

Abstract
The unique capability of germ cells to give rise to a new organism, allowing the transmission of primary genetic information from generation to generation, depends on their epigenetic reprogramming ability and underlying genomic totipotency. Recent studies have shown that genome-wide epigenetic modifications, referred to as “epigenetic reprogramming”, occur during the development of the gamete precursors termed primordial germ cells (PGCs) in the embryo. This reprogramming is likely to be critical for the germ line development itself and necessary to erase the parental imprinting and setting the base for totipotency intrinsic to this cell lineage. The status of genome acquired during reprogramming and the associated expression of key pluripotency genes render PGCs susceptible to transform into pluripotent stem cells. This may occur in vivo under still undefined condition, and it is likely at the origin of the formation of germ cell tumors. The phenomenon appears to be reproduced under partly defined in vitro culture conditions, when PGCs are transformed into embryonic germ (EG) cells. In the present paper, I will try to summarize the contribution that epigenetic modifications give to nuclear reprogramming in mouse PGCs.
1. Introduction
Nuclear reprogramming is generally defined as the process reverting the nucleus of a differentiated cell to a pluripotent or totipotent state. The formation in culture of embryonic stem (ES) or epiblast embryonic stem (EpiES) cells from the inner cell mass (ICM) of the blastocyst or the epiblast of postgastrulating embryo, respectively, of embryonic germ (EG) cells from primordial germ cells and more recently of induced pluripotent stem (iPS) cells from differentiated somatic cells are examples of nuclear reprogramming in vitro. As far as I know, in mammals, physiological nuclear reprogramming leading to totipotency occurs only at the onset of embryogenesis when the genome of the zygote and subsequently of the early blastomeres acquires totipotency. This reprogramming requires a genome status of gametes that originates from early processes of nuclear reprogramming occurring at the beginning of gametogenesis in the primordial germ cells (PGCs), the embryonic precursors of the gametes. The identification of the timing and the underlying molecular mechanisms of this early process in PGCs offers precious information not only about gametogenesis and reproduction, but also on the secretes of stemness and clues for a number of pathologies including cancer development.
Nuclear reprogramming involves a variety of genetic and epigenetic modulators. This latter include DNA methylation and a variety of posttranslational histone modifications. The emerging small regulatory RNA molecules can be also regarded as epigenetic regulators but will not be discussed here (for reviews, see [1, 2]).
During the last decades, with the relevant contribution of the ideas and the inspiration of the late Anne McLaren, her work and that of her disciples, important advancements have been done on the basic principles and mechanisms governing nuclear reprogramming in PGCs, mainly in the mouse. The present review, is an attempt to summarize the emerging information relative to the contribution of epigenetic changes, in particular DNA methylation and histone modification, to nuclear reprogramming in mouse PGCs.
2. DNA Methylation
In mammals, methylation to the 5 position of the cytosine pyrimidine ring represents the major epigenetic modification of DNA (for reviews, see [3, 4]). It occurs predominantly in regions containing high frequency of the sequence cytosine phosphate guanine (CpG), termed CpG islands. Transcription repression is generally associated to methylation of CpG islands located into or near gene promoters (5′flanking region). In almost all cell types, tissue-specific genes generally undergo demethylation of CpG islands specifically in their tissue of expression. In contrast, housekeeping genes contain CpG islands unmethylated in all cells.
Mammalian genome encodes three DNA methylases (DNMTs): the maintenance methyltransferase DNMT1 and the de novo methyltransferases DNMT3a and DNMT3b. Moreover, DNMT3L, another member of the DNMT3 family, does not possess DNA methyltransferase activity, but it is required for DNMT3a and DNMT3b functions. Once imposed by de novo DNMTs, DNA methylation is transmitted to the cell progeny by DNMT1 as long as demethylation processes take place.
DNA methylation/demethylation can be divided into global (genome wide) and specific (when just specific DNA sequences are methylated/demethylated). While it is thought that global methylation state, termed methylome, is relatively stable in differentiated cells, dynamic changes of methylome occur during cell differentiation. Global demethylation occurs at two specific times during development, namely, at the onset of embryogenesis and during the PGC development and is associated to nuclear reprogramming; specific demethylation seems typical of somatic cells responding to particular signals.
DNA demethylation can be achieved passively by the failure of the maintenance methylation by DNMT1 during DNA synthesis at the S stage of the cell cycle or by the active removal of methyl groups from cytosine, independently of DNA replication. Active DNA demethylation can be achieved basically by (1) direct removal of methyl group, (2) the removal of entire DNA patch followed by filling with new nucleotides by nucleotide excision repair (NER), and (3) the removal of methylated base either by direct removal of methylcytosine, or through previous cytosine modification. This latter can occur for example by 5-meC deamination to produce thymine (T) or hydroxylation to produce 5-hydroxymethylcytosine (5-hmeC) followed by the removal of T by T/G mismatch or 5-hmeC by several ways and the insertion of unmethylated cytosine using base excision repair (BER) machinery (for a review, see [5]). As discussed later, in mammals, active DNA demethylation seems mostly to employ mechanisms referred in point (3).
3. Histone Modifications
Histone modifications provide an additional and complex source of epigenetic modification of the genome. Many enzymes that regulate histone modifications, mainly occurring in their amino terminal tail, have been identified. They include histone acetyltransferases (HATs), deacetylases (HDACs), methyltransferases (HMTs), and demethylases (HDMases). Phosphorylation, ADP ribosylation, sumoylation, and ubiquitination are other possible histone modifications, but they will not be discussed here since, as far as I know, there is no evidence about their involvement in PGC nuclear reprogramming.
Generally, the histones are acetylated and deacetylated on lysine (K) residues. These reactions are catalyzed by enzymes with histone acetyltransferase (HAT) or histone deacetylase (HDAC) activity. In most cases, histone acetylation enhances gene transcription while histone deacetylation represses transcription. Histones may be methylated on either lysine (K) or arginine (R) residues by the addition of one, two, or three methyl groups. The process is catalyzed basically by three families of HMT enzymes: the protein arginine N-methyltransferase (PRMT) family, the lysine methyltransferases of the Su (var) 3–9, enhancer of zeste, trithorax (SET)-domain, or the disruptor of telomere silencing 1 (DOT1/DOT1L) protein families (for a review, see [6]). The regulative consequence of histone methylation on transcriptional activation or repression depends on the site and degree of methylation.
Among the best-characterized mediators of histone methylation are protein complexes of the polycomb (PcG) and trithorax (TRXG) groups containing a SET domain (for a review, see [7]). PcG and TRXG proteins form multimeric complexes that bind to DNA and direct posttranslational histone modifications. They are critical regulators of gene expression, repressors (PcG), or activators (TRXG), necessary for cell fate specification and maintenance. PcG proteins catalyze preferentially two distinct histone modifications: trimethylation of lysine 27 of histone 3 (H3K27me3) by polycomb repressive complex 2 (PRC2) and mono-ubiquitination of lysine 119 H2A (H2AK119ub1) by PRC1. H3K27 is trimethylated by the enhancer of zeste 2 (EZH2), which is the catalytic subunit of PRC2 that includes noncatalytic subunits suppressor of zeste 12 (SUZ12) and embryonic ectoderm development (EED). EEDs may also interact with HDACs and mediate repressive histone de-acetylation. Some TRXG proteins methylate histone H3 at lysine 4 (H3K4), a transcription permissive mark, while others posses demethylation or acetylation activities.
Elements of the histone code and some combinatorial pattern of histone marks are shown in Table 1. A particularly well-studied histone combination is the contemporary presence of the repressive H3K27me3 with the active H3K4me3, termed a “bivalent domain.” Bivalent domains maintain genes in a state that is repressed but ready for activation and have been recently discovered in ES cells (see below). Genomic regions that are associated with gene silencing, including transposons and repetitive elements, frequently possess the heterochromatin marks H3K9me3 and H4K20me3.
Table 1.
Summary of the main histone code in mammalian cells.
| Type of modification | Histone | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| H3K4 | H3K9 | H3K14 | H3K27 | H3K79 | H4K20 | H2BK5 | H3K4 + H3K27 | H3K9 + H4k20 | |
| Mono-methylation | Activation | Activation | Activation | Activation | Activation | Activation | |||
| Dimethylation | Activation | Repression | Repression | Activation | |||||
| Trimethylation | Activation | Aepression | Repression | Repression | Bivalent | Heterochromatin | |||
| Acetylation | Activation | Activation | |||||||
Histone methylation was believed to be a quite stable modification. After the discovery of histone demethylases (HDMases), histone methylation is now considered a dynamic modification. Two kinds of histone lysine demethylases have been identified, including lysine specific demethylase 1 (LSD1) and Jumonji C (JmjC) domain family proteins. Peptidyl-arginine deiminase 4 (PAD4/PADI4) antagonizes methylation on arginine residues by converting mono-methyl arginine in histone H3 and H4 to citrulline.
4. Basic Principle/s of Nuclear Reprogramming in PGCs
Before entering into the specific themes of the present review, a brief description of the PGC development in the mouse embryo is needed. For more detailed information excellent reviews are available on this topic [8–12].
The PGC precursors (about 6 cells) are specified in the proximal epiblast in the posterior region of the embryo around 6.25 days post coitum (dpc) [13]. While proliferating, PGC precursors move through the posterior primitive streak into the extraembryonic mesoderm at the basis of allantois. Here, around 7.25 dpc, a founder population of about 40 PGCs is determined [14]. Thereafter, these PGCs migrate and enter the gonadal ridges from 10.5 dpc onwards. By 13.5 dpc, PGCs enter meiotic prophase in the female becoming primary oocytes and mitotic arrest in the male gonad becoming prospermatogonia. In the present review, we will refer as early or pregonadal PGCs between 7.5 and 10.5 dpc before entering into the gonads, and late PGCs between 10.5 and 13.5 dpc after arrival and colonization of the gonadal ridges.
Three basic principles govern nuclear reprogramming in PGC development: first, the need to inhibit somatic cell lineage pathways, second, the establishment of a transcription regulatory network necessary to maintain differentiation plasticity, and third, the resetting of the genome epigenetic status to eliminate epimutations and erase the parental imprinting. These conditions are likely to be interrelated and require both genetic and epigenetic modulators. Alltogether, they are intrinsic to the germ cell differentiation pattern itself and necessary for setting the germ cell genome towards totipotency.
The inhibition of all cell differentiation pathways, including the germ cell lineage, and the presence of transcription regulatory network for pluripotency are typical features of the embryonic stem (ES) cells, derived from the culture of the inner cell mass (ICM) of the blastocyst. In addition, ES cells posses self-renewal, the capability of dividing while maintaining their undifferentiated and pluripotent status for long periods. PGCs, however, at least in the mouse, do not possess a long-lasting self-renewal capability, are lineage determined and do not manifest pluripotency in conditions in which ES cells normally do (i.e., chimeras [15, 16]). This means that in PGCs, some stem cell characteristics coexist with specific differentiation pathways, a unique feature of the germ cell lineage.
Before their final differentiation into oocytes in females and prospermatogonia in males, in several mammalian species, including humans, PGCs can be induced to deviate their normal differentiation pathway and give rise to true stem cell lines similar to ES cells termed embryonic carcinoma (EC) cells in vivo, and embryonic germ (EG) cells in vitro (for a review, see [17]). This process super imposes a nuclear reprogramming on PGCs that basically inhibits their germline differentiation pattern, confers them the self renewal capability and allows their latent pluripotency to manifest in suitable environments. This process offers a formidable model to understand the origin of certain tumours from germ cells and perhaps from other cell types and interesting clues about important aspects of stemness that will be discussed in a separate section below.
As reported above, when PGCs finally differentiate into oocytes, a very specialized differentiation pathway centred on meiosis begins. During meiosis prophase and up to meiotic block at the diplotene stage around birth, the genome of the oocyte is organized in condensed chromosomes, and all players of the transcription regulatory network for pluripotency (i.e., Oct4, Nanog, and Sox2 genes) are silenced. Thereafter, when the oocyte is included within a primordial follicle and meiosis is blocked, the genome remains relatively quiescent until the growing phase begins in selected primordial follicles. This signs the beginning of intense transcription activity and the reexpression of transcription factors typical of pluripotency (i.e., OCT4, SOX2). It is likely that during the oocyte growing phase, other genome changes complete nuclear reprogramming towards totipotency, but very little is known about. At the end of the maturation process, oocytes posses pluripotency as shown by their capability to give rise to teratomas, a kind of tumours composed of multiple cell types derived from one or more of the three germ layers [18].
In the male germ cell lineage, PGCs give rise to prospermatogonia, also called, gonocytes, which within the forming testicular cords of the fetal testis progressively undergo mitotic arrest. These cells remain quiescent until after birth reenter a mitotic cell cycle. Through unknown processes, a subpopulation of gonocytes give rise to a true stem cell populations the spermatogonia stem cells (SSCs) able to self-renewal and to give rise to waves of proliferating spermatogonia that enter meiosis as spermatocytes. It is now clear, that SSCs not only posses self renewal but, like PGCs, have an intrinsic pluripotency. In fact, under certain culture conditions, different from those causing PGCs transformation into EG cells, SSCs obtained from prepubertal or adult testes can give rise to pluripotent stem cell lines termed germ-like stem cells (GSCs, [19]), multipotent adult germline stem cells (maGSCs, [20]), and multipotent adult spermatogonial-derived stem cells (MASCs, [21]). There is no information about the mechanisms responsible for the nuclear reprogramming of SSCs into such stem cell types.
5. Timing and Mechanisms of Nuclear Reprogramming in PGCs
During development, nuclear reprogramming in PGCs occurs at several steps: when their precursors are specified, during their determination, the migratory phase, and after their arrival into the gonadal ridges. It is not known if it takes place under the influence of the different microenvironments or if it occurs following the activation of an autonomous program or both. It is known, however, that it is associated to epigenetic changes involving wide progressive DNA demethylation and several histone modifications. Moreover, complex genome-wide transcription dynamics are strictly related to these epigenetic modifications.
It is to be pointed out that the study of epigenetic modifications in PGCs mainly during pregonadal stages is made difficult by the small numbers of available cells. For example, methods as immunoprecipitation of methylated DNA (MeDIP) and chromatin immunoprecipitation technologies (ChIP) cannot be used with pregonadal PGCs. Changes in DNA methylation in pregonadal PGCs have been so far analysed only by immunohistochemistry using antibodies against 5-meC. In late PGCs, both immunohistochemistry and bisulphite methods followed by PCR have been used.
5.1. DNA Demethylation in Pregonadal PGCs
Early studies indicated that mouse PGCs isolated from the gonadal ridges possess relatively under methylated DNA [22, 23]. It is now known that just after specification, PGCs have a relatively high genome methylation status similar to that of the surrounding epiblast cells and that during the subsequent stages, they undergo various rounds of passive and active DNA demethylation [24]. This means that PGC precursors do not escape the progressive de novo methylation of extraembryonic and embryonic lineages that follows the loss of genomic methylation occurred at the onset of embryogenesis between the zygote and blastocyst stages.
According to immunohistochemistry, a genome-wide DNA demethylation begins in a portion of PGCs at around 8 dpc, soon after their determination [24]. This seems to arise mostly passively. In fact, at this time, several conditions in PGCs favour passive demethylation. They are proliferating, and the expression of three main methyltransferases, the maintenance methyltransferase DNMT1, the de novo methyltransferase DNMT3a, and DNMT3b, is repressed [25, 26]. In addition, the expression of Np95/Uhrf1, one of the DNMT1's cofactors, is also repressed [27]. It appears that the key transcription factors governing the germ-cell specification, BLIMP1/PRDM1 and PRDM14 (for a review, see [9, 28, 29]), are directly or indirectly involved in this repression [27]. DNMT3L that at later stages will be essential for the establishment of the primary imprinting both in male and female germ cells and DNA methylation of transposons in meiotic male germ cells [30], is not expressed in PGCs [31].
Subsequently, between 8.5 and 9.5 dpc, when PGCs begin to migrate towards the gonadal ridges and become temporarily mitotic quiescent, demethylation extends to the most part of PGCs [25]. At this time, because mitotic quiescence and the DNMT1 reexpression, demethylation is likely to be mostly active [25, 27]. Accumulating evidence supports the possibility that active DNA demethylation in PGCs employs DNA repair-mediated pathways [32, 33]. Such evidence actually comes from studies carried out in gonadal PGCs (see below), and no information is available for pregonadal PGCs. In a recent review, Mochizuki and Matsui [34] speculated that the expression in pregonadal PGCs of the growth arrest and DNA-damage-inducible protein 45 (GADD45) [27] and the DNA deaminase (AID) [35], suggest their involvement in active DNA demethylation in such cells. In this model, it is assumed that deamination by AID converts 5-meC into thymine and gives rise to a T-G mismatch followed by the insertion of unmethylated cytosine using BER; GADD45 might recruit AID and the DNA glycosidase methyl-CpG-binding domain protein 4 (MBD4) to 5-meC [36].
In this context of passive and active DNA demethylation, imprinted genes, repetitive DNA elements including retrotransposons and satellite centromeric sequences and some germ-cell specific genes (i.e., Vasa, Gcna1, Dazl, Scp3), remain largely protected from demethylation. An important unresolved question is how such selective epigenetic modifications can occur. At the onset of embryogenesis, in early PGCs, DNMT1 and STELLA might be responsible for such protection [37, 38].
Which might be the function of DNA demethylation at these stages? The wide-genome demethylation starting in pregonadal PGCs might favor the maintenance of one of the major processes of PGC specification, the inhibition of differentiation pathway towards somatic cell lineages (for a review, see [9, 28, 29]). At the same time, demethylation might favour the expression of pluripotency-related and germ cell-specific genes occurring just after PGC specification. Many pluripotency-related genes (i.e., Oct4, Nanog, Sox2, Rex1, Fbx15) and some germ cell-specific genes (i.e., Stella, Nanos3) are expressed specifically in PGCs at fate determination [26, 27]. Hypomethylation of promoters characterizes the expression of three key pluripotency genes such as Oct4, Nanog, and Sox2 in cell lines. Moreover, the suppression of Oct4 and Nanog expression associated to hypermethylation of these CpG islands has been reported in differentiating ES cells [39–41]. In the case of Oct4, de novo methylation of CpG islands mediated by DNMTs serves to stabilize the repression. In fact, during ES differentiation, the interaction of GCNF with the methyl-CpG-binding proteins MBD2 and MBD3 initiates Oct4 repression. The lysine-9 trimethylation of histone H3 mediated by the G9a histone methyltransferase also contributes to such early repression [42–44]. Due to the limited cell numbers, no such information are available for PGCs. However, a recent report has showed CpG hypomethylation of the flanking region of Oct4 during PGC-like cell induction from epiblast stem cells (EpiSCs) [45]. Similarly, the germ-like specific Stella and the pluripotency-related Rex1 and Fbx15 genes become hypomethylated in the CpGs of the flanking regions in PGC-like cells induced from EpiSCs in which hypermethylation of these CpG islands is associated to lack of expression of these genes [45].
Finally, demethylation events in female pregonadal PGCs might be involved in the initial reactivation of the inactive X. It is now known, that, contrary to the previous suggestions, reactivation of the inactive X already begins in nascent female PGCs and proceeds gradually [46]. The mechanism for X chromosome inactivation involves a nontranslated RNA transcript of the X-inactive specific transcript (Xist) gene located on the X chromosome inactivation centre (XIC) [47]. The control of Xist expression appears to be due to DNA methylation on the inactive X chromosome since the gene control region is unmethylated on the inactive X chromosome and methylated on the active X chromosome which does not express Xist [48]. X reactivation is accompanied by a progressive decrease of the Xist transcripts, reexpression of X-linked genes, and, quite surprisingly, by demethylation of the Xist promoter [49]. It seems, therefore, that reactivation of the inactive X chromosome is accompanied by epigenetic mechanisms other than Xist methylation.
5.2. DNA Demethylation in Gonadal PGCs
After 9.5 dpc, PGCs reenter the cell cycle and begin the colonization of the gonadal ridges. Around 13.5 dpc, in female, they enter meiosis as primary oocytes while in the male they undergo mitotic arrest in G2. During this period, PGCs undergo a further wave of DNA demethylation [24, 32]. The bulk of such demethylation takes place quite rapidly in about 24 h, mostly between 11.5 and 12.5 dpc, and despite the presence in the PGC nucleous of DNMT1. Thus suggesting that it mainly occurs by active demethylation. Such process preferentially affects single copy-imprinted, and nonimprinted genes, whereas demethylation of repetitive elements (especially of intracisternal A-particle (IAP) and long interspersed repeated (LINE-1) elements) is more protracted [49–51]. Genomic imprinting is a phenomenon by which certain genes are expressed in a parent-of-origin-specific manner. It is a typical epigenetic process that involves DNA methylation in order to achieve monoallelic gene expression (for a review, see [52, 53]). The erasure of imprinting in PGCs ensures the establishment of sex-specific new imprinting during the following stages of gametogenesis. On the other hand, the protracted demethylation of repetitive elements may be necessary to prevent dangerous transcriptional activation of the transposable elements, since this would increase the risk of germline mutations through deregulation of adjacent genes and through transposition.
When PGCs reach the gonadal ridges several, germline specific-genes are upregulated such as Mvh, Dazl, Gcna1, Mageb4, and Scp3. In Dmnt1-deficeint mice, however, premature expression of these genes occurs in pregonadal PGCs, indicating the possible importance of DNA demethylation for the expression of these genes [54].
Chromatin remodelling in the PGC nuclei seems another consequence of DNA demethylation. In fact, carefully observation by Hajkova et al. [55] showed that DNA demethylation precedes chromatin remodelling.
Recent studies have began to reveal the mechanisms possibly involved in the active demethylation in gonadal PGCs. Popp et al. [32], using unbiased sequencing of bisulphite-treated DNA by next generation sequencing (BS-Seq) method, investigated genome-wide DNA methylation in 13.5 dpc mouse PGCs. This analysis revealed that DNA from male and particularly female PGCs, was highly unmethylated (approximately 15% and 7% methylation of total DNA, resp.) compared to methylated DNA levels in sperm (about 85%), ES cells (about 80%), the foetus (about 75%), and placenta (about 45%). When this analysis was repeated with tissues obtained from mice depleted of the DNA deaminase (AID), it was found that while in most tissues examined, AID deficiency did not alter the level of DNA methylation; in PGCs a significant increase in methylation was observed (about 5% and 10%, resp.), thus, indicating a loss of DNA demethylation in these cells. In this model, deamination by AID converts C into T and gives rise to a glycosylase-mediated T-G mismatch followed by the insertion of unmethylated cytosine by BER. These results support the notion that in PGCs active AID-mediated demethylation occurs but also that other factors are involved. Another study has shown that at the time of DNA demethylation in 11.5 dpc PGCs, there is an upregulation of transcripts of genes involved in BER, including Parp1, Ape1, and Xrcc1. This increase was associated to the presence in the PGC nuclei of XRCC1, a primary component of BER, together with PARP1 and APE1 [33]. The authors favour the possibility that BER is activated by the conversion of 5-mC to 5-hmC, for which the enzymes of the ten-eleven translocation (TET) family are responsible, followed by the excision of this latter by a specific glycosylase; Tet1 expression was found indeed at high levels in 11.5 dpc PGCs [33].
The abundant presence of PARP1 and PAR polymers, a product of PARP1, in the PGC nuclei during DNA demethylation [33] (our unpublished observations) and the multifunctional actions of members of the PARP family on the genome (for a review, see [56]) suggest a role of this enzyme beyond than in BER. Surani and Hajkova [28] suggested that PARP1 might be responsible for the higher-order chromatin changes and the loss of chromocenters in PGCs. In addition, a direct involvement of PARP1 in DNA methylation is possible (for a review, see [57]). In this regard, high levels of PAR might inhibit DNMT1 during the about-five cell cycles occurring in PGCs between 10.5 and 13.5 dpc [58], the period of the imprinting erasure.
5.3. Histone Modifications in Pregonadal PGCs
Using immunohistochemistry with antibody specific for methylated or acetylated histones, it has been shown that the staining patterns of the PGC precursors and PGCs around 7.5 dpc for the modifications H3K4me2, H3K4me3, H3K9Ac, H3K9me1, H3K9me2, H3K9me3, H3K27me2, and H3K27me3 were indistinguishable from those of their somatic neighbours. It is of note that the lineage-restricted PGC precursors in female embryos showed prominent accumulation of H3K27me3 in a single spot, most likely the inactive X chromosome [59, 60]. This suggests that the initiation of the X-inactivation process in the germline is similar to that in the somatic lineages. Chuva de Sousa Lopes et al. [61], using immunohistochemistry for H3K27me3 and an X-located green fluorescent protein (GFP)-carrying transgene, actually confirmed that BLIMP1-positive PGC precursors were subjected to X inactivation like their somatic neighbors.
Around 8.0 dpc, a number of global histone changes begin in PGCs. These include erasure of H3K9me2, a repressive mark with high stability, and upregulation H3K27me3, a repressive mark with apparent plasticity. The first modification occurs despite the presence of G9a (also known as EHMT2), a histone methyltransferase with a strong HMTase activity towards H3-K9. The absence of glucagon-like peptide-2 (GLP-2 or EHMT1) necessary to form the active G9a-GLP2 complex and/or competition with the activation mark H3K9ac probably prevent G9a action. On the other hand, the enhancement of H3K27me3 might be due to the action of the EZH2, a polycomb group enzyme [24, 25, 55]. Interestingly, Seki et al. [25] found that these histone changes occur progressively in migrating PGCs, most likely depending on their developmental maturation. Moreover, they observed that before or concomitant with the erasure of H3K9m2, PGCs enter G2 arrest, which continued until they acquire high levels of H3K27me3. Notably, although the overall levels of nuclear H3K27me3 were increasing, female PGCs gradually lose H3K27me3 mark on the silent X chromosome [61]. Following the onset of H3K9me2 erasure, PGCs show also repression of RNA polymerase II-dependent transcription that is gradually relieved after the release from the G2 arrest and the acquisition of high levels of H3K27me3. The precise significance of repression of RNAP II-dependent transcription and G2 arrest is currently unknown. Although both events seem to occur through mechanisms independent from the histone modification state of PGCs [25], they are likely to be necessary for their efficient epigenetic reprogramming.
Histone modifications in pregonadal PGCs include at least two other changes: the enrichment of H3K4me2 and H3K4me3, generally associated to actively transcribed euchromatin, and the symmetrical methylation of arginine 3 on histones H4 and H2A (H4/H2AR3me2s), a repressive mark conferred by a complex between the transcriptional repressor BlIMP1 and protein Arginine methyl transferase 5 (PRMT5). This latter mark is likely important for maintaining the PGC lineage during migration [62]. So far, only the DEAH (Asp-Glu-Ala-His) box polypeptide 38 (Dhx38) gene encoding the pre-mRNA-splicing factor ATP-dependent RNA helicase PRP16 has been identified as target of this repressive mark [62]. Finally, another repressive mark H3K9m3 that specifically marks centromeric heterochromatin as well transposon and repetitive elements, is maintained relatively constant during this period up to 11.5 dpc. In female PGCs, the accumulation of repressive H3K27me3 gradually diminishes specifically on inactivated X [25]. This latter is accompanied by displacement of PcG repressor proteins EED and SUZ12 from the X despite the presence of high levels of these proteins in the nuclei of PGCs between 9.5 and 11.5 dpc [63]. As far as I know, no other information about the presence and role of PcG proteins in mammals PGCs are available.
These observations demonstrate that within euchromatin regions PGCs erase a repressive mechanism operated by H3K9me2 and replace it with H3K27me3 and H4/H2AR3me2s. The first modification may allow greater genome plasticity, the latter is possibly necessary to prevent differentiation towards somatic cell lineages. Transcriptionally permissive H3K4 methylations and H3K9ac are progressively increased. These marks might cooperate in events, discussed above, regarding DNA methylation, as the upregulation of pluripotency and germ cell-specific genes and might reflect the reprogramming of the PGC genome eventually necessary for totipotency at the onset of embryogenesis. Such chromatin status partly resembles that of ES cells and probably favours the transformation of PGCs into the pluripotent EG and EC cells (see below).
5.4. Histone Modifications in Gonadal PGCs
The first sign of histone modification in gonadal 11.5 dpc PGCs is a rapid loss of linker histone H1 accompanied by “loosening” of the chromatin and loss of chromocenters [55]. A concomitant downregulation of H3K9me3, H3K27me3, and H4/H2AR3me2s, and the disappearance or redistribution of factors are associated with facultative or constitutive heterochromatin, such as heterochromatin protein 1α (HP1α), HP1β, and HP1γ, the homologue of α-thalassaemia/mental retardation syndrome X-linked (ATRX) protein, and the polycomb-like protein M33 (also known as CBX2). Transcriptionally permissive H3K4 methylations and H3K9ac are also lost [24, 55]. Thus, it seems that the genome during PGC reprogramming undergos a phase of removing most epigenetic marks. It seems that this is achieved, at least in part, by histone replacement, potentially involving the histone chaperone and nucleosome assembly protein 1 (NAP1) [55].
In Figure 1, the main epigenetic changes accompanying mouse PGC development are schematically represented.
Figure 1.
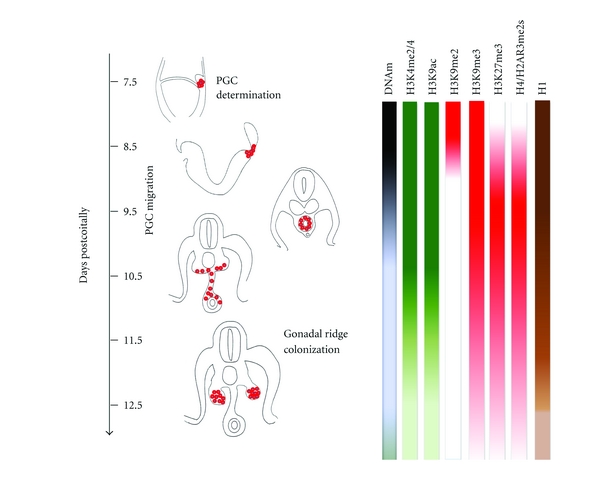
PGC development and associated epigenetic events in the mouse embryo. Changes in DNA methylation (DNAm) and histone modifications are represented by different shading intensity.
6. Comparing PGC and ES Cell Epigenetics
As reported above, some of the principles of nuclear reprogramming in pregonadal PGCs are common with those governing the formation and the maintenance of the undifferentiated state of ES cells, in particular the generalized inhibition state of somatic cell lineage pathways associated with the expression of key pluripotency genes. At the same time, however, PGCs show limited or absent self-renewal capability, and are committed to the germline lineage in which a unique DNA methylation resetting centred on the erasing of imprinting occurs. By considering the status of DNA methylation and the histone code in ES cells and how they are involved in the maintenance of pluripotency and self-renewal as well in the differentiation pathways of these cells, some clues about the contribution of these epigenetic changes and the underlying controlling mechanisms of the nuclear reprogramming in PGCs can be achieved.
Like somatic cells, ES cells show high global levels of DNA methylation, with about 60–80% of all CpG dinucleotides being methylated [64]. Although the global mCpG content is similar, the distribution of the mark is quite different from that of any other somatic cell type. In ES cells, promoters containing high level of CpG have low DNA methylation levels, whereas low CpG promoters are relatively high methylated [64–66]. CpG-rich promoters are almost invariably associated with the active H3K4me3 mark. Some of these promoters control constitutively expressed housekeeping genes, but others corresponding to developmental regulators also contain the repressive H3K27me3 mark. Methylated low CpG promoters are marked neither by H3K4me3 nor by H3K27me3 and are mostly repressed in ES cells [64–66]. Interestingly, the presence of two active XX chromosomes makes DNA globally hypomethylated both at repetitive and unique sequences [67].
From the information reported above on DNA methylation in PGCs, two main differences with ES cells are apparent. First, a progressive general demethylation was associated to PGC development in comparison to a more stable global methylation status of ES cells. Second, a DNA demethylation activity of PGCs was coupled to the erasure of the genome imprinting absent in ES cells. More detailed analyses of similarities and differences in DNA methylation have been performed only between ES cells and PGC-derived EG cell lines. Similarly to PGCs, EG cells derived from 11.5–12.5 dpc PGCs were shown to be grossly hypomethylated (mainly in EG cells with both active XX, [68]) and possess strong demethylation activity [69]. As for methylation of imprinted genes, EG cells derived soon after PGC specification (early EG cells) showed heterogeneous DNA methylation patterns in comparison with EG cells derived after PGC colonisation of the gonadal ridges (late EG cells) which were uniformly hypomethylated at these sites with the expected exception of H19 locus [68]. Direct comparison of the global methylation of ES and EG cells performed with different methods and on different lines revealed distinct DNA methylation patterns at various CpG islands [70] while substantial similarity in the methylation patterns of promoters with intermediate level of CpG content (between 2% and 9%) in which most dynamics changes in methylation occur [71].
Taken together, these results indicate that cell pluripotency, except for hypomethylation of the promoter of key pluripotency genes, cannot be associated with a particular status of global DNA methylation. Methylation of different CpG sites may vary in pluripotent cells probably depending on the different characteristics of their pluripotency. In addition, in the germ cells, the dynamics of DNA methylation is unique because functional to other process discussed above such as the removal of epimutations and erasing of the parental imprinting.
All four DNMTs are highly expressed in ES cells. While DNMTs are essential for embryonic development [72], loss of DNA methylation by ablation of single or various DNMTs does not influence the ES cell viability and self-renewal but impairs their differentiation properties (for a review, see [73]). Demethylating agent, 5-azacytidine (5-AZA), was reported to reverse the differentiation status of ES cells forming embryoid bodies (EBs) back to undifferentiated ES cells [74]. Both Oct4 and Nanog are devoid of CpG islands in promoter regions [75], whereas the promoter region of Sox2 is CpG rich. The promoter regions of Oct4 and Nanog are hypomethylated in ES cells but acquire significant methylation during cell differentiation process. However, the promoter region of Sox2 gene is almost completely unmethylated in both ES cells and differentiated cells.
All together, these observations suggest that DNA methylation in ES cells is passively and actively maintained at high levels by DNMT1, but loci of key pluripotent genes are maintained unmethylated. DNA methylation is not sufficient to repress differentiation nor is crucial for self-renewal and pluripotency. Differentiation towards certain cell lineages requires that a portion of DNA methylation is maintained. None of these suggestions is incompatible with the wide-genome demethylation accompanying PGC development and in contrast with the principles of their nuclear reprogramming. Despite high DNA methylation level, ES cells show that an open chromatin structure and active chromatin domains are widespread [76].
The histone code is probably the main responsible for the unique genome state of ES cells. Analyses of genome-wide chromatin-state maps have recently shown that many genes in mouse ES cells, including several that are involved in differentiation, are characterized by an unusual combination of histone marks, termed the “bivalent domains.” This includes H3K9ac and methylated H3K4, which are marks of active chromatin, and H3K27me3, which is typical of silent chromatin [77]. In most non stem cells, genes have either active or repressive marks, but not both. A similar epigenetic profile was also identified by others at genes encoding developmentally important transcription factors [78, 79]. These authors showed that loss of these epigenetic marks was correlated with differentiation and proposed that the presence of both active and repressive marks allowed differentiation-specific genes in ES cells to be repressed but also to be primed for activation when the right signals are received. Interestingly, about half of the identified bivalent domains in ES cells have binding sites for at least one of the three key pluripotency-associated transcription factors OCT4, NANOG, and SOX2.
As reported above, histone acetylation depends on the activities of several HATs or HDACs while histone methylation/demethylation is catalyzed by families of HMTs and HDMases, respectively.
In general, it appears that ES cell differentiation is accompanied by a global increase of histone acetylation except in specific loci of key pluripotent genes [80], followed by differentiation-specific deacetylation [81]. In line with this notion, increased level of acetylation, induced by the inhibition of HDACs by trichostatin (TSA), results in a rapid repression of pluripotent genes and the induction of differentiation-associated genes [82, 83]. A sustained inhibition of HDACs favours differentiation towards certain cell lineages such a cardiomyocytes and neurons [83].
Recently, a number of general global-wide analyses have identified large numbers of binding sites for the HMTs of the PcG family across the ES cell genome [84, 85]. The PcG proteins are essential for early mammalian embryo development [86–89], but not for maintaining ES cell pluripotency. PcG mutant ES cells can still self-renewal, maintain normal morphology, and express pluripotent genes [90–93]. In addition, although the PcG knockout ES cells do not differentiate efficiently into the three germ layers, they can still contribute to their formation [90, 92, 93]. However, loss of individual PRC proteins in ES cells results in increased expression of diverse lineage-associated genes and spontaneous differentiation [90, 92, 93], an effect that is more pronounced in ES cells carrying targeted deletions of both Prc1 and Prc2 genes [93]. Notably, the promoter regions that are occupied by PcG proteins in ES cells contain the “bivalent” H3K4me3 and H3K27me3 marks [76, 78]. These genes were also found to be generally transcriptionally silent. This suggests that PcG proteins help to maintain the silencing of these genes in undifferentiated ES cells. Because some results suggest that the repressive H3K27me3 mark can be heritably transmitted to daughter cells to maintain specific gene expression programs [94–96], the expression of developmentally regulated genes would necessitate the removal of the H3K27me3 mark. Demethylation of H3K27me3/me2 by the UTX and JMJD3 might be the mechanism by which PcG-repressed promoters are activated [96–98].
Alltogether, these results suggest that epigenetic regulation may be dispensable for maintaining ES cell identity. It appears that epigenetic mechanisms of gene silencing contribute to the overall stability of pluripotency but are downstream in this setting. ES cell characteristics might principally be regulated by transcription factors and intrinsic molecular pathways activated by exogenous signals. Epigenetic chromatin-based repressive and activating modifiers may serve transcriptional corepressor and coactivating functions in this process. On the other hand, the stability of a given cell state relies on the silencing of genes encoding players of other cell states. In this context, the major ES cell transcription factors appear to activate both programs of self-renewal and pluripotency through an autoregulatory circuit. In some cases, they could possess also the ability to alter chromatin structure [99–102].
As in ES cells, the activity of key pluripotency transcription factors such as OCT4, NANOG, and SOX2 is likely crucial for nuclear reprogramming in the emerging PGCs. Before this, however, the first process governing nuclear reprogramming in PGCs seems exerted by the repressive action of the transcription factor BLIMP1/PRDM1 on somatic Hox genes [103]. This is closely followed by the expression of PRDM14 that together with BLIMP1 is critical for the activation of pluripotency genes [104]. In PGCs, these events, however, do not activate a long-lasting self-renewal capability. This likely requires the contemporary expression of other transcription factors (i.e., c-MYC, KLF4) and the activity of intracellular signalling pathways (i.e., STAT3) necessary for self-renewal. At the same time, in PGCs, the expression of genes and activities of molecular pathways specific of the germline could restrain the self-renewal circuit. BMPs, namely, BMP-4, BMP-2, and BMP-8b and probably adhesion molecules such as E-Cadherin and Fragilis are exogenous signals regulating such initial reprogramming (for review, see [8–12]). In this contest, as discussed above, epigenetic changes might favour and stabilize the germ cell differentiation programme. Moreover, in PGCs, they serve to begin the genome resetting for the sex- specific gamete imprinting and towards totipotency.
The first detectable epigenetic modification (the beginning of genome-wide DNA demethylation, erasure of H3K9me2, upregulation H3K27me3, H3K9ac, H3K4me2, and H3K4me3 and H4/H2AR3me2s), are seen around the period when PGCs are determined 7-8 dpc, suggesting that they exert as in ES cells, transcriptional corepressor, and coactivating functions. At least in part, these modifications appear to be directly or indirectly activated by the BLIMP1/PRMT5 complex and PRDM14 [27, 103, 104]. Interestingly, the pattern of histone marks established in pregonadal PGCs with the contemporary presence of H3K27me3 and H3K4 methylations and H3K9ac partly resembles the “bivalent domain” code of ES cells, thus, suggesting that this chromatin structure might represent the base for the maintenance of latent PGC pluripotency at this stage. How HATs or HDACs and HMTs and HDMases participate to the histone modifications in PGCs is not known. In fact, apart from the observations reported above that the progressive X reactivation in female PGCs is accompanied by displacement of PcG repressor proteins EED and SUZ12 and that high levels of these proteins are present in the nuclei of PGCs between 9.5 to 11.5 dpc [62], no other information about the presence and role of PcG proteins in mammals PGCs are yet available.
7. Reprogramming on Reprogramming: EG Cell Formation
During migration and for some time after the arrival into the gonadal ridges, PGCs from several mammalian species, including humans, can be induced in vitro to deviate from their normal differentiation pattern and transform or transdifferentiate into EG cells, cell lines showing self-renewal, and pluripotency characteristics very similar to ES cells (for a review, see [17]). After PGCs begin to differentiate into oocyte and prospermatogonia, around 12.5 dpc in the mouse, they lose this capability. As reported above, however, descendents of PGCs, the SSCs in male and the mature oocytes in female, show again the capability to give rise multipotent stem cells in culture and teratomas in vivo, respectively, indicating the presence of latent pluripotency like PGCs.
The formation of EG cells from PGCs means that a new nuclear reprogramming can be superimposed on the ongoing nuclear reprogramming until a certain developmental stages. This basically renders PGCs able to manifest their latent pluripotency and acquire self-renewal.
Early studies showed that a fraction of pregonadal mouse PGCs (around 2–20% of 8.5 dpc PGCs) transformed into EG cells when they were cultured in vitro onto cell monolayers in serum-supplemented medium and in continuous presence for 7–10 days of three exogenous growth factors, namely, the kit ligand (KL), leukemia inhibitory factors (LIFs), and basic fibroblast growth factors (bFGFs) [105, 106]. Each of the growth factors required for EG cell derivation activates unique signal transduction pathways partly overlapping in the downstream effectors. Several studies by us and others have established that KL and LIF act as antiapoptotic factors and comitogens to control PGC survival and proliferation whereas bFGF appeared mostly mitogenic for PGCs (for a review, see [107]). This latter notion was also supported by the observation that in the growth factors cocktail, bFGF can be replaced with potent PGC mitogens such as retinoic acid (RA) or agents that activate cAMP such as forskolin (FRSK) [108, 109]. More recently, it has been shown that bFGF and partly KL can be also substituted by hyperactivation of AKT [110], a potent survival and proliferative stimulus downstream phosphoinositide 3-kinase (PI3K). This can be activated by a number of growth factors including bFGF and KL. In line with this, EG cell colony formation was enhanced in PGCs depleted of phosphatase and tensin homolog (PTEN) which antagonizes the actions of PI3K [109, 111]. Thus, sustained PGC survival and proliferation in culture appear a prerequisite for EG cell formation.
At the same time, genetic and epigenetic events of nuclear reprogramming must take place. It has been proposed that following bFGF-binding upregulation of the FGFR-3 receptor and its translocation into the nucleus is a crucial event for inhibiting PGC differentiation PGCs and beginning their reprogramming in culture as EG cells [16]. Exposure of PGCs to bFGF for just the first 24 hours of culture is necessary and sufficient for EG cell formation providing that LIF is present in the medium after 1 day of culture [16, 31]. Since as reported above, bFGF can be substituted by RA, FRSK, or hyperactivated AKT, several signalling pathways in collaboration with LIF can induce the reprogramming of cultured PGCs.
A number of genetic events involved in such process have been identified. A key early event during the culture of PGCs in the presence of bFGF is the downregulation of Blimp1 and the consequent upregulation of its target genes Dhx38, c-Myc, and Klf4 [31]. While Dhx38 repression has been proposed to be necessary for maintaining early germ cells [62], the latter represents two of key transcription factors known to promote reprogramming of somatic cells to the pluripotent state of the iPS cells [112–115].
It has been proposed that the AKT promoting action on EG cell derivation is exerted through two targets: direct phosphorylation and inhibition of GSK-3 and inhibition of p53 activity mediated by GSK-3-dependent phosphorylation of MDM2, an E3 ubiquitin ligase for p53. In line with these, both inhibition of GSK-3 and absence of p53 favour the EG cell derivation [110, 116]. GSK-3 participates in various signaling pathways, such as WNT/β-catenin, Hedgeog proteins, Notch, and protein kinase A (PKA) signals. However, it seems unlikely that the activation of the WNT/β-catenin, usually a consequence of the GSK-3 inhibition, plays a role in nuclear reprogramming of PGCs in culture. In fact, the suppression of WNT/β-catenin signalling by GSK-3 appears to be necessary for normal PGC proliferation [117]. The p53 deficiency in the cultured PGCs, that only partly, however, substitute bFGF, might favour the acquisition of dedifferentiate state. Recent studies have identified the roles of p53 in suppressing pluripotency and cellular dedifferentiation. In this context, p53 suppresses the self-renewal of embryonic stem cells and blocks the reprogramming of somatic cells into iPSCs by inducing the expression of hundreds of genes, leading to cell cycle arrest, apoptosis, and senescence (for a review, see [118]). These new studies support the idea that molecules critically involved in genome stability function as not only guardians of the genome, but also barriers to pluripotency. It is interesting to note that, as reported above, in addition to p53 deficiency also that of PTEN, another important tumour suppressor, greatly facilitates EG cell derivation.
Whatever are the GSK-3 targets, its inhibition is alone not sufficient for PGC conversion. In fact, it requires the contemporary inhibition of the extracellular signal-regulated kinases (ERKs). Notably, EG can be obtained from 8.5 dpc PGCs in serum-free culture in 2i-LIF medium supplemented only with LIF and containing an inhibitor of GSK-3 and of the upstream kinase MAP ERK kinases (MEK1/2) [119]. Recently, it has been shown that the mouse ES cells can be self-renewal in basal medium if autocrine-activated ERK signalling is eliminated and GSK-3 activity reduced. Moreover, the 2i-LIF medium allowed efficient derivation and expansion of ES cells [119].
Finally, trichostatin (TSA), an inhibitor of HDACs not only replaced bFGF but it also accelerated and increased the efficiency of PGC reprogramming into EG cells [31]. This indicates that epigenetic modification is crucial for reprogramming PGCs in culture and suggests that it is the chromatin organization that prevents establishing the OCT4, NANOG, and SOX2 circuit of active pluripotency and prolonged self-renewal in such cells. These results are difficult to reconcile with a previous study by Maatouk and Resnick [120] in which the authors reported that under similar culture conditions TSA and 5-AZA as well accelerated the differentiation of 8.5 dpc PGCs as estimated by a more rapid downregulation of alkaline phosphatase and upregulation of GCNA1 after three days of culture. A possible explanation might be found in the fact that nuclear reprogramming leading to dedifferentiation actually occurs only in a subpopulation of PGCs, as reported above around 2–20% of 8.5 dpc PGCs, and/or that in the short period of culture analysed by Maatouk and Resnick [120], the drugs might induce temporary changes in the expression of some genes irrelevant for nuclear reprogramming.
In this context, it is likely that LIF action is necessary in the longer time for stabilizing and maintaining the full reprogramming process after the main reprogramming events are occurred. In fact, LIF is not required during day one of culture when bFGF or hyperactivated AKT exerted their action [16, 31, 108], but its presence is obligatory after this time in any EG cell derivation conditions. Notably, pregonadal PGCs do not express STAT3 ([121], our unpublished observation), the transcriptional activator that mediates the LIF-dependent self-renewal and maintenance of pluripotency in ES cells, but it appears after 4 days of culture [31], likely as a consequence of the previous nuclear reprogramming.
In Figure 2, the hypothetical multistep process of the nuclear reprogramming of PGCs in culture is shown.
Figure 2.
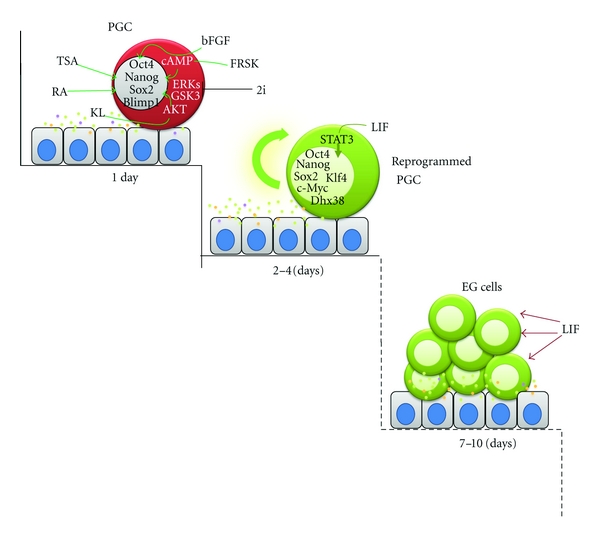
PGC transformation in EG cells. The process is schematically subdivided in to three main steps: rapid 1 day reprogramming processes involving TSA-induced epigenetic modifications and resetting of various gene and intracellular signalling pathways, a mid-time (2–4 days) establishment of a LIF/STAT3-dependent self-renewal circuit, and the formation of EG cell colonies requiring 7–10 days of culture. Various growth factors and compounds controlling each steps are reported; a cell monolayer producing KL and other not identified compounds is also represented.
8. Concluding Remarks
Nuclear reprogramming occurs progressively in PGCs during migration and gonadal ridge colonization. Epigenetic mechanisms of gene silencing and activation contribute to such nuclear reprogramming, but they are likely downstream genetic players. In this regard, PGC characteristics might principally be regulated by transcription factors whereas the most part of the epigenetic modifications are mainly associated with resetting the genome for subsequent parental imprinting on the gametes and regaining zygote/blastomeres totipotency. Nuclear reprogramming in nascent PGCs centred on BLIMP1/PRDM1- and PRMD14-dependent inhibition of somatic cell lineages and activation of core transcriptional circuits of pluripotency is activated by growth factors of the BMP family. Future studies will clarify whether epigenetic changes are also controlled by transcription factors and/or exogenous signals or occur by intrinsic regulators. Improvements of epigenomic analysis technologies are needed to identify the whole normal epigenomic setting of PGCs and epigenomic changes occurring in mutants and disease conditions. The possibility to apply single-cell gene expression analysis to PGCs (see, [103]) will probably facilitate such studies.
A further nuclear reprogramming can be superimposed on PGCs in vivo and in vitro leading to their transformation into pluripotent stem cells. Reprogramming of PGCs in vitro only requires the addition of exogenous growth factors and provides important insights into the molecular mechanisms regulating the stemness characteristics. The expression in PGCs of key pluripotency transcription factors such as OCT4, NANOG, and SOX2 is not sufficient to confer them a long lasting self-renewal capability and a manifest pluripotency. This might be a consequence of the lack of expression of other transcription factors (i.e., c-MYC, KLF4) and intracellular signalling pathways (i.e., STAT3) necessary for self-renewal. The derivation of EG cells in vitro from PGCs is actually triggered by downregulation of BLIMP1/PRDM1 and activation of c-Myc and Klf4 as well the establishment of a LIF/STAT3 signalling. Interestingly, epigenetic modifications induced by a TSA-dependent DNA iper-acetylation or inhibition of ERK and GSK-3 pathways are able to replace the reprogramming action of growth factors on PGCs. Besides highlighting the crucial relevance of such processes for nuclear reprogramming, these results suggest that the development of the germline relies on the maintenance of a definite epigenetic state and the activity of certain intracellular pathways.
Acknowledgment
The author's research is supported by the Italian Ministry of the University and Research, Grant COFIN PRIN 2008.
References
- 1.Robinson R. siRNA and DNA methylation do a two-step to silence tandem sequences. PLos Biology. 2006;4(11, article e407) doi: 10.1371/journal.pbio.0040407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawasaki H, Taira K. Transcriptional gene silencing by short interfering RNAs. Current Opinion in Molecular Therapeutics. 2005;7(2):125–131. [PubMed] [Google Scholar]
- 3.Prokhortchouk E, Defossez PA. The cell biology of DNA methylation in mammals. Biochimica et Biophysica Acta. 2008;1783(11):2167–2173. doi: 10.1016/j.bbamcr.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 4.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics. 2010;11(3):204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nature Reviews Molecular Cell Biology. 2010;11(9):607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nature Reviews Molecular Cell Biology. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 7.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128(4):735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 8.McLaren A, Lawson KA. How is the mouse germ-cell lineage established? Differentiation. 2005;73(9-10):435–437. doi: 10.1111/j.1432-0436.2005.00049.x. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi K, Lopes SMCDS, Surani MA. Germ cell specification in mice. Science. 2007;316(5823):394–396. doi: 10.1126/science.1137545. [DOI] [PubMed] [Google Scholar]
- 10.Saga Y. Mouse germ cell development during embryogenesis. Current Opinion in Genetics & Development. 2008;18(4):337–341. doi: 10.1016/j.gde.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Western P. Foetal germ cells: striking the balance between pluripotency and differentiation. International Journal of Developmental Biology. 2009;53(2-3):393–409. doi: 10.1387/ijdb.082671pw. [DOI] [PubMed] [Google Scholar]
- 12.De Felici M. Primordial germ cell biology at the beginning of the XXI Century. International Journal of Developmental Biology. 2009;53(7):891–894. doi: 10.1387/ijdb.082815mf. [DOI] [PubMed] [Google Scholar]
- 13.Ohinata Y, Payer B, O’Carroll D, et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature. 2005;436(7048):207–213. doi: 10.1038/nature03813. [DOI] [PubMed] [Google Scholar]
- 14.Ginsburg M, Snow MHL, McLaren A. Primordial germ cells in the mouse embryo during gastrulation. Development. 1990;110(2):521–528. doi: 10.1242/dev.110.2.521. [DOI] [PubMed] [Google Scholar]
- 15.McLaren A, Durcova-Hills G. Germ cells and pluripotent stem cells in the mouse. Reproduction, Fertility and Development. 2001;13(7-8):661–664. doi: 10.1071/rd01080. [DOI] [PubMed] [Google Scholar]
- 16.Durcova-Hills G, Adams IR, Barton SC, Surani MA, McLaren A. The role of exogenous fibroblast growth factor-2 on the reprogramming of primordial germ cells into pluripotent stem cells. Stem Cells. 2006;24(6):1441–1449. doi: 10.1634/stemcells.2005-0424. [DOI] [PubMed] [Google Scholar]
- 17.Donovan PJ, De Miguel MP. Turning germ cells into stem cells. Current Opinion in Genetics & Development. 2003;13(5):463–471. doi: 10.1016/j.gde.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Stevens LC, Varnum DS. The development of teratomas from parthenogenetically activated ovarian mouse eggs. Developmental Biology. 1974;37(2):369–380. doi: 10.1016/0012-1606(74)90155-9. [DOI] [PubMed] [Google Scholar]
- 19.Kanatsu-Shinohara M, Inoue K, Lee J, et al. Generation of pluripotent stem cells from neonatal mouse testis. Cell. 2004;119(7):1001–1012. doi: 10.1016/j.cell.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Guan K, Nayernia K, Maier LS, et al. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature. 2006;440(7088):1199–1203. doi: 10.1038/nature04697. [DOI] [PubMed] [Google Scholar]
- 21.Seandel M, James D, Shmelkov SV, et al. Generation of functional multipotent adult stem cells from GPR125+ germline progenitors. Nature. 2007;449(7160):346–350. doi: 10.1038/nature06129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development. 1987;99(3):371–382. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 23.Kafri T, Ariel M, Brandeis M, et al. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes and Development. 1992;6(5):705–714. doi: 10.1101/gad.6.5.705. [DOI] [PubMed] [Google Scholar]
- 24.Seki Y, Hayashi K, Itoh K, Mizugaki M, Saitou M, Matsui Y. Extensive and orderly reprogramming of genome-wide chromatin modifications associated with specification and early development of germ cells in mice. Developmental Biology. 2005;278(2):440–458. doi: 10.1016/j.ydbio.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 25.Seki Y, Yamaji M, Yabuta Y, et al. Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development. 2007;134(14):2627–2638. doi: 10.1242/dev.005611. [DOI] [PubMed] [Google Scholar]
- 26.Yabuta Y, Kurimoto K, Ohinata Y, Seki Y, Saitou M. Gene expression dynamics during germline specification in mice identified by quantitative single-cell gene expression profiling. Biology of Reproduction. 2006;75(5):705–716. doi: 10.1095/biolreprod.106.053686. [DOI] [PubMed] [Google Scholar]
- 27.Kurimoto K, Yabuta Y, Ohinata Y, Shigeta M, Yamanaka K, Saitou M. Complex genome-wide transcription dynamics orchestrated by Blimp1 for the specification of the germ cell lineage in mice. Genes and Development. 2008;22(12):1617–1635. doi: 10.1101/gad.1649908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Surani MA, Hajkova P. Epigenetic rprogramming of mouse germ cells toward totipotency. Cold Spring Harbor Symposia on Quantitative Biology. 2010;75:211–218. doi: 10.1101/sqb.2010.75.010. [DOI] [PubMed] [Google Scholar]
- 29.Surani MA, Hayashi K, Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128(4):747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 30.Kato Y, Kaneda M, Hata K, et al. Role of the Dnmt3 family in de novo methylation of imprinted and repetitive sequences during male germ cell development in the mouse. Human Molecular Genetics. 2007;16(19):2272–2280. doi: 10.1093/hmg/ddm179. [DOI] [PubMed] [Google Scholar]
- 31.Durcova-Hills G, Tang F, Doody G, Tooze R, Surani MA. Reprogramming primordial germ cells into pluripotent stem cells. PLoS One. 2008;3(10) doi: 10.1371/journal.pone.0003531. Article ID e3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popp C, Dean W, Feng S, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463(7284):1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329(5987):78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mochizuki K, Matsui Y. Epigenetic profiles in primordial germ cells: global modulation and fine tuning of the epigenome for acquisition of totipotency. Development Growth and Differentiation. 2010;52(6):517–525. doi: 10.1111/j.1440-169X.2010.01190.x. [DOI] [PubMed] [Google Scholar]
- 35.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. Journal of Biological Chemistry. 2004;279(50):52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 36.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135(7):1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Branco MR, Oda M, Reik W. Safeguarding parental identity: Dnmt1 maintains imprints during epigenetic reprogramming in early embryogenesis. Genes and Development. 2008;22(12):1567–1571. doi: 10.1101/gad.1690508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura T, Arai Y, Umehara H, et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nature Cell Biology. 2007;9(1):64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- 39.Hattori N, Nishino K, Ko YG, et al. Epigenetic control of mouse Oct-4 gene expression in embryonic stem cells and trophoblast stem cells. Journal of Biological Chemistry. 2004;279(17):17063–17069. doi: 10.1074/jbc.M309002200. [DOI] [PubMed] [Google Scholar]
- 40.Hattori N, Imao Y, Nishino K, et al. Epigenetic regulation of Nanog gene in embryonic stem and trophoblast stem cells. Genes to Cells. 2007;12(3):387–396. doi: 10.1111/j.1365-2443.2007.01058.x. [DOI] [PubMed] [Google Scholar]
- 41.Farthing CR, Ficz G, Ng RK, et al. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genetics. 2008;4(6) doi: 10.1371/journal.pgen.1000116. Article ID e1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu P, Le Menuet D, Chung ACK, Cooney AJ. Differential recruitment of methylated CpG binding domains by the orphan receptor GCNF initiates the repression and silencing of Oct4 expression. Molecular and Cellular Biology. 2006;26(24):9471–9483. doi: 10.1128/MCB.00898-06. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Sato N, Kondo M, Arai K-I. The orphan nuclear receptor GCNF recruits DNA methyltransferase for Oct-3/4 silencing. Biochemical and Biophysical Research Communications. 2006;344(3):845–851. doi: 10.1016/j.bbrc.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 44.Feldman N, Gerson A, Fang J, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nature Cell Biology. 2006;8(2):188–194. doi: 10.1038/ncb1353. [DOI] [PubMed] [Google Scholar]
- 45.Hayashi K, Lopes SM, Tang F, Surani MA. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell. 2008;3(4):391–401. doi: 10.1016/j.stem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugimoto M, Abe K. X chromosome reactivation initiates in nascent primordial germ cells in mice. PLoS Genetics. 2007;3(7, article e116) doi: 10.1371/journal.pgen.0030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herzing LBK, Romer JT, Horn JM, Ashworth A. Xist has properties of the X-chromosome inactivation centre. Nature. 1997;386(6622):272–275. doi: 10.1038/386272a0. [DOI] [PubMed] [Google Scholar]
- 48.Sado T, Hoki Y, Sasaki H. Tsix silences Xist through modification of chromatin structure. Developmental Cell. 2005;9(1):159–165. doi: 10.1016/j.devcel.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 49.Hajkova P, Erhardt S, Lane N, et al. Epigenetic reprogramming in mouse primordial germ cells. Mechanisms of Development. 2002;117(1-2):15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 50.Lane N, Dean W, Erhardt S, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis. 2003;35(2):88–93. doi: 10.1002/gene.10168. [DOI] [PubMed] [Google Scholar]
- 51.Lees-Murdock DJ, De Felici M, Walsh CP. Methylation dynamics of repetitive DNA elements in the mouse germ cell lineage. Genomics. 2003;82(2):230–237. doi: 10.1016/s0888-7543(03)00105-8. [DOI] [PubMed] [Google Scholar]
- 52.Bartolomei MS. Genomic imprinting: employing and avoiding epigenetic processes. Genes and Development. 2009;23(18):2124–2133. doi: 10.1101/gad.1841409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hudson QJ, Kulinski TM, Huetter SP, Barlow DP. Genomic imprinting mechanisms in embryonic and extraembryonic mouse tissues. Heredity. 2010;105(1):45–56. doi: 10.1038/hdy.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maatouk DM, Kellam LD, Mann MRW, et al. DNA methylation is a primary mechanism for silencing postmigratory primordial germ cell genes in both germ cell and somatic cell lineages. Development. 2006;133(17):3411–3418. doi: 10.1242/dev.02500. [DOI] [PubMed] [Google Scholar]
- 55.Hajkova P, Ancelin K, Waldmann T, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452(7189):877–881. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Molecular Cell. 2010;39(1):8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caiafa P, Guastafierro T, Zampieri M. Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. The FASEB Journal. 2009;23(3):672–678. doi: 10.1096/fj.08-123265. [DOI] [PubMed] [Google Scholar]
- 58.Tam PPL, Snow MHL. Proliferation and migration of primordial germ cells during compensatory growth in mouse embryos. Journal of Embryology and Experimental Morphology. 1981;64:133–147. [PubMed] [Google Scholar]
- 59.Plath K, Fang J, Mlynarczyk-Evans SK, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300(5616):131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 60.Silva J, Mak W, Zvetkova I, et al. Establishment of histone H3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Developmental Cell. 2003;4(4):481–495. doi: 10.1016/s1534-5807(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 61.Chuva de Sousa Lopes SM, Hayashi K, Shovlin TC, Mifsud W, Surani MA, McLaren A. X chromosome activity in mouse XX primordial germ cells. PLoS Genetics. 2008;4(2, article e30) doi: 10.1371/journal.pgen.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ancelin K, Lange UC, Hajkova P, et al. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nature Cell Biology. 2006;8(6):623–630. doi: 10.1038/ncb1413. [DOI] [PubMed] [Google Scholar]
- 63.de Napoles M, Nesterova T, Brockdorff N. Early loss of Xist RNA expression and inactive X chromosome associated chromatin modification in developing primordial germ cells. PLoS One. 2007;2(9, article e860) doi: 10.1371/journal.pone.0000860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fouse SD, Shen Y, Pellegrini M, et al. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2(2):160–169. doi: 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohn F, Weber M, Rebhan M, et al. Lineage-specific Polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Molecular Cell. 2008;30(6):755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 67.Zvetkova I, Apedaile A, Ramsahoye B, et al. Global hypomethylation of the genome in XX embryonic stem cells. Nature Genetics. 2005;37(11):1274–1279. doi: 10.1038/ng1663. [DOI] [PubMed] [Google Scholar]
- 68.Shovlin TC, Durcova-Hills G, Surani A, McLaren A. Heterogeneity in imprinted methylation patterns of pluripotent embryonic germ cells derived from pre-migratory mouse germ cells. Developmental Biology. 2008;313(2):674–681. doi: 10.1016/j.ydbio.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 69.Tada T, Tada M, Hilton K, et al. Epigenotype switching of imprintable loci in embryonic germ cells. Development Genes and Evolution. 1998;207(8):551–561. doi: 10.1007/s004270050146. [DOI] [PubMed] [Google Scholar]
- 70.Shiota K, Kogo Y, Ohgane J, et al. Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes to Cells. 2002;7(9):961–969. doi: 10.1046/j.1365-2443.2002.00574.x. [DOI] [PubMed] [Google Scholar]
- 71.Farthing CR, Ficz G, Ng RK, et al. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genetics. 2008;4(6) doi: 10.1371/journal.pgen.1000116. Article ID e1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 73.Latham T, Gilbert N, Ramsahoye B. DNA methylation in mouse embryonic stem cells and development. Cell and Tissue Research. 2008;331(1):31–55. doi: 10.1007/s00441-007-0537-9. [DOI] [PubMed] [Google Scholar]
- 74.Tsuji-Takayama K, Inoue T, Ijiri Y, et al. Demethylating agent, 5-azacytidine, reverses differentiation of embryonic stem cells. Biochemical and Biophysical Research Communications. 2004;323(1):86–90. doi: 10.1016/j.bbrc.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 75.Yeo S, Jeong S, Kim J, Han JS, Han YM, Kang YK. Characterization of DNA methylation change in stem cell marker genes during differentiation of human embryonic stem cells. Biochemical and Biophysical Research Communications. 2007;359(3):536–542. doi: 10.1016/j.bbrc.2007.05.120. [DOI] [PubMed] [Google Scholar]
- 76.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130(1):77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Azuara V, Perry P, Sauer S, et al. Chromatin signatures of pluripotent cell lines. Nature Cell Biology. 2006;8(5):532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 78.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 79.Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karantzali E, Schulz H, Hummel O, Hubner N, Hatzopoulos AK, Kretsovali A. Histone deacetylase inhibition accelerates the early events of stem cell differentiation: transcriptomic and epigenetic analysis. Genome Biology. 2008;9(4, article R65) doi: 10.1186/gb-2008-9-4-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nature Reviews Molecular Cell Biology. 2006;7(7):540–546. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 82.McCool KW, Xu X, Singer DB, Murdoch FE, Fritsch MK. The role of histone acetylation in regulating early gene expression patterns during early embryonic stem cell differentiation. Journal of Biological Chemistry. 2007;282(9):6696–6706. doi: 10.1074/jbc.M609519200. [DOI] [PubMed] [Google Scholar]
- 83.Saraiva NZ, Oliveira CS, Garcia JM. Histone acetylation and its role in embryonic stem cell differentiation. World Journal of Stem Cells. 2010;2(6):121–126. doi: 10.4252/wjsc.v2.i6.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee TI, Jenner RG, Boyer LA, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125(2):301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boyer LA, Plath K, Zeitlinger J, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441(7091):349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 86.O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Molecular and Cellular Biology. 2001;21(13):4330–4336. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part II: ATP-dependent chromatin remodeling. Trends in Molecular Medicine. 2007;13(9):373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Voncken JW, Roelen BAJ, Roefs M, et al. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2468–2473. doi: 10.1073/pnas.0434312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pasini D, Bracken AP, Jensen MR, Denchi EL, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. The EMBO Journal. 2004;23(20):4061–4071. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Molecular and Cellular Biology. 2007;27(10):3769–3779. doi: 10.1128/MCB.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pasini D, Hansen KH, Christensen J, Agger K, Cloos PAC, Helin K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and polycomb-repressive Complex 2. Genes and Development. 2008;22(10):1345–1355. doi: 10.1101/gad.470008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chamberlain SJ, Yee D, Magnuson T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells. 2008;26(6):1496–1505. doi: 10.1634/stemcells.2008-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes and Development. 2010;24(3):265–276. doi: 10.1101/gad.544410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morin-Kensicki EM, Faust C, LaMantia C, Magnuson T. Cell and tissue requirements for the gene eed during mouse gastrulation and organogenesis. Genesis. 2001;31(4):142–146. doi: 10.1002/gene.10017. [DOI] [PubMed] [Google Scholar]
- 95.Hansen KH, Bracken AP, Pasini D, et al. A model for transmission of the H3K27me3 epigenetic mark. Nature Cell Biology. 2008;10(11):1291–1300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 96.Margueron R, Justin N, Ohno K, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461(7265):762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Agger K, Cloos PAC, Christensen J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449(7163):731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 98.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130(6):1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 99.Lan F, Bayliss PE, Rinn JL, et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449(7163):689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- 100.Lee MG, Villa R, Trojer P, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318(5849):447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 101.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Molecular Cell. 2002;9(2):279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 102.Lin CH, Jackson AL, Guo J, Linsley PS, Eisenman RN. Myc-regulated microRNAs attenuate embryonic stem cell differentiation. The EMBO Journal. 2009;28(20):3157–3170. doi: 10.1038/emboj.2009.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saitou M, Barton SC, Surani MA. A molecular programme for the specification of germ cell fate in mice. Nature. 2002;418(6895):293–300. doi: 10.1038/nature00927. [DOI] [PubMed] [Google Scholar]
- 104.Yamaji M, Seki Y, Kurimoto K, et al. Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nature Genetics. 2008;40(8):1016–1022. doi: 10.1038/ng.186. [DOI] [PubMed] [Google Scholar]
- 105.Resnick JL, Bixler LS, Cheng L, Donovan PJ. Long-term proliferation of mouse primordial germ cells in culture. Nature. 1992;359(6395):550–551. doi: 10.1038/359550a0. [DOI] [PubMed] [Google Scholar]
- 106.Matsui Y, Zsebo K, Hogan BLM. Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell. 1992;70(5):841–847. doi: 10.1016/0092-8674(92)90317-6. [DOI] [PubMed] [Google Scholar]
- 107.De Felici M, Farini D, Dolci S. In or out stemness: comparing growth factor signalling in mouse embryonic stem cells and primordial germ cells. Current Stem Cell Research and Therapy. 2009;4(2):87–97. doi: 10.2174/157488809788167391. [DOI] [PubMed] [Google Scholar]
- 108.Koshimizu U, Taga T, Watanabe M, et al. Functional requirement of gp130-mediated signaling for growth and survival of mouse primordial germ cells in vitro and derivation of embryonic germ (EG) cells. Development. 1996;122(4):1235–1242. doi: 10.1242/dev.122.4.1235. [DOI] [PubMed] [Google Scholar]
- 109.Moe-Behrens GHG, Klinger FG, Eskild W, Grotmol T, Haugen TB, De Felici M. Akt/PTEN signaling mediates estrogen-dependent proliferation of primordial germ cells in vitro. Molecular Endocrinology. 2003;17(12):2630–2638. doi: 10.1210/me.2003-0006. [DOI] [PubMed] [Google Scholar]
- 110.Kimura T, Tomooka M, Yamano N, et al. AKT signaling promotes derivation of embryonic germ cells from primordial germ cells. Development. 2008;135(5):869–879. doi: 10.1242/dev.013474. [DOI] [PubMed] [Google Scholar]
- 111.Kimura T, Suzuki A, Fujita Y, et al. Conditional loss of PTEN leads to testicular teratoma and enhances embryonic germ cell production. Development. 2003;130(8):1691–1700. doi: 10.1242/dev.00392. [DOI] [PubMed] [Google Scholar]
- 112.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 113.Maherali N, Sridharan R, Xie W, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1(1):55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 114.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 115.Wernig M, Meissner A, Foreman R, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448(7151):318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 116.Leitch HG, Blair K, Mansfield W, et al. Embryonic germ cells from mice and rats exhibit properties consistent with a generic pluripotent ground state. Development. 2010;137(14):2279–2287. doi: 10.1242/dev.050427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kimura T, Nakamura T, Murayama K, et al. The stabilization of β-catenin leads to impaired primordial germ cell development via aberrant cell cycle progression. Developmental Biology. 2006;300(2):545–553. doi: 10.1016/j.ydbio.2006.06.038. [DOI] [PubMed] [Google Scholar]
- 118.Deng W, Xu Y. Genome integrity: linking pluripotency and tumorgenicity. Trends in Genetics. 2009;25(10):425–427. doi: 10.1016/j.tig.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ying QL, Wray J, Nichols J, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453(7194):519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maatouk DM, Resnick JL. Continuing primordial germ cell differentiation in the mouse embryo is a cell-intrinsic program sensitive to DNA methylation. Developmental Biology. 2003;258(1):201–208. doi: 10.1016/s0012-1606(03)00110-6. [DOI] [PubMed] [Google Scholar]
- 121.Murphy K, Carvajal L, Medico L, Pepling M. Expression of Stat3 in germ cells of developing and adult mouse ovaries and testes. Gene Expression Patterns. 2005;5(4):475–482. doi: 10.1016/j.modgep.2004.12.007. [DOI] [PubMed] [Google Scholar]