

Abstract
The roles of the NAT2 genotype and enzyme maturation on isoniazid pharmacokinetics were investigated in South African infants with perinatal HIV exposure enrolled in a randomized, double-blind, controlled trial of isoniazid for prevention of tuberculosis disease and latent infection. Plasma concentration-time measurements of isoniazid from 151 infants (starting at 3-4 months of age) receiving isoniazid 10 to 20 mg/kg/d orally during the course of the 24-month study were incorporated in a population analysis along with NAT2 genotype, body weight, age, and sex. The results showed a different NAT2 enzyme maturation profile for each of the 3 acetylation groups, with the 70-kg body weight–normalized typical apparent clearance for the fast and intermediate acetylators increasing from 14.25 L/h and 10.88 L/h at 3 months of age to 22.84 L/h and 15.58 L/h at 24 months of age, respectively, with no significant change in the apparent clearance of the slow group during this period. A hypothesis is proposed to explain the genotype-dependent enzyme maturation processes for the NAT2 enzyme.
Keywords: Infants, pharmacogenetics, population pharmacokinetics, enzyme maturation, genetic polymorphism
Pediatric tuberculosis (TB) and human immunodeficiency virus (HIV) co-infection results in considerable childhood morbidity and mortality.1,2 Isoniazid (INH) has been a cornerstone of regimens for the treatment and prevention of TB for more than 30 years.3 INH is primarily metabolized through hepatic arylamine N-acetyltransferase type 2 (NAT2) to an inactive form.4 N-acetylation activity is genetically determined, and a trimodal distribution of INH elimination has been demonstrated.5 Several single nucleotide polymorphisms have been identified by genetic analysis of NAT2, some of which code for NAT2 proteins with impaired enzyme activity and are classified as the slow acetylator allele group (S allele), while others having enzyme activity similar to the wild-type (NAT2*4) are designated as the fast acetylator allele group (F allele).6
INH pharmacokinetic data are very limited in infants and children, as is information on the developmental expression of NAT2 in the fetal, neonatal, and early infant periods.7 Pharmacokinetic studies of INH in children have demonstrated a relationship between INH clearance and postnatal age8-10; however, none have separated the effects of body size change and enzyme maturation on the clearance of INH. The latter requires studies in infants, the focus of the study reported herein, because considerable NAT2 enzyme maturation is expected to occur during this stage of life.
The goal of this work was to quantify the roles of body size change and enzyme maturation on INH pharmacokinetics in infants. This was accomplished by developing a population pharmacokinetic model that separated the effects of size and enzyme maturation of the 3 NAT2 genotypes of INH metabolism, fast, intermediate, and slow acetylation. The work represents the first pharmacogenetic population model of INH pharmacokinetics in infants. The INH pharmacokinetic and pharmacogenetic data were from P1041, a phase II/III prospective, randomized, double-blind, placebo-controlled trial of INH for prevention of tuberculosis disease and latent infection in South African infants with perinatal HIV exposure conducted by the International Maternal, Pediatric, Adolescent, AIDS Clinical Trials Network (IMPAACT).
METHODS
Study Design and Infant Population
The study design for the IMPAACT P1041 trial is summarized below, focusing on the objectives for this work. Infants from Cape Town and Durban, South Africa, who were perinatally exposed to HIV were eligible for the study following informed consent by the primary caregivers. The study protocol was approved by the human subjects committee at the 2 study sites: the Medicines Control Council in South Africa and the Division of AIDS of the NIH. In the treatment arm of the trial, infants received weight-normalized oral doses of INH from 50 to 200 mg once daily from age 3 through 24 months. Infants were randomized into 2 different blood-sampling strategies. Blood samples were collected at either 1 or 3 hours postdose or at 2 and 4 hours postdose. Table I summarizes the blood-sampling design as well as demographic and pharmacogenetic information. Two study participants had data at 33 to 34 months of age (week 120) instead of week 84.
Table I.
Data Summary and Demographic and Pharmacogenetic Characteristics of Model Development, Validation, and Complete Data Set for Population Pharmacokinetic Analysis
| Complete Data Set | Model Development Data Set | Validation Data Set | P | |
|---|---|---|---|---|
| Number of subjects | ||||
| Total | 151 | 98 | 53 | |
| Only week 0 | 62 | 42 | 20 | .90a |
| Only week 12 | 48 | 32 | 16 | |
| Only week 84 | 7 | 4 | 3 | |
| Week 0 and 84 | 19 | 11 | 8 | |
| Week 12 and 84 (or 120) | 15 | 9 | 6 | |
| Number of samples | 368 | 234 | 134 | |
| Age, mo | 8.84 ± 8.03 (3.03-33.47) | 8.49 ± 7.78 (3.03-33.47) | 9.47 ± 8.47 (3.03-33.27) | .53b |
| Body weight, kg | 7.78 ± 2.65 (3.5-18.1) | 7.67 ± 2.56 (3.5-16.7) | 7.97 ± 2.83 (3.8-18.1) | .57b |
| NAT2 genotype, SS/FS/FF | 48/67/36 | 34/43/21 | 14/24/15 | .36a |
| Sex, M/F | 71/80 | 45/53 | 26/27 | .88a |
Data are presented as mean ± SD (range).
Chi-square test for comparison of model development and validation data sets.
Mann-Whitney test for comparison of model development and validation data sets.
INH Assay and NAT2 Genotyping
Plasma concentrations of INH were determined by HPLC.11,12 DNA was purified from blood samples using a salting out procedure.13 A 1000-base pair NAT2 sequence was amplified by the polymerase chain reaction (PCR) as described previously.14 This 1000-bp segment was then analyzed using the restriction fragment length polymorphism technique, with appropriate cleavage by the restriction enzymes BamHI, KpnI, MpI, and TaqI; these enzymes delineate the NAT2*7, NAT2*5, NAT2*14, and NAT2*6 alleles, respectively. An additional cleavage of the BamHI, KpnI, and MspI products was also performed using PstI, to facilitate the accurate analyses of these restriction profiles. Additional analyses of the NAT2-PCR product with the restriction enzymes DdeI and FokI as well as the inclusion of an allele-specific PCR reaction (for the T341C mutation) allowed for the subclassification of these NAT2 alleles. DNA cleavage profiles, generated by the restriction enzymes, were analyzed via nondenaturing polyacrylamide gel electrophoresis, and the profiles were visualized by silver staining.15 NAT2 polymorphisms NAT2*4, NAT2*12, and NAT2*13 were considered as fast (F) acetylator alleles, and NAT2*5, NAT2*6, NAT2*7, and NAT2*14 were slow (S) acetylator alleles (Supplementary Table S1). Infants were classified into 1 of 3 NAT2 acetylator phenotypes: fast (FF), intermediate (FS), and slow (SS), according to their NAT2 genotypes. Distributions of NAT2 genotypes (allelic combination) in the 3 NAT2 phenotype groups are summarized in Supplementary Table S2.
Base Model
The pharmacokinetic base model (ie, model without covariates) was a one-compartment model with first-order absorption and first-order elimination. Population analysis was performed using the first-order conditional estimation method in NONMEM version VI.16 Since no plasma samples were obtained during the absorption phase, the absorption rate constant (Ka) was fixed at 3 h−1 based on previous relevant literature reports.17,18 Plasma INH samples obtained during week 12 and week 84 were assumed to be in steady state, while week 0 data were not. A combined proportional/additive error model was used to describe the residual error. The parameters of the apparent clearance (CL/F, L/h) and apparent volume of distribution (V/F, L) were assumed to be log-normally distributed in the population with nonzero covariance. Since some subjects were sampled on 2 different occasions (week 0 and week 84 or week 12 and week 84), inter-ccasion variability (IOV) was included in the model.
Pharmacogenetic Enzyme Maturation Model
During infancy, both growth and development can influence the pharmacokinetics of a drug (see Anderson and Holford19 for a detailed review). The objective of the enzyme maturation model is to separate the effect of growth (body size change) and development (enzyme maturation). Body weight and age were used as primary covariates to reflect body size change and enzyme maturation effects, respectively.
Motivated by the results of exploratory data analysis, the final population model incorporated effects of body size change, NAT2 enzyme maturation, and NAT2 genotype on the disposition of INH in the population of infants studied. The influence of size (fsize) was incorporated using an allometric power model of body weight (WT, kg).
Body weight was normalized for a typical adult (70 kg) to allow for extrapolation and comparison with adult values, and the power exponents were fixed at 0.75 for CL/F and 1 for V/F, as is conventionally done in population studies involving children.20,21
Separate sigmoidal models were used to represent the effect of enzyme maturation (fEM) for each of the 3 NAT2 genotypes (GT) as follows:
The extent of enzyme maturation (MAT) was represented by the chronological age (AGE) plus gestational age (full-term gestation assumed, 9.33 months) to better reflect the time period of enzyme maturation.19 In the above equation, AgeCL50 is the MAT at which CL/F is 50% of the mature CL/F value. H is the hill coefficient for CL/F.
The final model also incorporated an increase in bioavailability during the course of the study. The following covariate model was used to account for relative bioavailability change:
where AgeF50 is the AGE at which bioavailability is 50% of the final bioavailability value.
The complete covariate models for apparent clearance (CL/F) and apparent volume of distribution (V/F) are as follows:
where (CL/F)Std and (V/F)Std are the 70-kg body weight standardized asymptotic (mature) apparent clearance and distribution volume values, and CL/F and V/F are the true typical apparent clearance and distribution volume values.
Model Validation Procedure
Model validation via data splitting was implemented to evaluate the ability of the population model to predict the observed concentrations from an independent data set. Following the guidance from the Food and Drug Administration,22 the complete data set (n = 151) was randomly divided into a model development data set (n = 98) and a validation data set (n = 53), as summarized in Table I. Statistical methods were used to test for differences in the model development and model validation data sets (Mann-Whitney rank-sum test for continuous covariates and χ2 test for categorical covariates) with regard to the subject-specific covariates.
The population model derived from the model development data set was then used to predict the INH plasma concentrations for each subject from the validation data set, using that subject’s dosing, sampling history, and covariates. The predicted INH plasma concentrations were compared with the measured INH concentrations in the validation data set. The standardized mean prediction error was calculated for each subject in the validation data set.23 A Shapiro-Wilk test for normality was performed, and a one-sample Wilcoxon signed-rank test was then used to assess whether the plasma INH predictions error had a mean of zero.23
Validation of pharmacokinetic parameters was based on a qualitative and quantitative comparison of pharmacokinetic parameters predicted from the model development data set with the pharmacokinetic parameters determined from the validation data set subjects. The model development data set population model predictions of the validation data set subjects’ parameters (based on covariate values of the validation data set subjects) were compared with individual pharmacokinetic parameter estimates after fitting the base model (without covariates) to the validation data set (naive predictor).24 In the qualitative assessment, the prediction errors of the pharmacokinetic parameters were plotted against covariates in the validation data set to evaluate any residual dependence of pharmacokinetic parameters on the covariates. Quantitative assessment involved calculation of the prediction error of each pharmacokinetic parameter (model development data set prediction, naive prediction) and evaluation of the prediction error bias (median prediction error, mpe) and precision (median absolute prediction error, mae).24
Following the validation, the data from the model development and validation data sets were pooled, and the population model obtained from the model development data set was then fitted to the complete data set and refined to attain the final population model.22 The model refinement included considering other previously unexplanatory covariates, as well as alternative forms of the covariate models. The likelihood ratio test and standard error of parameter estimates were compared to achieve the final pharmacogenetic enzyme maturation model.
RESULTS
Patient Demographics and NAT2 Genotype
Data from 151 infants enrolled in the treatment arm of the trial were available for pharmacokinetic analysis. INH plasma concentration-time profiles are shown in Figure 1. Infants received INH orally from 3 to 24 months of age, and plasma INH concentrations were measured on 3 study weeks: week 0 (infants 3-4 months of age), week 12 (infants 6-7 months), and week 84 (infants 22-24 months). This information is summarized in Table I (complete data set column), along with demographic information. Plasma samples were obtained from 19 infants at both study week 0 and study week 84, while 15 infants had plasma INH measurements at both study week 12 and study week 84. The NAT2 genotype was determined in all infants (Table I).
Figure 1.
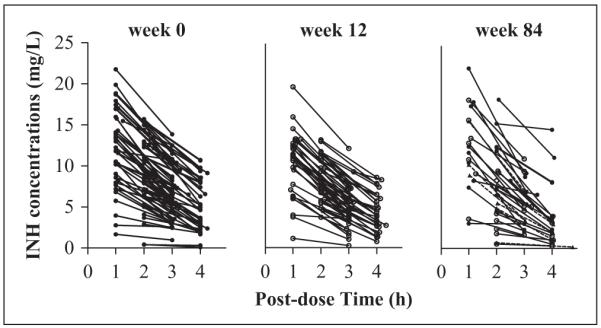
INH concentration measurements for all the study subjects (N = 151). Solid lines connecting filled circles represent subjects having week 0 data. Solid lines connecting open circles represent subjects having week 12 data. Dashed lines connecting triangles represent subjects having only week 84 data.
Pharmacogenetic Enzyme Maturation Model
A one-compartment model with first-order absorption and first-order elimination was used as the stage 1 pharmacokinetic model, with a proportional error variance model. A stage 2 population model was developed that incorporated the effects of size (body weight) change, enzyme maturation, NAT2 genotype, and change of relative bioavailability. Interoccasion variability was also included in the model. To evaluate goodness of fit, both the individual predictions and population predictions of the final model were plotted against the observed concentrations in Figure 2a and 2b, resulting in R2 values (from the Spearman correlation) of 0.97 and 0.44, respectively. Taken together with the weighted residual versus postdose time and population predictions plots shown in Figure 2c and 2d, these results indicate reasonably good fits of the final model to INH plasma concentration-time data. None of the other tested covariates were able to explain any additional interindividual variability (IIV).
Figure 2.

Goodness-of-fit plots from the final pharmacogenetic enzyme maturation model. (a) Individual predicted concentrations and (b) population predicted concentrations versus observed concentrations, and population weighted residuals versus (c) postdose time and (d) population predicted concentrations.
The parameter estimates for the final pharmacogenetic enzyme maturation model developed using the complete data set are presented in Table II. Typical apparent oral clearance (CL/F) and apparent distribution volume (V/F) for each of the 3 NAT2 genotypes were affected by size (body weight) and relative bioavailability change. As shown in Figure 3, the typical CL/F depended on enzyme maturation for both the FS and FF genotypes, while there was no detectable change in the typical CL/F over the course of the study for the SS NAT2 genotype. With the relative bioavailability fixed at 1, the 70-kg body weight standardized typical CL/F for NAT2 FF and FS group increased from 14.25 L/h and 10.88 L/h at 3 months old to 22.84 L/h and 15.58 L/h at 24 months old, respectively. For the SS group, the 70-kg body weight–normalized typical CL/F was estimated at 7.35 L/h, and no significant change was found during the study period. Relative bioavailability changed from 0.72 at 3 months to 0.95 at 24 months.
Table II.
Summary of Final Population Enzyme Maturation Model Pharmacokinetic and Covariate Parameter Estimates for Complete Data Set
| Parameter (Unit) | Model Parameter Estimate (%RSE) |
|---|---|
| CL/F = (CL/F)Std_FF*fSize*fEM(HFF, AgeCL50FF)/fF | |
| (CL/F)Std_FF (L/h) | 23.7a (11.0) |
| HFF | 2.88 (23.5) |
| AgeCL50FF (mo) | 10.7 (6.4) |
| CL/F = (CL/F)Std_FS*fSize*fEM(HFS, AgeCL50FS)/fF | |
| (CL/F)Std_FS (L/h) | 23.7a (11.0) |
| HFS | 0.82 (37.5) |
| AgeCL50FS (mo) | 15.1 (20.9) |
| CL/F = (CL/F)Std_SS*fSize/fF | |
| (CL/F)Std_SS (L/h) | 7.35a (9.3) |
| V/F = (V/F)Std*fSize/fF | |
| (V/F)Std (L) | 53.3a (7.7) |
| fSize = (WT/70)Power | |
| Power for CL/F | 0.75 fixed |
| Power for V/F | 1 fixed |
| fF(AgeF50) | |
| AgeF50 (mo) | 1.18 (37.5) |
| Ka | 3 fixed |
| Residual variability | |
| Proportional (CV%) | 13.4 |
(CL/F)Std and (V/F)Std estimates in the final model are 70-kg body weight–normalized values. %RSE is percentage relative standard error. fSize, fEM, and fF are defined in the Methods section.
Figure 3.
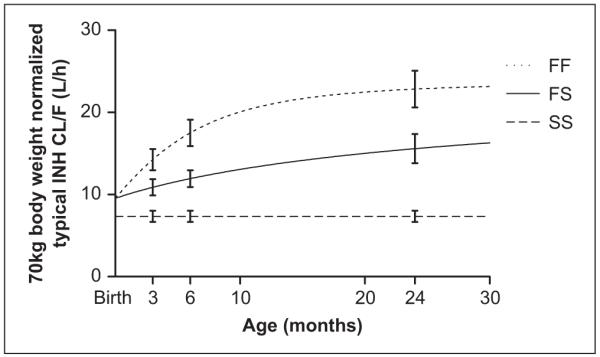
Seventy-kilogram body weight–normalized typical value of isoniazid apparent clearance versus age plot from the final enzyme maturation model with relative bioavailability fixed at 1. Error bars represent standard error of the mean. The curves were obtained from the population covariate model functions with relative bioavailability effect (fF) fixed at 1.
The interindividual variability for CL/F and V/F in the final model was 42.2% and 29.0%, versus 57.5% and 29.2% in the base model, and the objective function value decreased from 1040 in the base model to 886 in the final model. Moreover, interoccasion variability for CL/F and V/F also decreased from 61.4% and 35.2% in the base model to 50.1% and 34.4% in the final model. The results of the visual predictive check shown in Figure 4 indicate that the final model reflected the overall variability in the observed data.
Figure 4.
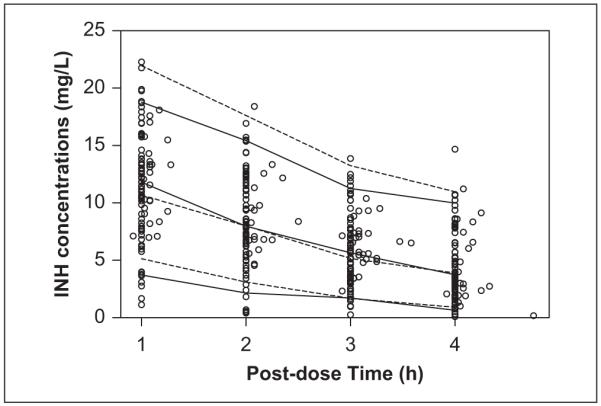
Visual predictive check of the final model. Open circles are the observed isoniazid concentrations. Solid lines are the 5th, 50th, and 95th percentiles of the observations. Dashed lines are the 5th, 50th, and 95th percentiles of the simulated predictions.
Model Validation
There were no statistically significant differences in subject-specific covariates between the model development data set and model validation data set (Table I). The population pharmacokinetic model derived from the model development data set successfully predicted the measured INH plasma concentrations in the validation data set (ie, mean standardized mean prediction error not significantly different from zero, P = .35). Regarding validation through pharmacokinetic parameters, both the qualitative (Supplementary Figure S1) and quantitative (Supplementary Table S3) assessments showed that the covariate model predictions were better than the naive predictions. After validation, the 2 data sets were combined, and the model refinement did not produce any changes to the form of the pharmacogenetic enzyme maturation model obtained using the model development data set (ie, all covariates and covariate models were retained).
DISCUSSION
A population model is presented based on a randomized controlled trial of INH for prevention of tuberculosis disease in South African infants with perinatal HIV exposure. The model quantifies the role of size (body weight) and enzyme maturation for each of the 3 NAT2 genotype groups on INH CL/F, providing insights into the pharmacogenetics of NAT2 enzyme maturation in infants.
In the population model, the absorption rate constant (Ka) was fixed at 3 h−1 based on previous literature reports17,18 due to the absence of INH plasma concentration data during the absorption phase (Figure 1). A test of the sensitivity of the model results to this assumption found no notable changes in the final model parameter estimates for several values of Ka in the range of 1.5 h−1 to 3.9 h−1. In the final population model, it was also assumed that CL/F for the NAT2 FF genotype group is greater than or equal to that of the FS group, which in turn is greater than or equal to that of the SS group.
For the visual predictive check results displayed in Figure 4, the median value of the observations agrees with the median simulated predictions of the predictive check; however, the 5th and 95th percentiles of the observations are overpredicted. This may be the result of some extremely low INH plasma concentrations (<3 mg/L) measured at 1 hour and 2 hours postdose in 9 of the 151 infants. When the final model was reanalyzed using a reduced data set without those 9 subjects’ data, the resulting 5th and 95th percentile predictions were in close agreement with the corresponding values calculated from the observations (Supplementary Figure S2). We also noted that the estimate of interoccasion variability of CL/F and V/F decreased from 50.1% and 34.4% to 38.3% and 26.7% using the reduced data set, because 4 of the 9 subjects removed had inconsistent concentration values between their week 0 or week 12 and week 84 values.
The final pharmacogenetic enzyme maturation model indicates that CL/F is associated with the following 4 factors: NAT2 genotype, enzyme maturation, size (body weight), and relative bioavailability changes. Concordance between acetylator activity and NAT2 genotype is evident from the resulting model, which is supported by a number of INH pharmacokinetic studies performed in both adults and older children.5,10 Our finding that INH acetylation capability significantly increases with age is consistent with the earlier work of Pariente-Khayat et al8 on the maturation of NAT2 enzyme. By using NAT2 genotypic information in infants during the first 2 years of age, our population model allows the quantification of different NAT2 enzyme maturation processes of the 3 NAT2 genotypes. As shown in Figure 3, with relative bioavailability fixed at 1, the 70-kg body weight–normalized typical CL/F values for the FF and FS groups increase with age (enzyme maturation), while there was no significant change in the SS group. In addition, the faster CL/F increase in FF group compared with the FS group suggests that maturation of the NAT2 enzyme in infants with the FF genotype is faster than those with the FS genotype. We tested a model that included a maturation process for the apparent CL/F of the SS group, which did show some early increase in clearance. The results of this model, however, were not significantly different from the simpler constant clearance model for the SS group. This may suggest the NAT2 enzyme with SS genotype matures before 3 months of age or that any effect of maturation on clearance, which occurs in the SS genotype during the study period 3 months to 24 months of age, is undetectable via a population model approach given the study design.
Based on the observation that NAT2 enzymes with 3 genotypes have different enzyme maturation processes, we hypothesize that each NAT2 genotype has its own gene expression pattern. During development, certain genes can be turned on and off and can be up- and down-regulated. The maturation in the NAT2 enzyme with different genotypes may depend on the regulation of gene expression at certain levels, such as transcriptional and posttranslational levels. Related research has been extensively conducted on the ontogeny of some of the major cytochrome P450 enzymes, but information on the developmental expression of the NAT2 gene in fetal, neonatal, and early infant periods is still limited.7,25-30 Further investigation into the molecular genetics of the developmental expression of NAT2 gene is needed to test this hypothesis.
In addition to enzyme maturation, size change is also responsible for the variation in INH pharmacokinetics from birth to adulthood. Once size is standardized, as recommended,19 the effects of other factors such as enzyme maturation, NAT2 genotype, and relative bioavailability can be investigated. By normalizing all pharmacokinetic parameters to typical adult body weight (70 kg), we can compare the extrapolated results from our model to adult values and thereby assess the predictive ability of the enzyme maturation model. The resulting model predicted a 70-kg body weight–standardized typical mature CL/F of 23.7 L/h and 7.35 L/h for the NAT2 FF and SS groups, respectively, and a typical V/F of 53.3 L, which are comparable with the adult values estimated in a previous INH population pharmacokinetic model.17
In the final model, relative bioavailability was found to increase with age, which may be explained in part by dose regimen adherence that is expected to improve with the age of the infants. In contrast to improved dose adherence, other factors would be expected to decrease the oral bioavailability, such as increased first-pass effect caused by NAT2 enzyme maturation in the liver and the gut.31 However, the NAT2 enzyme maturation’s effect on oral bioavailability is likely to be negligible compared with dose adherence since INH absolute bioavailability is high and not significantly different among the 3 NAT2 genotypes based on adult data.17 Change in diet over 3 to 24 months of age could also have affected bioavailability, but we do not have the data to investigate this possibility further. The model with a relative bioavailability effect had a better fit and better validation results than did the model without a change in relative bioavailability.
There are several pharmacokinetic candidate markers that can be used to describe enzyme maturation, including 70-kg body weight–normalized absolute clearance using the allometric 3/4 power model (CLwn), 70-kg body weight–normalized apparent clearance using the allometric 3/4 power model (CLwn/F), elimination rate constant (kel), and half-life (t1/2). While CLwn is a more appropriate marker than CLwn/F, for many orally administered drugs, such as INH, CLwn/F can also be used to describe enzyme maturation if any bioavailability change is well characterized. Although the other 2 markers, kel and t1/2, can successfully separate the effect of bioavailability, they cannot be used to characterize enzyme maturation processes due to the inconsistent and independent changes of clearance and distribution volume in infancy and childhood.19
The pharmacogenetic enzyme maturation model presented here provides useful insights into INH pharmacokinetics in infants and NAT2 enzyme maturation. Based on the modeling results, we propose a hypothesis to explain the genotypically polymorphic maturation patterns of the NAT2 enzyme, which could be confirmed by further molecular genetics research on developmental expression of NAT2 gene. With those well-characterized explanatory covariates and clinical outcomes, the resulting model may serve as a guide to pediatric INH dose selection in infants. In addition, the modeling framework presented has potential application to other drugs used in neonates and infants.
Supplementary Material
Acknowledgments
The authors acknowledge Drs Madhi and McSherry, protocol co-chairs; Dr Nachman and Violari, protocol vice-chairs; S. Kim, protocol statistician; the entire P1041 protocol team and the sites that conducted this study, particularly Dr Cotton and Tygerberg Children’s Hospital at the Stellenbosch University, Tygerberg, South Africa; Dr. Bobat of the University of Kwazulu-Natal and her study team who enrolled subjects into this pharmacokinetic study; and all of the study subjects and their families who participated in this trial.
Financial disclosure: Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID; U01 AI068632), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). Support of the sites was provided by the NIAID and the NICHD International and Domestic Pediatric and Maternal HIV Clinical Trials Network funded by NICHD (contract number N01-DK-9-001/HHSN267200800001C). This work was supported in part by grant NIH/NIBIB P41-EB001978 (to D.Z.D.) and grants NIH/NIAID UO1-AI068632 (to C.V.F.).
Footnotes
Supplementary data for this article are available at http://jcp.sagepub.com/supplemental/.
REFERENCES
- 1.Wilkinson D, Davies GR. Pediatric tuberculosis in rural South Africa—value of directly observed therapy. J Trop Pediatr. 1998;44:266–269. doi: 10.1093/tropej/44.5.266. [DOI] [PubMed] [Google Scholar]
- 2.Lalloo UG, Pillay S. Managing tuberculosis and HIV in sub-Sahara Africa. Curr HIV/AIDS Rep. 2008;5:132–139. doi: 10.1007/s11904-008-0021-5. [DOI] [PubMed] [Google Scholar]
- 3.Robitzek EH, Selikoff IJ. Hydrazine derivatives of isonicotinic acid (rimifon marsilid) in the treatment of active progressive caseous-pneumonic tuberculosis; a preliminary report. Am Rev Tuberc. 1952;65:402–428. doi: 10.1164/art.1952.65.4.402. [DOI] [PubMed] [Google Scholar]
- 4.Weber WW, Hein DW. Clinical pharmacokinetics of isoniazid. Clin Pharmacokinet. 1979;4:401–422. doi: 10.2165/00003088-197904060-00001. [DOI] [PubMed] [Google Scholar]
- 5.Parkin DP, Vandenplas S, Botha FJ, et al. Trimodality of isoniazid elimination: phenotype and genotype in patients with tuberculosis. Am J Respir Crit Care Med. 1997;155:1717–1722. doi: 10.1164/ajrccm.155.5.9154882. [DOI] [PubMed] [Google Scholar]
- 6.Fretland AJ, Leff MA, Doll MA, Hein DW. Functional characterization of human N-acetyltransferase 2 (NAT2) single nucleotide polymorphisms. Pharmacogenetics. 2001;11:207–215. doi: 10.1097/00008571-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Butcher NJ, Tiang J, Minchin RF. Regulation of arylamine N-acetyltransferases. Curr Drug Metab. 2008;9:498–504. doi: 10.2174/138920008784892128. [DOI] [PubMed] [Google Scholar]
- 8.Pariente-Khayat A, Rey E, Gendrel D, et al. Isoniazid acetylation metabolic ratio during maturation in children. Clin Pharmacol Ther. 1997;62:377–383. doi: 10.1016/S0009-9236(97)90115-6. [DOI] [PubMed] [Google Scholar]
- 9.Rey E, Gendrel D, Treluyer JM, et al. Isoniazid pharmacokinetics in children according to acetylator phenotype. Fundam Clin Pharmacol. 2001;15:355–359. doi: 10.1046/j.1472-8206.2001.00044.x. [DOI] [PubMed] [Google Scholar]
- 10.Schaaf HS, Parkin DP, Seifart HI, et al. Isoniazid pharmacokinetics in children treated for respiratory tuberculosis. Arch Dis Child. 2005;90:614–618. doi: 10.1136/adc.2004.052175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seifart HI, Gent WL, Parkin DP, Donald PR. HPLC determination of isoniazid, acetylisoniazid, and hydrazine concentrations in plasma samples from TB patients. Tuber Lung Dis. 1994;75(suppl 1):61–62. [Google Scholar]
- 12.Seifart HI, Gent WL, Parkin DP, van Jaarsveld PP, Donald PR. High-performance liquid chromatographic determination of isoniazid, acetylisoniazid and hydrazine in biological fluids. J Chromatogr B Biomed Appl. 1995;674:269–275. doi: 10.1016/0378-4347(96)82886-6. [DOI] [PubMed] [Google Scholar]
- 13.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hickman D, Sim E. N-acetyltransferase polymorphism: comparison of phenotype and genotype in humans. Biochem Pharmacol. 1991;42:1007–1014. doi: 10.1016/0006-2952(91)90282-a. [DOI] [PubMed] [Google Scholar]
- 15.Bassam BJ, Caetano-Anollés G, Gresshoff PM. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal Biochem. 1991;196:80–83. doi: 10.1016/0003-2697(91)90120-i. [DOI] [PubMed] [Google Scholar]
- 16.Beal SL, Sheiner LB, NONMEM Project Group . NONMEM User’s Guide. University of California; San Francisco: 1992. [Google Scholar]
- 17.Kinzig-Schippers M, Tomalik-Scharte D, Jetter A, et al. Should we use N-acetyltransferase type 2 genotyping to personalize isoniazid doses? Antimicrob Agents Chemother. 2005;49:1733–1738. doi: 10.1128/AAC.49.5.1733-1738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peloquin CA, Jaresko GS, Yong CL, Keung AC, Bulpitt AE, Jelliffe RW. Population pharmacokinetic modeling of isoniazid, rifampin, and pyrazinamide. Antimicrob Agents Chemother. 1997;41:2670–2679. doi: 10.1128/aac.41.12.2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson BJ, Holford NH. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol. 2008;48:303–332. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 20.Anderson BJ, Allegaert K, Van den Anker JN, Cossey V, Holford NH. Vancomycin pharmacokinetics in preterm neonates and the prediction of adult clearance. Br J Clin Pharmacol. 2007;63:75–84. doi: 10.1111/j.1365-2125.2006.02725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menon-Andersen D, Mondick JT, Jayaraman B, et al. Population pharmacokinetics of imatinib mesylate and its metabolite in children and young adults. Cancer Chemother Pharmacol. 2009;63:229–238. doi: 10.1007/s00280-008-0730-x. [DOI] [PubMed] [Google Scholar]
- 22.US Food and Drug Administration. Center for Drug Evaluation and Research . Guidance for Industry: Population Pharmacokinetics. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); Rockville, MD: 1999. [Google Scholar]
- 23.Vozeh S, Maitre PO, Stanski DR. Evaluation of population (NONMEM) pharmacokinetic parameter estimates. J Pharmacokinet Biopharm. 1990;18:161–173. doi: 10.1007/BF01063558. [DOI] [PubMed] [Google Scholar]
- 24.Bruno R, Vivier N, Vergniol JC, De Phillips SL, Montay G, Sheiner LB. A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinet Biopharm. 1996;24:153–172. doi: 10.1007/BF02353487. [DOI] [PubMed] [Google Scholar]
- 25.Burk O, Wojnowski L. Cytochrome P450 3A and their regulation. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:105–124. doi: 10.1007/s00210-003-0815-3. [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Goldstein JA. The transcriptional regulation of the human CYP2C genes. Curr Drug Metab. 2009;10:567–578. doi: 10.2174/138920009789375397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu MH, Savas U, Griffin KJ, Johnson EF. Human cytochrome p450 family 4 enzymes: function, genetic variation and regulation. Drug Metab Rev. 2007;39:515–538. doi: 10.1080/03602530701468573. [DOI] [PubMed] [Google Scholar]
- 28.Oesch-Bartlomowicz B, Oesch F. Phosphorylation of cytochromes P450: first discovery of a posttranslational modification of a drug-metabolizing enzyme. Biochem Biophys Res Commun. 2005;338:446–449. doi: 10.1016/j.bbrc.2005.08.092. [DOI] [PubMed] [Google Scholar]
- 29.Stevens JC. New perspectives on the impact of cytochrome P450 3A expression for pediatric pharmacology. Drug Discov Today. 2006;11:440–445. doi: 10.1016/j.drudis.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Choudhary D, Jansson I, Sarfarazi M, Schenkman JB. Xenobiotic-metabolizing cytochromes P450 in ontogeny: evolving perspective. Drug Metab Rev. 2004;36:549–568. doi: 10.1081/dmr-200033447. [DOI] [PubMed] [Google Scholar]
- 31.Husain A, Zhang X, Doll MA, States JC, Barker DF, Hein DW. Identification of N-acetyltransferase 2 (NAT2) transcription start sites and quantitation of NAT2-specific mRNA in human tissues. Drug Metab Dispos. 2007;35:721–727. doi: 10.1124/dmd.106.014621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.