EPSC amplitude is decreased, but remains more consistent across a train of stimuli after administration of the CB1 agonist WIN 55,212-2. Time scale is adjusted for each rate; the scale bar equals 50, 10, 5 or 4 ms, respectively, as stimulation rate increases.
The effect of WIN on EPSC amplitude was readily seen on the first EPSC of the stimulus train, isolating the drug effect from the possible confounding influence of activity-dependent synaptic depression. Figure 4 displays the average change in first EPSC amplitude before and after WIN (or control) application. In control conditions, first EPSC amplitude decreased approximately 13% over time. WIN application, however, resulted in a much greater reduction of first EPSC amplitude (approximately 34%) at both doses tested. There was no difference in the effects of WIN when picrotoxin was added to the bath. Statistical analyses confirmed these conclusions. A WIN dose X Picrotoxin between-subjects analysis of variance revealed a reliable effect of WIN [F(2,23)= 5.1, p < 0.05] but no effect of picrotoxin, nor a WIN X Picrotoxin interaction [Fs < 1.0]. Post-hoc Newman-Keuls pairwise comparisons (p < 0.05) revealed that all WIN groups were different than both control groups, but did not reliably differ from each other.
Figure 4.

Amplitude of the initial EPSC of a train is reduced by cannabinoid agonist both with and without blockade of GABAA receptors (n = 4-6 for each group). There was a slight reduction in the amplitude of the initial EPSC with the passage of time in control conditions. The percentage change in amplitude was much larger, however, when WIN 55,212-2 was bath applied during this time period. This effect appears to be at the glutamatergic auditory nerve – NM synapse since the WIN was equally effective when GABAA receptors were blocked with picrotoxin.
Since statistical analyses (detailed below) revealed no reliable effects of picrotoxin, the groups were pooled for the graphical presentation of the synaptic depression data. To examine the effects of drug treatment on depression, EPSC amplitudes were first transformed to their relative size compared to the first EPSC in the train. The drug-induced changes in these relative sizes were then computed by subtraction of the post-drug relative size from the pre-drug relative size. Consequently a negative number reflects less depression post-drug compared to pre-drug. These changes in depression across the train are presented in Figure 5. As can be seen, WIN treatment reduced synaptic depression observed at higher rates of stimulation. Only slight changes in depression were observed simply because of the passage of time and repeated stimulation (control conditions).
Figure 5.
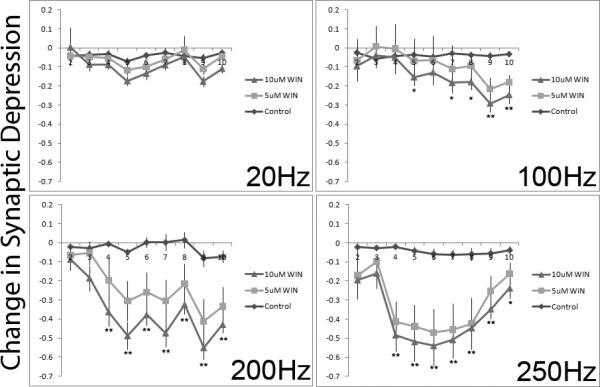
The CB1 agonist WIN 55,212-2 significantly reduces depression observed later in the pulse train when stimulated at high rates. Depression was measured across a train of 10 pulses before and after bath application of WIN. Amplitudes of EPSCs 2-10 were standardized to the amplitude of the 1st EPSC. The change in depression before and after WIN (standardized post-WIN amplitude – standardized pre-Win amplitude) administration is plotted by pulse number and separated by rate of stimulation. Lower numbers represent reduced depression after application of the CB1 agonist. Note that activation of the CB1 receptor reduced synaptic depression, particularly at the two highest rates of stimulation. Error bars represent standard error of the mean. N = 9-11 cells for each drug condition. ** indicates both WIN doses differ from control, * indicates only the high dose of WIN differs reliably from control.
Statistical analyses confirmed these conclusions. First, a full-design 2-between, 3-within mixed ANOVA was performed using picrotoxin treatment (2 levels) and Drug treatment (3 levels) as between-subjects variables. Pulse number (9 levels), Pre-post drug treatment (2 levels) and Stimulation rate (4 levels) were the within-subjects variables. The dependent measure was the relative size of the EPSC compared to the first EPSC of the 10-pulse train. There was no main effect of Drug treatment or picrotoxin (Fs < 1.0). There was also no reliable interaction of any of the other terms with picrotoxin (Fs < 1.0). As WIN was only present post-drug, this lack of main effect for WIN treatment was not surprising since all baseline recordings should be equal. Picrotoxin, however, was present throughout the entire experiment. A lack of any effect suggests that GABAergic activation is not involved in this protocol and any effects of WIN treatment are not acting on GABAergic signaling.
The main effects of the 3 within-subjects variables were, however, statistically reliable (Pulse number, F(8,136)=159; Rate, F(3, 51)=74; Pre-Post, F(1, 17)=33; p < 0.001 for each). As noted, depression was observed across the pulse train and was more apparent at higher stimulation rates (see Figure 3). The rate-dependent depression was also confirmed by the reliable Pulse number X Rate interactions (F(1,17)=174, p < 0.001).
The reliable Pre-Post effect suggests that time and/or WIN treatment had an effect. This is clarified by examination of the interaction terms. There was a reliable Pre-Post × WIN Group interaction (F(2, 17)=6.3, p < 0.01), revealing that the effect of time was dependent on whether or not cells were exposed to WIN. This effect depended on the rate of stimulation and was only apparent at later pulses in the train (reliable Pre-Post × Rate × Pulse number x WIN interaction, F(48, 408)=2.1, p < 0.001).
Given the complexity of the full design, separate ANOVAs were carried out for each of the 4 pulse rates. Since picrotoxin was without effect, we combined subjects with and without picrotoxin treatment resulting in 4, 3-way mixed ANOVAs using WIN dose as a between-subjects variable. Pulse number and Pre-Post drug were used as within-subjects variables. The results for the most important terms are presented in Table 1. At all rates, Pulse number was statistically reliable, indicating that depression was apparent at all rates. Pre-Post also had reliable effects in all but the 100Hz group where the effect was only marginal (F(1,26) = 2.8, p = 0.11). The interaction effects involving WIN treatment were found only at higher rates of stimulation. At 200 and 250Hz rates of stimulation the Pre-Post x Group interaction was reliable as were the 3-way Pre-Post × Pulse number × Group interactions. At 100Hz, only the 3-way interaction reached statistical significance. At the lowest rate of stimulation there was no apparent effect of WIN on the depression observed. Together, these results show that WIN has reliable effects on the synaptic depression observed at higher rates of stimulation.
Table 1.
Summary of statistical results for the analyses of synaptic depression at each stimulation rate.
| Stimulation Rate | 20Hz | 100Hz | 200Hz | 250Hz | ||||
|---|---|---|---|---|---|---|---|---|
| F(df) | p | F(df) | p | F(df) | p | F(df) | p | |
| Pre-Post | (8,208)=16 | <0.001 | (1, 26)=2.8 | 0.11 | (1,23)=17.6 | <0.001 | (1, 21)=15 | <0.001 |
| Pulse | (1,26)=8.4 | <0.01 | (8, 208)=51 | <0.001 | (2, 23)=3.6 | <0.05 | (2, 21)=4.4 | <0.05 |
| Pre-Post × Pulse | (8,208)= 1.6 | 0.14 | (8, 208)=2.0 | <0.05 | (8, 184)=5.4 | <0.001 | (8, 168)=4.1 | <0.001 |
| Pre-Post × Pulse × Group | (16,208)<1.0 | NS | (16, 208)=1.7 | <0.05 | (16, 184)=2.9 | <0.001 | (2, 168)=2.9 | <0.001 |
4. Discussion
CB1 is one of the most abundant neuromodulatory receptors found in the CNS. The variety of processing in which CB1 has been shown to be important grows steadily, but includes memory, motor function, and sensory processing. The present set of experiments demonstrated that CB signaling may also regulate the encoding of auditory information at its earliest stages in the brain. These studies first demonstrated the presence of CB1 in the cochlear nucleus of the chick and then showed that activation of this receptor modulated the response to a train of stimuli.
CB1 expression in the chick brain appears to follow a pattern described in mammalian literature both in concentration and localization with few exceptions (Mailleux et al., 1992, Mailleux and Vanderhaeghen, 1992, Matsuda et al., 1993, Stincic and Hyson, 2008). Using ISH and IHC in a complementary fashion, CG cells were revealed to be the likely source of CB1 localized to the calyceal terminals synapsing with NM. As such, the present data are consistent with the common finding that CB1 protein is localized in presynaptic terminals. While immuno-EM analyses would be necessary to definitively prove the calyceal localization, the comparison of the ISH and IHC data make a compelling case, particularly when combined with the evidence of physiological effects of CB1 agonist at this synapse. The annular appearance of CB1 immunoreactivity is similar to the known presynaptic marker SV2. The two antibodies did not show overlapping co-immunolabeling, but this would not be expected since CB1 is membrane bound and is not coupled directly to the synaptic vesicles. A possible alternative hypothesis is that CB1 is expressed by glial cells in NM. If this were the case, however, one would expect to observe substantial CB1 mRNA in and around NM, but this was not the case. Instead, the present study demonstrated dramatic levels of CB1 mRNA in the CG. A second alternative hypothesis for this pattern of immunolabeling is that CB1 receptors are located on GABAergic terminals, which originate primarily from neurons in the superior olivary nucleus (SON). If this were the case, however, one would expect punctate immunolabeling in NM rather than the more expansive labeling reminiscent of calyceal synapses. Additionally, one would expect significant CB1 mRNA to be localized in the SON. Such labeling in the SON was not observed (Stincic and Hyson, 2008). Consequently, the most parsimonious explanation of the combined data is that CB1 mRNA is produced by CG cells, and the receptor is inserted at the terminals in NM.
Based on the standard roles of CB signaling in other systems, it was hypothesized that CB1 activation may modulate the strength of the auditory nerve-NM synapse. In particular, CB signaling was considered to be potentially useful for aiding the consistent, often phase-locked, activity necessary for downstream, bilateral ITD comparison. Activity-dependent CB modulation may be an additional adaptation assisting NM in accurately responding to a wide range of acoustic stimuli while remaining relatively insensitive to sound intensity. Previous work examining the calyceal synapse of the MNTB (Kushmerick et al., 2004) are consistent with the hypothesis that CB signaling may be useful to regulating precisely-timed synaptic activity.
The ability of CB1 activation to increase the consistency of synaptic transmission was demonstrated by examining synaptic depression across a train of stimuli. High rates of stimulation led to synaptic depression at the cnVIII-NM synapse and application of an agonist to CB1 receptors attenuated this synaptic depression. The increase in depression with both pulse number and stimulation rates matched the patterns of depression previously observed (Brenowitz et al., 1998) in NM. Despite minor differences from other depression protocols, the observed depression during baseline recordings was typical for NM. Importantly, CB1 activation with WIN reduced the degree of synaptic depression observed at higher rates of stimulation.
The combined anatomical and physiological data suggest that CB1 is localized at the glutamatergic cnVIII-NM synapse. To be confident of this conclusion, however, we must address the possibility that other receptors systems or technical details may have been influenced by our treatment. Only glutamatergic and GABAergic signaling has been described at the cnVIII-NM synapse. In order to determine which of these systems were influenced by CB1 activation, picrotoxin was included during a portion of the recordings. Picrotoxin blocks GABAA receptors which are known to mediate inhibitory postsynaptic currents in NM (Kuo et al., 2009). As picrotoxin had no effect, CB1-activation is most likely influencing glutamatergic signaling. It may be noted, however, that GABAB receptors are present in NM and do regulate GABA and glutamate release (Lu et al., 2005). Activation of GABAB receptors during high rates of stimulation has the same beneficial effect as that proposed for CB1 activation, in that it is thought to reduce synaptic depression by lowering neurotransmitter release probability (Brenowitz et al., 1998). The similarity in the pattern of results may, at first, raise the possibility that the CB effects observed in the present studies are mediated through changes in GABA release and consequent changes in GABAB receptor activation. This possibility, however, is not logical. If there was CB-mediated retroinhibition of GABA release, then it would decrease GABAB activation and this would presumably have the opposite effect on synaptic depression across the train of stimuli (i.e., it would produce greater depression). Together, the data suggest that the observed effects of WIN are most likely on glutamate/AMPA signaling by a mechanism that is independent of GABA signaling.
One technical issue that could have potentially influenced the results of these experiments was time-dependent degradation throughout the course of the day. Slices were typically kept for up to 4 hours, but there appeared to be no reliable differences over time. In the case of individual cells the average length of an experiment was around 45 minutes with a maximum duration of 70 minutes. Rundown of cells and an accompanying decrease in EPSC amplitude and increase in series resistance were potentially confounding variables. Large increases in series resistance resulted in rejection of cells from our analysis. Still, small changes in cell responsiveness and recording could potentially mask or even mimic the hypothesized result (i.e. decreased overall EPSC amplitude and degree of depression). Control subjects that were held for an equal period of time experienced a 13% average reduction in the amplitude of the 1st EPSC in any given train. Treatment with WIN for at least 10 minutes, however, resulted in an average 33-35% reduction in the amplitude of the 1st EPSC. Statistical comparisons confirmed that the reduction in the 1st EPSC amplitude was greater following WIN than in control conditions over the course of a recording.
Even a 35% reduction may not cause an increase in action potential failure rate due to the high-safety factor of the cnVIII-NM synapse. What is notable is that CBs do indeed reduce the degree of depression seen across cnVIII pulse trains. Based on the commonly accepted mode of action of CBs on presynaptic terminals (reduced neurotransmitter release), the decreased EPSC amplitude coincides with a conservation of neurotransmitter release, and decrease in vesicle depletion, receptor desensitization, and autoreceptor activation. Consequently, the decreased release from cnVIII may provide for a more consistent response during a long duration stimulus.
Depression increased with rate, but some degree of depression was observed with all 4 stimulation frequencies. WIN, however, only had statistically reliable effects at the higher rates of stimulation. This may be because depression at the lower rates was not great enough to observe a reliable reduction (i.e., a floor effect), or it may be because different mechanisms contribute to depression. Consequently, it is possible that a CB1 sensitive mechanism of synaptic depression is only engaged by high rates of stimulation. Endogenous CB production is activity-dependent; hence eCBs would be most active and potentially beneficial when the synapse is highly active. A more uniform EPSC could preserve the consistent activation of NM neurons by maintaining suprathreshold currents even when cnVIII is firing at high rates. The auditory system could then continue to code temporal events throughout the duration of a stimulus and not just at the onset.
The effects of exogenously applied CBs to the slice preparation may mimic the ongoing level CB1 activation that would occur in vivo. Spontaneous rates in NM, even with no acoustic stimuli, can reach 100Hz in vivo (Warchol and Dallos, 1990). Since eCB production appears to be activity-dependent, little eCB production would be expected in an unstimulated slice, but some basal level of production should exist at all times with intact tissue. Higher release should also follow rises in activity above this baseline. eCBs may therefore act to maintain a favorable signal to noise ratio, dampening depression by reining in the amount of glutamate present in the cleft as release rises, without actually inhibiting firing. Even if eCBs were significantly present at low rates, little effect would be expected. Our findings show WIN had at best minor effects on EPSCs from 20Hz pulse trains. Further studies should focus on the mechanisms by which eCBs are produced in NM to clarify the role played here.
NM must fulfill a physiologically demanding role in the temporal coding of acoustic stimuli. Details of adaptations of the ionotropic and metabotropic receptors with excitatory and inhibitory signaling in NM have emerged over the last couple decades. CB signaling now must be added to the list of synaptic adaptations. The present studies demonstrate the anatomical and physiological presence of the CB1 receptors. The role of these receptors in acoustic processing will require additional studies, but the present studies suggest that they may help maintain consistent response amplitudes across a long duration stimulus.
Highlights.
We demonstrate the location and action of CB1 receptors in the avian cochlear nucleus.
CB1 mRNA is expressed in the cochlear ganglion neurons.
CB1 protein is located in the nucleus magnocellularis.
CB1 agonist decreased EPSC amplitude and also reduced synaptic depression.
Consistent activation could be important for coding prolonged acoustic stimuli.
Acknowledgements
The authors thank Dr. Zhijun Shen for helping begin this line of investigation, Dr. Ken Soderstrom for providing CB1 antibody, and Katie Carzoli and Melissa Masicampo for critical evaluation of this manuscript. This research was supported by NIDCD grant DC000858.
Abbreviations
- ABC
Avidin-Biotin Complex
- ACSF
Artificial Cerebral Spinal Fluid
- BSA
Bovine Serum Albumin
- CB
Cannabinoid
- CB1
Cannabinoid Receptor One
- CG
Cochlear Ganglion
- cnVIII
Vestibulocochlear Nerve
- DAB
Diaminobenzidine
- DMSO
Dimethyl Sulfoxide
- E(n)
Embryonic Day (n)
- eCB
Endocannabinoid
- EPSC
Excitatory Post Synaptic Current
- EPSP
Excitatory Post Synaptic Potential
- IHC
Immunohistochemistry
- ISH
In Situ Hybridization
- MNTB
Medial Nucleus of the Trapezoid Body
- NGS
Normal Goat Serum
- NHS
Normal Horse Serum
- NL
Nucleus Laminaris
- NM
Nucleus Magnocellularis
- P(n)
Posthatch Day (n)
- PBS
Phosphate Buffered Saline
- SON
Superior Olivary Nucleus
- SSC
Standard Sodium Citrate
- WIN
WIN 55,212-2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barnes-Davies M, Forsythe ID. Pre- and postsynaptic glutamate receptors at a giant excitatory synapse in rat auditory brainstem slices. The Journal of physiology. 1995;488(Pt 2):387–406. doi: 10.1113/jphysiol.1995.sp020974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science (New York, NY) 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- Brenowitz S, David J, Trussell L. Enhancement of synaptic efficacy by presynaptic GABA(B) receptors. Neuron. 1998;20:135–141. doi: 10.1016/s0896-6273(00)80441-9. [DOI] [PubMed] [Google Scholar]
- Brenowitz S, Trussell LO. Minimizing synaptic depression by control of release probability. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:1857–1867. doi: 10.1523/JNEUROSCI.21-06-01857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr CE, Boudreau RE. Central projections of auditory nerve fibers in the barn owl. The Journal of comparative neurology. 1991;314:306–318. doi: 10.1002/cne.903140208. [DOI] [PubMed] [Google Scholar]
- Carr CE, Fujita I, Konishi M. Distribution of GABAergic neurons and terminals in the auditory system of the barn owl. The Journal of comparative neurology. 1989;286:190–207. doi: 10.1002/cne.902860205. [DOI] [PubMed] [Google Scholar]
- Carr CE, Konishi M. A circuit for detection of interaural time differences in the brain stem of the barn owl. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1990;10:3227–3246. doi: 10.1523/JNEUROSCI.10-10-03227.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Brown DA. Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. British journal of pharmacology. 1992;106:231–232. doi: 10.1111/j.1476-5381.1992.tb14321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Code RA, Burd GD, Rubel EW. Development of GABA immunoreactivity in brainstem auditory nuclei of the chick: ontogeny of gradients in terminal staining. The Journal of comparative neurology. 1989;284:504–518. doi: 10.1002/cne.902840403. [DOI] [PubMed] [Google Scholar]
- Cook DL, Schwindt PC, Grande LA, Spain WJ. Synaptic depression in the localization of sound. Nature. 2003;421:66–70. doi: 10.1038/nature01248. [DOI] [PubMed] [Google Scholar]
- Czesnik D, Schild D, Kuduz J, Manzini I. Cannabinoid action in the olfactory epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2967–2972. doi: 10.1073/pnas.0609067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh S, Onozuka K, Bender KJ, Bender VA, Lutz B, Mackie K, Feldman DE. Postnatal development of cannabinoid receptor type 1 expression in rodent somatosensory cortex. Neuroscience. 2007;145:279–287. doi: 10.1016/j.neuroscience.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, Briley EM, Axelrod J, Simpson JT, Mackie K, Devane WA. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7656–7660. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JT, Jackson H, Rubel EW. Synaptic excitation of the second and third order auditory neurons in the avian brain stem. Neuroscience. 1982;7:1455–1469. doi: 10.1016/0306-4522(82)90257-3. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB. Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. Journal of neurochemistry. 1997;69:631–638. doi: 10.1046/j.1471-4159.1997.69020631.x. [DOI] [PubMed] [Google Scholar]
- Howlett AC. Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Molecular pharmacology. 1985;27:429–436. [PubMed] [Google Scholar]
- Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Molecular pharmacology. 1984;26:532–538. [PubMed] [Google Scholar]
- Howlett AC, Qualy JM, Khachatrian LL. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Molecular pharmacology. 1986;29:307–313. [PubMed] [Google Scholar]
- Hyson RL, Reyes AD, Rubel EW. A depolarizing inhibitory response to GABA in brainstem auditory neurons of the chick. Brain research. 1995;677:117–126. doi: 10.1016/0006-8993(95)00130-i. [DOI] [PubMed] [Google Scholar]
- Krause EG, Curtis KS, Stincic TL, Markle JP, Contreras RJ. Oestrogen and weight loss decrease isoproterenol-induced Fos immunoreactivity and angiotensin type 1 mRNA in the subfornical organ of female rats. The Journal of physiology. 2006;573:251–262. doi: 10.1113/jphysiol.2006.106740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Koyano K, Ohmori H. Synaptic depression improves coincidence detection in the nucleus laminaris in brainstem slices of the chick embryo. The European journal of neuroscience. 2002;15:984–990. doi: 10.1046/j.1460-9568.2002.01933.x. [DOI] [PubMed] [Google Scholar]
- Kuo SP, Bradley LA, Trussell LO. Heterogeneous kinetics and pharmacology of synaptic inhibition in the chick auditory brainstem. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:9625–9634. doi: 10.1523/JNEUROSCI.0103-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmerick C, Price GD, Taschenberger H, Puente N, Renden R, Wadiche JI, Duvoisin RM, Grandes P, von Gersdorff H. Retroinhibition of presynaptic Ca2+ currents by endocannabinoids released via postsynaptic mGluR activation at a calyx synapse. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:5955–5965. doi: 10.1523/JNEUROSCI.0768-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachica EA, Rübsamen R, Rubel EW. GABAergic terminals in nucleus magnocellularis and laminaris originate from the superior olivary nucleus. The Journal of comparative neurology. 1994;348:403–418. doi: 10.1002/cne.903480307. [DOI] [PubMed] [Google Scholar]
- Lu Y, Burger RM, Rubel EW. GABA(B) receptor activation modulates GABA(A) receptor-mediated inhibition in chicken nucleus magnocellularis neurons. Journal of neurophysiology. 2005;93:1429–1438. doi: 10.1152/jn.00786.2004. [DOI] [PubMed] [Google Scholar]
- Mackie K, Devane WA, Hille B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Molecular pharmacology. 1993;44:498–503. [PubMed] [Google Scholar]
- Mailleux P, Parmentier M, Vanderhaeghen JJ. Distribution of cannabinoid receptor messenger RNA in the human brain: an in situ hybridization histochemistry with oligonucleotides. Neuroscience letters. 1992;143:200–204. doi: 10.1016/0304-3940(92)90265-9. [DOI] [PubMed] [Google Scholar]
- Mailleux P, Vanderhaeghen JJ. Distribution of neuronal cannabinoid receptor in the adult rat brain: a comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience. 1992;48:655–668. doi: 10.1016/0306-4522(92)90409-u. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Bonner TI, Lolait SJ. Localization of cannabinoid receptor mRNA in rat brain. The Journal of comparative neurology. 1993;327:535–550. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- McCullumsmith RE, Stincic TL, Agrawal SM, Meador-Woodruff JH. Differential effects of antipsychotics on haloperidol-induced vacuous chewing movements and subcortical gene expression in the rat. European journal of pharmacology. 2003;477:101–112. doi: 10.1016/j.ejphar.2003.08.018. [DOI] [PubMed] [Google Scholar]
- Monsivais P, Yang L, Rubel EW. GABAergic inhibition in nucleus magnocellularis: implications for phase locking in the avian auditory brainstem. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:2954–2963. doi: 10.1523/JNEUROSCI.20-08-02954.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyilas R, Gregg LC, Mackie K, Watanabe M, Zimmer A, Hohmann AG, Katona I. Molecular architecture of endocannabinoid signaling at nociceptive synapses mediating analgesia. The European journal of neuroscience. 2009;29:1964–1978. doi: 10.1111/j.1460-9568.2009.06751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholt EM, Rubel EW, Hyson RL. A circuit for coding interaural time differences in the chick brainstem. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1992;12:1698–1708. doi: 10.1523/JNEUROSCI.12-05-01698.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks TN, Rubel EW. Organization and development of the brain stem auditory nuclei of the chicken: primary afferent projections. The Journal of comparative neurology. 1978;180:439–448. doi: 10.1002/cne.901800303. [DOI] [PubMed] [Google Scholar]
- Penzo MA, Peña JL. Endocannabinoid-mediated long-term depression in the avian midbrain expressed presynaptically and postsynaptically. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:4131–4139. doi: 10.1523/JNEUROSCI.5466-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes AD, Rubel EW, Spain WJ. Membrane properties underlying the firing of neurons in the avian cochlear nucleus. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1994;14:5352–5364. doi: 10.1523/JNEUROSCI.14-09-05352.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends in pharmacological sciences. 2001;22:565–572. doi: 10.1016/s0165-6147(00)01805-8. [DOI] [PubMed] [Google Scholar]
- Stincic TL, Hyson RL. Localization of CB1 cannabinoid receptor mRNA in the brain of the chick (Gallus domesticus). Brain research. 2008;1245:61–73. doi: 10.1016/j.brainres.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Stella N, Piomelli D, Mackie K, Karten HJ, Maguire G. Cannabinoid CB1 receptors and ligands in vertebrate retina: localization and function of an endogenous signaling system. Proceedings of the National Academy of Sciences of the United States of America. 1999a;96:14565–14570. doi: 10.1073/pnas.96.25.14565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker AJ, Maguire G, Mackie K, Lindsey J. Localization of cannabinoid CB1 receptors in the human anterior eye and retina. Investigative ophthalmology & visual science. 1999b;40:2442–2448. [PubMed] [Google Scholar]
- Sullivan WE, Konishi M. Segregation of stimulus phase and intensity coding in the cochlear nucleus of the barn owl. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1984;4:1787–1799. doi: 10.1523/JNEUROSCI.04-07-01787.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Moiseff A, Konishi M. Time and intensity cues are processed independently in the auditory system of the owl. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1984;4:1781–1786. doi: 10.1523/JNEUROSCI.04-07-01781.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton-Jones ZD, Kennett GA, Vickers SP, Clifton PG. A comparison of the effects of the CB(1) receptor antagonist SR141716A, pre-feeding and changed palatability on the microstructure of ingestive behaviour. Psychopharmacology. 2007;193:1–9. doi: 10.1007/s00213-007-0745-8. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E. Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:8137–8146. doi: 10.1523/JNEUROSCI.17-21-08137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warchol ME, Dallos P. Neural coding in the chick cochlear nucleus. Journal of comparative physiology A, Sensory, neural, and behavioral physiology. 1990;166:721–734. doi: 10.1007/BF00240021. [DOI] [PubMed] [Google Scholar]
- Wilkinson BL, Jeromin A, Roder J, Hyson RL. Activity-dependent regulation of the subcellular localization of neuronal calcium sensor-1 in the avian cochlear nucleus. Neuroscience. 2003;117:957–964. doi: 10.1016/S0306-4522(02)00928-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Trussell LO. A characterization of excitatory postsynaptic potentials in the avian nucleus magnocellularis. Journal of neurophysiology. 1994a;72:705–718. doi: 10.1152/jn.1994.72.2.705. [DOI] [PubMed] [Google Scholar]
- Zhang S, Trussell LO. Voltage clamp analysis of excitatory synaptic transmission in the avian nucleus magnocellularis. The Journal of physiology. 1994b;480(Pt 1):123–136. doi: 10.1113/jphysiol.1994.sp020346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Rubio ME, Tzounopoulos T. Distinct functional and anatomical architecture of the endocannabinoid system in the auditory brainstem. Journal of neurophysiology. 2009;101:2434–2446. doi: 10.1152/jn.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]