

Summary
IL-2, a cytokine linked to human autoimmune diseases, limits IL-17 production. We show that deletion of STAT3 in T cells abrogates IL-17 production and attenuates autoimmunity associated with IL-2 deficiency. While STAT3 induces IL-17 and RORγt and inhibits FOXP3, IL-2 inhibited IL-17 independently of FOXP3 and RORγt. We found that STAT3 and STAT5 bound to multiple common sites across the Il17 genetic locus. The induction of STAT5 binding by IL-2 was associated with a reduction in STAT3 binding at these sites and the inhibition of associated active epigenetic marks. Titrating the relative activation of STAT3 and STAT5 modulated Th17 cell specification and thus, the balance rather than the absolute magnitude of these signals determine the propensity of cells to make a key inflammatory cytokine.
Introduction
Optimal host defense requires that CD4+ T helper cells adapt distinct cellular fates efficiently to eliminate microbial pathogens1, 2. CD4+ T cells also play critical roles in the pathogenesis of immune-mediated diseases and Th17 cells in particular have emerged as key players in a range of autoimmune disorders including: inflammatory bowel disease, psoriasis and ankylosing spondylitis3. IL-17 contributes to inflammation by a number of mechanisms, including myeloid cell expansion and recruitment. Along with the Th17 associated cytokine, IL-21, IL-17 provides help to B cells4. Thus, defining the factors that govern the regulation of IL-17 in T cells is of great importance with respect to the pathogenesis of autoimmune disease.
IL-6, IL-21 and IL-23 in conjunction with IL-1 and/or TGF-β1 promote IL-17 production5-8. The former cytokines all activate STAT3, which is critical for Th17 cell differentiation. STAT3 directly regulates the Il17 gene and is necessary for the expression of multiple transcription factors involved in Th17 differentiation9, 10. Mice that lack Stat3 in T cells are unable to generate Th17 cells11-13 and are resistant to a number of models of autoimmunity10, 14. In humans, patients with Hyper-immunoglobulin E syndrome exemplify the contribution of STAT3 to the immune system15. Further evidence for the relevance of STAT3 is made by the identification of polymorphisms associated with increased risk of autoimmune disease 16.
Given the highly inflammatory nature of IL-17, it is not surprising that many factors serve to constrain its expression. Cytokines such as IFN-γ, IL-27 and IL-4 inhibit Th17 differentiation7, 17. A second category of factors that inhibit IL-17 expression in T cells shares the ability to induce FOXP3 expression in vitro. These include high doses of TGF-β118, retinoic acid19, 20 and IL-2. Genetic deletion or antibody blockade of IL-2 results in increased IL-17 production in vitro13. Moreover, a feature of the autoimmune disease associated with IL-2 deficiency is the overproduction of IL-17 in vivo13. The effects of IL-2 appear to be largely due to STAT5 as deficiency of this transcription factor in T cells mimics many aspects of IL-2 deficiency. For example, mice in which Stat5a/b have been deleted from their T cells, exhibit widespread autoimmune disease21, which is associated with overproduction of IL-1713.
In the light of these findings, we set out to dissect the mechanisms by which IL-2 negatively regulates IL-17, and to identify the participation of STAT3 and STAT5 in this process. We found that absence of STAT3 in the context of IL-2 deficiency ameliorated autoimmunity associated with this cytokine. However, the inhibitory effect of IL-2 on IL-17 was independent of FOXP3. Instead, the data supported a model in which the relative activation of STAT3 and STAT5 directly dictates the outcome of IL-17 production. Consistent with this prediction, we found that efficient Th17 differentiation occurred at very low doses of IL-6, as long as STAT5 activation was antagonized. Thus, our findings point to a new model of T helper cell specification and reveal the opposing effects of two closely related transcription factors that act on the same genetic element.
Results
STAT3 in T cells mediates inflammatory colitis observed in IL-2 deficient mice
Mutations of the Il2 gene, or the genes encoding its receptor subsets, are associated with severe autoimmune disease in both mouse and man22. This pathology is associated with a reduction in T regulatory (Treg) cells and elevated numbers of Th1 and Th17 cells. To explore the contribution Th17 cells make to the inflammatory disease associated with IL-2 deficiency, we bred CD4-Cre;Stat3fl/fl (S3K) mice with Il2-/- animals to generate doubly-deficient mice (Il2-/-S3K). Il2-/-S3K mice, like Il2-/- mice, have a significant reduction in the proportion of FOXP3+CD4+ T cells in the spleen, mesenteric lymph nodes (MLN) and lymphocytes of the colonic lamina propria (LPL) at three months old (Fig. 1a, Supplemental Fig. 1a). Consistent with this reduction in the proportion of FOXP3+ CD4+ T cells, there was an expansion in the proportion of activated CD4+ T cells (Supplemental Fig. 1b) and an increase in the total numbers of CD4+ T cells (Supplemental Fig. 2c) in both Il2-/- and Il2-/-S3K mice compared with control animals. The lymphocytosis seen in Il2-/- and Il2-/-S3K mice resulted in similar absolute numbers of CD4+ FOXP3+ cells in all four groups of mice, (Supplemental Fig. 1c), despite the differences seen in the proportion of FOXP3+CD4+ T cells.
Figure 1. The inflammatory colitis in IL-2 deficient mice is dependent on T cell STAT3.
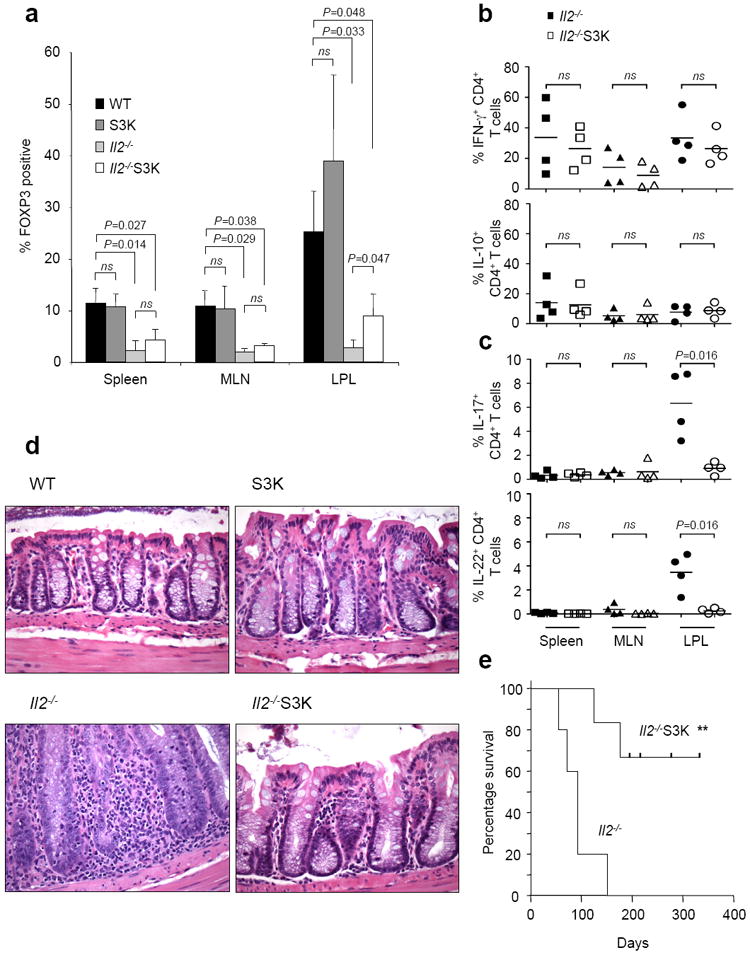
(a) CD4+ T cells were isolated from the spleen, mesenteric lymph nodes (MLN) and colonic lamina propria (LPL) of three month old Stat3fl/fl (WT), CD4-Cre;Stat3fl/fl (S3K), Il2-/- or CD4-Cre;Stat3fl/fl;Il2-/- (Il2-/-S3K) mice. FOXP3 expression was determined by intracellular staining. Histograms representing data from four separate experiments are shown. Paired t tests were used to determine statistical significance (ns: not significant). (b, c) Cells were stimulated for two hours with PMA, ionomycin and brefeldin A and expression of IFN-γ, IL-10 (b) and IL-17, IL-22 (c) was determined by intracellular staining. Data are representative of four separate experiments; Statistics were determined by paired t testing (ns: not significant). (d) Formaldehyde fixed colon sections of WT, S3K, Il2-/- and Il2-/-S3K mice were stained with haematoxylin and eosin. (e) Kaplan-Meyer survival curve comparing mice with IL-2 deficiency (Il2-/-) and mice with both STAT3 and IL-2 deficiency (Il2-/-S3K) (**P=0.003).
We next compared T cell cytokine expression. The proportions and total numbers of IFN-γ positive cells were not significantly different between Il2-/- and Il2-/-S3K mice (Fig. 1b, Supplemental Fig. 2a, Supplemental Fig. 2c), but there was a profound reduction in proportions of IL-17 and IL-22 production in T cells lacking STAT3 in the colonic lamina propria (Fig. 1c, Supplemental Fig. 2a, Supplemental Fig. 2b). Importantly, despite the residual impairment in Treg cells, deletion of Stat3 reduced the pathology associated with IL-2 deficiency and significantly prolonged lifespan (P=0.003) (Fig. 1d and 1e). Thus, T cell STAT3 is necessary for the inflammatory colitis associated with IL-2 deficiency, irrespective of Treg cell deficits.
IL-2 and STAT5 inhibit IL-17 independently of FOXP3
The failure to induce FOXP3 has been considered a major reason for the autoimmunity underlying IL-2 deficiency23. In dissecting the possible mechanisms through which IL-2 inhibits IL-17, it was logical to consider that the induction of FOXP3 might be a major factor. To test whether the inhibition of IL-17 by IL-2 was causally related to FOXP3 induction, we assessed the effect of IL-2 in cells lacking FOXP3. Using naïve cells from TCR transgenic, FOXP3 mutant (Scurfy: Foxp3sf;Rag1-/-;OT2) mice, we measured in vitro production of IL-17 in the presence and absence of IL-2. As expected, addition of IL-2 induced FOXP3 expression in control T cells but not in Scurfy T cells (Fig. 2a). To our surprise, IL-2 inhibited IL-17 expression equivalently regardless of the presence or absence of FOXP3, suggesting that the induction of FOXP3 plays only a small part, if any, in the ability of IL-2 to inhibit IL-17.
Figure 2. IL-2 inhibits IL-17A expression in the absence of FOXP3.
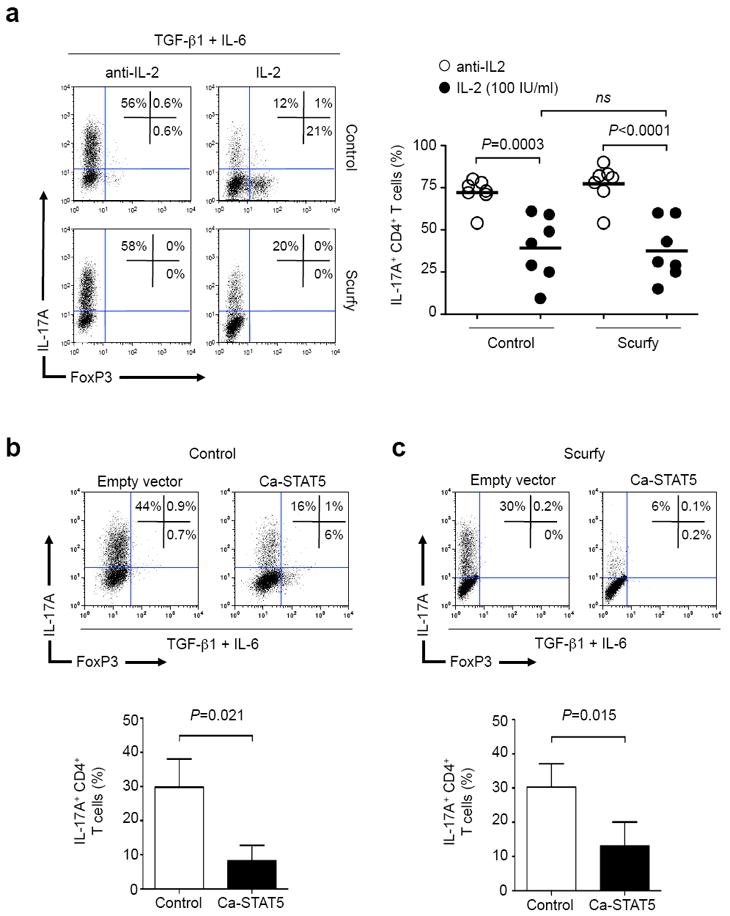
(a) Naïve CD4+ T cells from control (Rag1-/-;OT2) and Scurfy (Foxp3sf;Rag1-/-;OT2) mice were stimulated with TGF-β1, IL-6, anti-IFN-γ, anti-IL-4 and anti-IL-2 (left panels) or hIL-2 (right panels). IL-17A and FOXP3 expression were determined by intracellular staining. Representative flow cytometry is shown (left panel a) together with cumulative results of seven biological replicates from four independent experiments (right panel a), and significance was determined by unpaired t testing (ns: not significant). (b, c) Naïve CD4+ T cells from control (Rag1-/-;OT2) mice (b) and Scurfy (Foxp3sf;Rag1-/-;OT2) mice (c) were activated in media alone. Twenty-four and 48 hours later, cells were infected with retrovirus expressing human NGF-receptor (hNGFR) (empty vector) or hNGFR together with a constitutively active STAT5 (Ca-STAT5). After the second virus transduction, TGF-β1, IL-6, anti-IFN-γ, anti-IL-4 and anti-IL-2 were added to the culture. Two days later, IL-17A and FOXP3 expression were determined by intracellular staining. Flow cytometry plots of a single representative experiment are shown above; the histogram representing three independent experiments is below. Significance was determined using a paired t test.
We previously showed that inhibition of IL-17 by IL-2 is dependent upon STAT513. We next sought to determine if expression of STAT5 alone was sufficient to inhibit IL-17 production by transducing naïve CD4+ T cells with a retrovirus expressing constitutively active STAT5 (Ca-STAT5). As shown in figure 2b, this was the case; the proportion of IL-17-producing cells was significantly reduced by expression of Ca-STAT5. Since STAT5 regulates FOXP3 expression24, it was important to assess whether the ability of Ca-STAT5 to inhibit IL-17 was also independent of FOXP3 induction. We found that Th17 differentiation was equally inhibited by Ca-STAT5 in both control (Rag1-/-;OT2) (P=0.021) and Scurfy (Foxp3sf;Rag1-/-;OT2) T cells (P=0.015) (Fig. 2b, 2c). Thus, contrary to our expectations, FOXP3 induction was unnecessary for IL-2 and STAT5 to inhibit IL-17 production.
IL-2 inhibition of IL-17 is not rescued by forced expression of RORγt
Reduction of RORγt expression mediates the inhibition of Th17 differentiation by the action of the anti-inflammatory cytokine IL-2725. We therefore next assessed whether IL-2 acted by a similar mechanism. We found that addition of IL-2 caused a marked reduction in the proportion of IL-17-producing cells and a modest reduction in both the magnitude (as determined by mean fluorescence intensity) and the proportion of cells that expressed RORγt (Fig. 3a, top panels, Fig. 3b, left graph, Fig. 3c, Fig. 3d, left graph).
Figure 3. Over-expression of RORγt does not abrogate the inhibitory effect of IL-2.
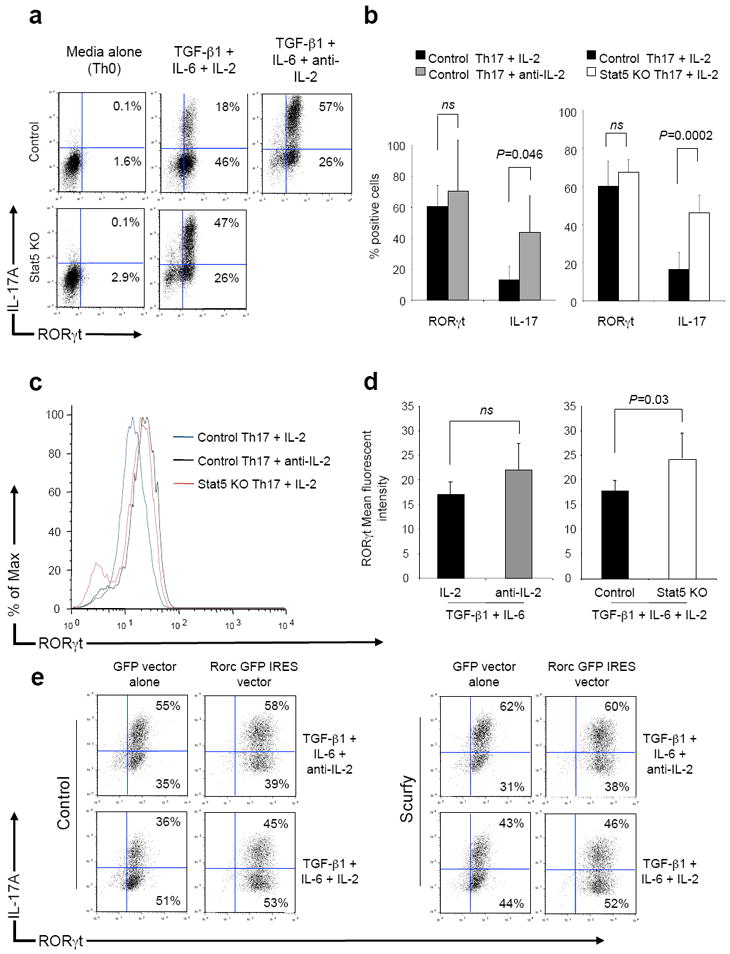
(a,b) Naïve CD4+ T cells from control (Stat5fl/fl;OT2) mice were stimulated in media (Th0), the presence of TGF-β1, IL-6 and IL-2 or the presence of TGF-β1, IL-6 and anti-IL-2. Naïve T cells from STAT5 KO (CD4-Cre;Stat5fl/fl;OT2) mice were stimulated in media (Th0) or the presence of TGF-β1, IL-6 and IL-2. After three days, RORγt and IL-17 expression was determined by intracellular staining. Data from a single representative experiment (a) and three independent experiments (b) are shown. Significance was determined using a paired t test (ns: not significant). (c,d) Mean fluorescence intensity for RORγt was determined for CD4+ T cells from control mice stimulated under Th17 conditions in the presence of anti-IL-2 or IL-2 or from STAT5 KO mice stimulated under Th17 conditions in the presence of IL-2, a single representative experiment is shown (c) together with cumulative data from three independent experiments (d). P values were determined by paired t testing (ns: not significant). (e) Naïve CD4+ T cells from control (Rag1-/-;OT2) or Scurfy (Foxp3sf;Rag1-/-;OT2) mice were stimulated in the presence of TGF-β1, IL-6 and anti-mIL-2 for two days. On day 1 and day 2, cells were infected with retrovirus containing GFP alone or GFP together with RORγt. After two days, IL-2 was added (lower panels) and the cells were incubated for a further two days. RORγt and IL-17 expression was determined in the GFP positive cell population by intracellular staining in cells stimulated again for 2 hours with PMA, ionomycin and brefeldin A.
The former was significant but the latter was not, and was unaffected by the absence of FOXP3 (Supplemental Fig. 3a). As expected, absence of STAT5 reversed the inhibitory effect of IL-2, resulting in enhanced IL-17 production (Fig. 3a). This had no significant effect on the proportions of cells expressing RORγt (Fig. 3b, right graph), but did have a small and statistically significant effect on the mean fluorescence intensity of RORγt expression (Fig 3c, Fig. 3d, right graph) (P=0.03).
These small changes in RORγt expression appeared not to explain the larger alternation in IL-17 production. However, to assess the role of RORγt more directly, we infected cells with a retrovirus encoding RORγt, to see if forced expression of RORγt would abrogate IL-2-mediated inhibition of IL-17 expression. To do this, it was necessary to delay addition of IL-2 to allow time for the expression of transduced RORγt. The data in supplemental figure 3b demonstrate that the earlier IL-2 was added, the more pronounced was its effect. However, there was still a 30% reduction in IL-17-producing cells, even if IL-2 was added after a 48-hour delay. Next, naïve T cells from Rag1-/-;OT2 (Control) and Foxp3sf;Rag1-/-;OT2 (Scurfy) mice were stimulated under Th17 conditions during which they were infected with retrovirus encoding GFP or GFP and RORγt genes. After 48 hours, IL-2 was added to the cell cultures (Fig. 3e, lower panels) and after a further 48 hours RORγt and IL-17 expression were determined in the GFP positive cells. The data in figure 3e demonstrate that cells stimulated under Th17 conditions and infected with GFP virus expressed physiological amounts of RORγt whereas Th17 cells infected with RORγt retrovirus expressed supra-physiological amounts of RORγt. The inhibitory effect of IL-2 on the proportion of IL-17+ cells was similar irrespective of whether RORγt was over-expressed. IL-2 also inhibited IL-17 expression to a similar extent in cells that both expressed supra-physiological amounts of RORγt and lacked FOXP3. Thus, IL-2 was able to inhibit IL-17 expression by a mechanism that was independent of expression of either transcription factor.
IL-2 does not impair IL-6-dependent STAT3 activation
Since the effect of IL-2 appeared not to be explained predominantly by regulation of expression of FOXP3 or RORγt, we next considered whether IL-2 might interfere with IL-6-dependent STAT3 activation. Upon IL-6 stimulation, SOCS proteins limit STAT3 activation. SOCS3 in particular has been identified as an inhibitor of STAT3 and Th17 development9, 26. Thus, we considered the possibility that prior activation of T cells with IL-2 may have an enhancing effect on SOCS proteins, which would interfere with STAT3 phosphorylation. We therefore first measured SOCS3 mRNA expression in Th17 cells polarized for three days in the presence or absence of IL-2, but found no difference (Supplemental Fig. 4a). We also compared the ability of IL-2 to inhibit Th17 polarization in cells lacking SOCS3 (CD4-Cre;Socs3fl/fl), but found that IL-2 inhibited Th17 polarization to a comparable extent in SOCS3-deficient and control T cells (Supplemental Fig. 4b). Finally, we explored whether the activation of cells by IL-2 inhibited subsequent IL-6-dependent STAT3 phosphorylation. However, the magnitude of STAT3 phosphorylation as determined by immunoblotting (Supplemental Fig. 4c) or by flow cytometry (Supplemental Fig. 4d) was not affected by pre-incubation with IL-2. The experiment was also repeated in STAT5-deficient cells (Supplemental Fig. 4e) and the presence or absence of STAT5 had no influence on the level of phosphorylated STAT3 detected in response to IL-6 stimulation.
STAT5 is sufficient to inhibit IL-17 and acts directly in repressing the Il17 locus
Since indirect modes of negative regulation of the IL-17 failed to explain the effects of IL-2, we next turned our attention to the possibility that STAT5 has direct effects on the genetic locus spanning the Il17a - Il17f genes. Our previous data showed that STAT5 bound the Il17a promoter region; however, at that time, other aspects of the architecture of this locus had not been elucidated27, 28. To investigate the possibility that STAT5 might function as a direct transcriptional repressor of the Il17a gene, we first mapped its binding sites using chromatin immunoprecipitation with massive parallel sequencing (ChIP-seq) and compared these with STAT3 binding sites within the Il17a-f locus. From this comparison, we concluded that STAT5 bound not only at the Il17a promoter, but also at distal sites (-5K, +10K, +23K, +28K and +36K) (Fig. 4a, lower panel). These sites all co-localised to regions where STAT3 bound (Fig. 4a, upper panel), to predicted STAT binding site motifs (Fig. 4a, blue triangles) and to DNase hypersensitivity sites in stimulated Th17 cells27.
Figure 4. STAT3 and STAT5 compete for the same binding sites within the Il17a-f locus.
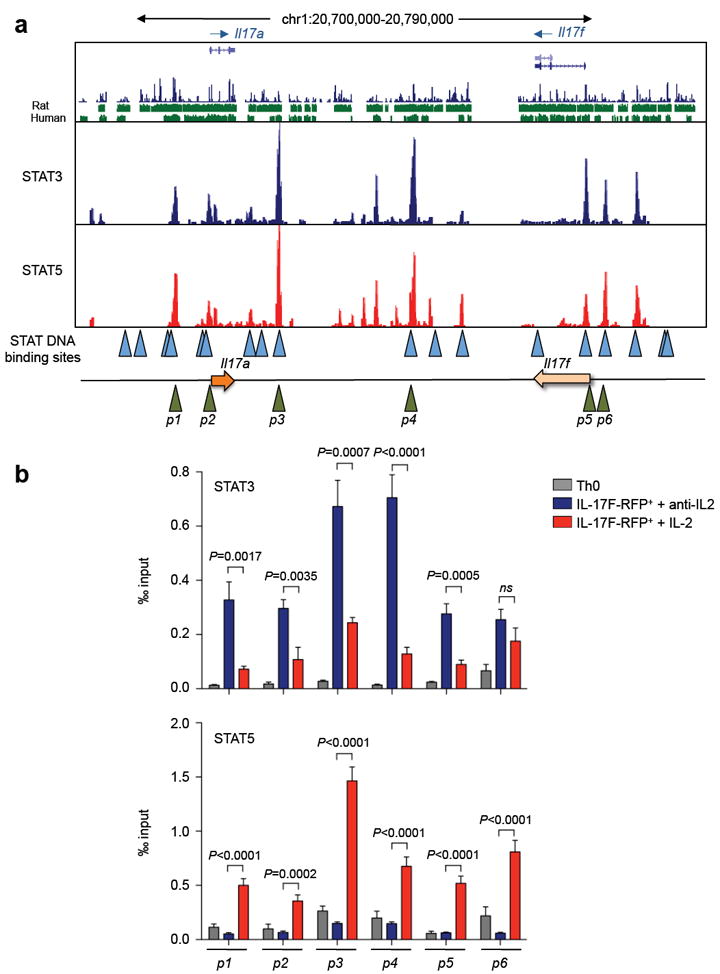
(a) Naïve CD4+ T cells were stimulated for three days under Th17 conditions. On the third day cells were either stimulated with IL-6, crosslinked with formaldehyde and immunoprecipitated with anti-STAT3 (upper panel a) or stimulated with IL-2 and immunoprecipitated with anti-STAT5 (lower panel a). Immunoprecipitated DNA was sequenced by massive parallel sequencing. The Il17a-f locus is illustrated; the locations of primer sites p1 – p6 are depicted by green triangles and candidate STAT binding sites are illustrated by blue triangles. (b) Naïve CD4+ T cells from IL-17F-RFP reporter mice were cultured for three days in media alone (Th0), or with TGF-β1, IL-6 and anti-IL-2 (Th17+anti-IL-2) or with TGF-β1, IL-6 and IL-2 (Th17+IL-2). On the third day, RFP+ cells from both groups were isolated using flow cytometry cell sorting. Cells were restimulated with IL-6 in the presence or absence of IL-2, crosslinked with formaldehyde, and immunoprecipitated with anti-STAT3 (b, upper panel) and anti-STAT5 (b, lower panel). Bound DNA was amplified by quantative-PCR for primer sites p1 – p6 and expressed as a permil of input DNA. Data are pooled from two independent experiments. Error bars denote s.e.m and P values were determined by unpaired t testing (ns: not significant).
Given that IL-2, acting through STAT5, had direct effects on the Il17a gene, we next wondered how this signal would interfere with the positive actions of STAT3. We were struck by the finding that STAT3 and STAT5 appeared to bind at the same regions of the Il17a-f locus (Fig. 4a), suggesting that STAT5 could directly interfere with the ability of STAT3 to bind the locus. To address this point, chromatin immuno-precipitation (ChIP) and quantitative PCR was used to interrogate whether the presence of bound STAT5 inhibited STAT3 binding. Primers were made corresponding to six sites (p1 – p6, green triangles) and each site was associated with both STAT3 and STAT5 binding and contained single discrete DNA STAT binding sites (Fig. 4a, blue triangles). To be certain that IL-17-producing cells were selectively interrogated, naïve Th cells from IL-17F-RFP reporter mice were stimulated under Th0 conditions or Th17 conditions in the presence of IL-2 or anti-IL-2. RFP+ cells were sorted from the cells stimulated under Th17 conditions and STAT3 and STAT5 binding was measured by ChIP and quantitative PCR. The results in figure 4b show that the presence of IL-2 led to enhanced STAT5 binding across the Il17 locus at all the sites identified by ChIP-seq (Fig. 4b, lower panel). Evidence of STAT5 binding correlated with a significant reduction in STAT3 binding at all the sites associated with the Il17a gene and within the intergenic region but only at one of the two sites associated with the Il17f gene (Fig. 4b, upper panel, p5 and p6 sites).
Recent genome-wide studies have identified p300 binding as a marker of gene enhancer elements29, 30. As shown in figure 5a (upper left panel), we saw evidence of p300 recruitment at all the interrogated STAT binding sites. The most prominent binding occurred at the intergenic sites p3 and p4, suggesting that these two sites represent classical enhancer elements. Of note, the presence of IL-2 resulted in reduced p300 binding to sites associated with the Il17a promoter, but had little effect on sites associated with the Il17f promoter.
Figure 5. STAT5 binding is associated with a reduction in active epigenetic marks across the Il17a promoter region and associated enhancer elements.
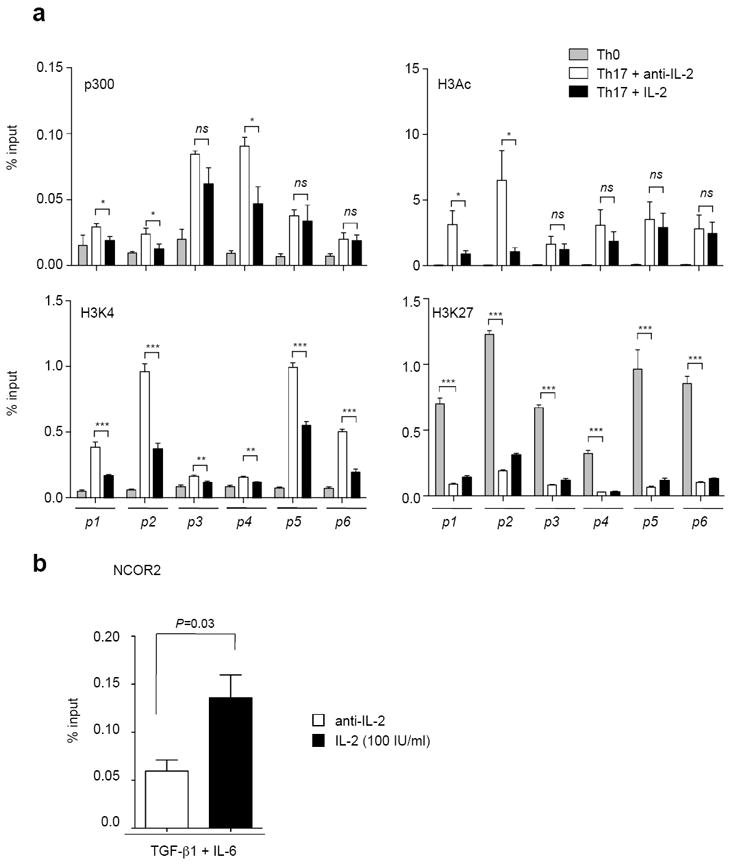
Naïve CD4+ T cells were stimulated for three days in the presence of media with anti-IFN-γ, anti-IL-4 alone (Th0), anti-IFN-γ, anti-IL-4, TGF-β1, IL-6 and anti-IL-2 (Th17+anti-IL-2) or anti-IFN-γ, anti-IL-4, TGF-β1, IL-6 and IL-2 (Th17+IL-2). Cells were crosslinked and immunoprecipitated with anti-p300, anti-H3Ac, anti-H3K4me3, anti-H3K27me3. DNA was amplified as described in Fig. 5 and expressed as a percentage of input DNA. The data are representative of two independent experiments. (b) Naïve CD4+ T cells were stimulated under Th17 condition with anti-IL-2 or IL-2. Cells were crosslinked and immunoprecipitated with anti-NCoR2. Immunoprecipitated DNA was determined for the p4 region by quantitative-PCR and expressed as a percentage of input DNA. Data are representative of three independent experiments. Error bars denote s.e.m and P values were determined by unpaired t testing (* P<0.05, ** P<0.01, *** P<0.001, ns: not significant)
In order for efficient and stable gene transcription to occur, the locus must be held in an open conformation, which can be monitored by the presence of characteristic histone modifications10, 27, 31. The finding that STAT5 bound the putative regulatory sites suggested to us that the IL-2/STAT5 signal might alter the epigenetic modifications within the extended Il17a-f locus. We therefore next explored the effect of IL-2 on total acetylation of histone 3 (H3Ac, Fig. 5a, upper right panel) and histone 3 lysine 4 trimethylation status (H3K4me3, Fig. 5a, lower left panel), both associated with transcriptional activation. The data show that IL-2 stimulation resulted in a reduction in H3Ac modification primarily at the Il17a promoter (sites p1 and p2). In contrast, there was little effect on the Il17f promoter (sites p5 and p6). The addition of IL-2 was associated with a reduction in H3K4me3 status throughout the Il17a-f locus. Histone 3 lysine 27 trimethylation (H3K27me3) is typically associated with an inhibition of gene transcription. We therefore next asked whether STAT5 binding to the Il17 locus was associated with the acquisition of H3K27me3. The data demonstrate that cells stimulated under Th17 conditions had significantly less H3K27me3 at all of the sites examined within the Il17a-f locus compared with cells stimulated under non-polarizing (Th0) conditions. The addition of IL-2 resulted in a modest elevation in the levels of H3K27me3 across the Il17a-f locus (Fig. 5a, lower right panel).
It has been previously reported that STAT5 is able to recruit the histone deacetylator adaptor protein, NCoR232. To assess whether this was relevant for the regulation of IL-17, we polarized cells under Th17 conditions in the presence or absence of IL-2 and measured the recruitment of NCoR2. In preliminary experiments, we found that the major site of NCoR2 recruitment occurred within the intergenic region (p4) of the Il17 locus. As shown in figure 5b, IL-2 significantly increased binding of NCoR2 binding to this site. Thus, in response to IL-2, STAT5 displaced STAT3, which resulted in loss of permissive histone modifications and promoted recruitment of the NCoR2 repressor.
The balance of STAT5/STAT3 activation regulates Th17 specification
If STAT3 and STAT5 compete for binding to the same sites within the Il17a-f genetic locus, this suggests a model of IL-17 expression that would depend less on the absolute concentration of IL-6 or IL-2, but more on the relative ratios. Furthermore, the epigenetic data predict that IL-17F would be more resistant to inhibition by IL-2 compared with IL-17A. To explore these possibilities, we polarized naïve T cells in TGF-β1 and anti-mouse IL-2 with varying concentrations of IL-6 and human IL-2 and assessed intracellular expression of IL-17A and IL-17F (Fig. 6). In the absence of IL-2, the proportions of T cells that expressed IL-17A were near maximal, even at very low doses of IL-6. In the presence of a low concentration of IL-2, increasing the concentration of IL-6 was able to reverse its inhibitory action leading to the proportion of IL-17A+ cells that approximated the percentage seen at the lowest concentration of IL-6 in the absence of IL-2 (59% vs. 65%) (Fig. 6a, top left panel vs. middle right panel). At high concentrations of IL-2, Th17 differentiation was abrogated even in the presence of maximal doses of IL-6 (Fig. 6b). At the lowest concentration of IL-6, IL-2 potently inhibited IL-17F production. By contrast to the inhibition of IL-17A by IL-2, the inhibition of IL-17F was completely reversed with increasing concentrations of IL-6 irrespective of the concentration of IL-2 (Fig. 6c). At the highest concentration of IL-6 and IL-2, the majority of the Th cells that were IL-17F+ were IL-17A-. In summary, Th17 commitment represents a dynamic balance between the binding of STAT3 and STAT5 to sites along the Il17a-f genetic locus, although IL-17A and IL-17F gene expression are not equally affected.
Figure 6. Th17 generation is dynamically regulated by opposing effects of IL-2 and IL-6: differential regulation of IL-17A and IL-17F.
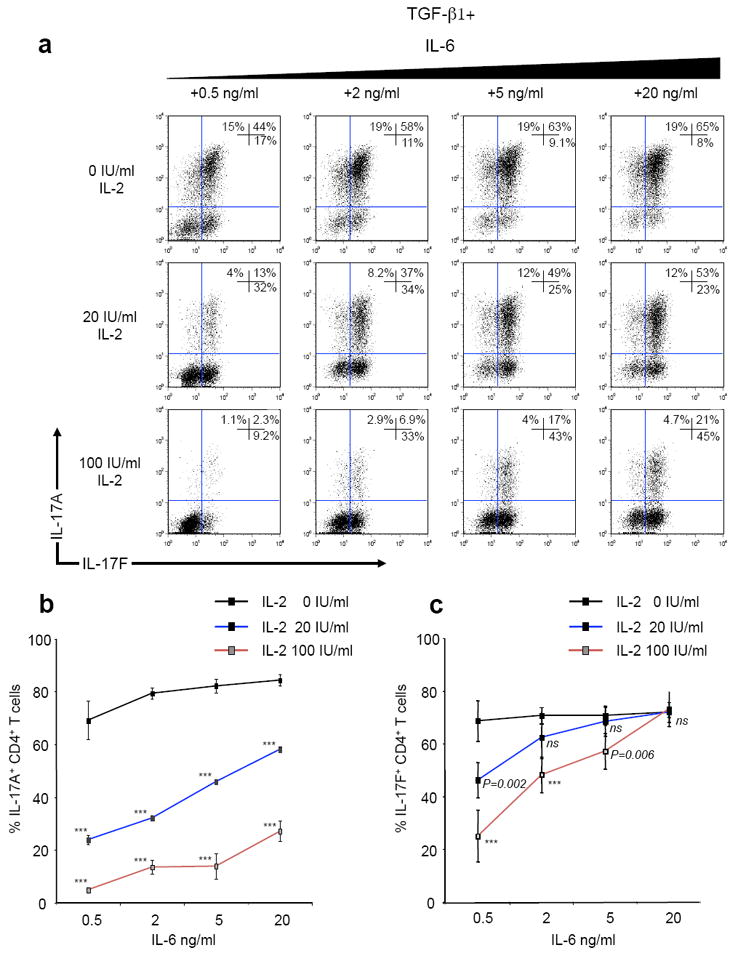
(a-c) Naïve CD4+ T cells were polyclonally stimulated for three days with anti-IFN-γ, anti-IL-4, anti-mIL-2, TGF-β1, varying concentrations of hIL-2 (0 to 100 IU/ml) and IL-6 (0.5 to 20 ng/ml). IL-17A and IL-17F expression was determined by intracellular staining. (a) Flow cytometry plots showing a single representative experiment. (b,c) Summary of four biological replicates from two separate experiments showing the effect of altered IL-2 and IL-6 doses on (b) IL-17A expression and (c) IL-17F expression. Statistics were determined using an unpaired t test (*** P<0.001, ns: not significant).
Discussion
We previously showed that IL-2 inhibits the differentiation of Th17 cells. Herein, we show that IL-2 inhibits IL-17 gene expression, largely independent of FOXP3 or RORγt. Instead, our data suggest an alternative explanation: that the major mode of action of IL-2 is directly to repress the Il17 locus. STAT5 was necessary and sufficient for this action, inhibiting transcription and removing accessible histone marks, and displacing STAT3 occupancy. Consistent with this competition model, the proportion of naïve T cells that became Th17 cells depended on the relative concentrations of IL-2 and IL-6, such that blockade of IL-2 permitted IL-17A production at very low levels of IL-6. Conversely, increasing the concentration of IL-6 overcame the inhibitory effects of IL-2. Thus, we propose a model in which Th17 differentiation represents a continuum that reflects the balance between extrinsic factors that both inhibit and promote fate determination. Specification is not simply due to the instructive effects of a single cytokine, but rather represents the equilibrium of these opposing signals. Strikingly, these signals activate two highly related transcription factors, which act on the same locus but have opposite functional effects.
Complete lack of IL-2 in mice results in autoimmune disease that is modified by the genetic background33, 34. Accordingly, humans with inherited deficiency of IL-2 receptor or STAT5b suffer from autoimmune disease35, 36. Recent work devoted to identifying genetic predictions to autoimmunity has revealed that polymorphisms within the Il2ra genetic locus are a relevant factor for Type I diabetes and multiple sclerosis37, 38. Taken together, these data point to a critical role of IL-2 in limiting immune responses. Unquestionably, an important aspect of IL-2’s anti-inflammatory action is its ability to induce FOXP3. The ability of IL-2 to inhibit Th17 differentiation may also be relevant, but the extent to which IL-17 is an independent driver of autoimmunity in the setting of IL-2 deficiency is not clear. While deletion of IL-17 alone did not reverse the autoimmune hemolytic anemia (AIHA) associated with IL-2 deficiency in BALB/c mice39, its role in mediating the colitis was not explored. We found that the inflammatory colitis associated with IL-2 deficiency was dependent on STAT3 in T cells, culminating in a significant prolongation of survival in Il2-/-S3K mice compared with Il2-/- animals.
Our findings contrast with that of Hoyer et al, but it must be emphasized that there are a number of differences between our studies. The background of the mice studied was different, with Il2-/- BALB/c animals dying at an earlier age of AIHA, whereas Il2-/- C57BL/6 mice die later of colitis33, 34. Hoyer et al studied the effect of IL-17A alone, whereas our study looked at T cell STAT3, critical for development of the Th17 lineage11, 12, which is characterized by a unique repertoire of chemokines and cytokines for which IL-17A is but one member.
Originally identified as mediators of interferon action, STATs typically function as positive transactivating factors. Several lines of evidence indicate that for some genes the actions of one STAT can be compensated by a second, with both acting in a positive manner40. Prior to the advent of ChIP-seq, the number of known STAT-regulated genes was limited. However, with this new technology the identification of STAT-bound genes has expanded41. These data confirm the notion that many genes are bound and positively regulated by more than one STAT41.
By contrast prior to this new approach, there were few examples of STATs serving as repressors42. However, an unexpected finding revealed by these data sets was that STATs serve as transcriptional repressors for a considerable proportion of genes41. Furthermore, a third category of genes, identified by ChIP-seq analysis, can be characterized by circumstances in which more than one STAT binds a given gene, and yet the individual STATs may have opposing actions. This was quite evident in the analysis of STAT4 and STAT6 targets in Th1 and Th2 cells41. In fact, of 265 genes bound and inhibited by STAT4, 150 were oppositely regulated under Th2 conditions in a STAT6-dependent manner. In these cases, there was little evidence that this competition was directed to the same elements. On a genome-wide scale, binding of a transcription factor that acts in a positive manner is enriched in promoter regions. By contrast, when the factor is acting as a repressor, it is more likely to be enriched in regions outside the promoter43. In the case of STAT4 and STAT6, the binding motifs are also distinct; thus, in many circumstances activators and repressors seem to have spatially distinct localization.
In this context the actions of STAT3 and STAT5 on the Il17a-f locus seemed particularly intriguing to explore. STAT3 and STAT5 can induce expression of the same genes and their actions can be complimentary; both factors induce Bcl-xl and Cyclin D1 expression44. Furthermore, STAT3 and STAT5 bind the same DNA motif. Then how would the action of these two transcription factors divergently regulate Il17 gene transcription? As indicated, we initially considered that indirect modes of regulation would be the most likely scenario; however, our data indicated this not to be the case. On the contrary, our data clearly indicate that STAT5 is recruited to the Il17 locus where its presence is associated with a reduction in gene transcription, the histone H3 acetylation and K4 trimethylation status and the recruitment of the transcriptional repressor NCoR2.
If STAT3 and STAT5 functioned like other factors that act as activators and repressors of gene transcription, we expected that there would be enrichment of STAT3 and STAT5 in different parts of the Il17 locus; however, this was also not the case. Both factors bound to the same regions 5’ and 3’ of the gene, and the presence of STAT5 was associated with a displacement of the positive transactivating factor STAT3 in the Il17a promoter and associated gene enhancers. It was notable that STAT5 binding also served to recruit the inhibitor NCor2 predominantly in the intergenic region. To our knowledge, our data represent the first example in which two STATs bind the same elements and have opposing functions. However, we do not believe that this is singular to the Il17 gene. There are other genes in which STAT3 and STAT5 have opposing roles in their expression in tumor cell lines44; however, with the exception of the present work, the precise modes of negative regulation of the endogenous genes have yet to be investigated. We compared ChIP-seq data with differential gene expression by microarray in STAT5- and STAT3-deficient T cells stimulated under Th17 conditions. We identified more than 300 genes that showed evidence of direct STAT binding within their loci whose expression was both diminished in the absence of STAT3 and enhanced in the absence of STAT5. Within this group we found that 95% of these genes contained co-localised STAT3 and STAT5 binding peaks. This contrasts with our previous work in Th1/Th2 development, in which only 24% of genes bound and counter-regulated by STAT4 and STAT6 showed co-localization of STAT4 and STAT6 binding peaks41. It will therefore be important to carefully delineate genes for which these factors have complimentary versus opposing functions and to assess the mechanisms of regulation.
The idea that related transcription factors can bind the same elements and have opposing functions is not without precedent. For instance, E2F4 is recruited to the nucleus in cells resting in G1 phase and serves to recruit the histone deacetylator adaptor protein mSin3B45. The initiation of cells into S phase from G1 is associated with the replacement of E2F4 with the related factors E2F1, E2F2 and E2F346, 47. In this case, one family member, E2F4, serves uniquely as a repressor; it does not have a dual role. More analogous to the situation with STAT3 and STAT5 is the NF-κB family. It comprises five members that form multiple homo- and heterodimers but recognize the same consensus sequence. Initially, p65/p50 bind the Il12p40 promoter, but they are displaced by RelB/p52, which correlates with extinction of transcription48. In this case, however, the same stimulus induces the formation of both complexes and the regulation is temporal. This contrasts with our data in which two distinct extrinsic signals activate different family members. However, it is also important to recognize that individual cytokines are capable of activating multiple STAT proteins to varying degrees. The possibility that for some genes two STATs will have complementary functions, while others will have opposing ones, is a subtlety that is not generally recognized. Thus, cytokines should not be viewed as acting in a single linear pathway, activating a single STAT: instead, the net effect of cytokines acting on their targets represents the integration of actions of multiple STAT proteins, which can act co-operatively at some gene loci and competitively at others.
Methods
Mice and media
C57BL/6 mice and Il2-/- mice were purchased from Jackson Laboratory (Bar Harbor, Maine). Rag1-/-;OT2 mice and Foxp3sf mice were purchased from Taconic (Hudson, New York). CD4-Cre;Stat5fl/fl mice were published previously, CD4-Cre;Stat3fl/fl mice were obtained from Dr Levy (New York university)49. IL-17F-RFP reporter mice were obtained from Dr Dong (MD Anderson) and have been described previously50. All animal studies were performed according to the NIH guidelines for the use and care of live animals and were approved by the Institutional Animal Care and Use Committee of NIAMS. All cell cultures were performed in RPMI with 10% fetal calf serum, 2 mM glutamine, 100 IU/ml penicillin, 0.1 mg/ml streptomycin, Hepes buffer (all Invitrogen, CA) and 2 mM β-mercaptoethanol (Sigma, MO).
T cell isolation and differentiation
CD4+ T cells from spleens and lymph nodes of 6- to 8-week-old mice were purified by negative selection and magnetic separation (Miltenyi Biotec, Germany) followed by sorting of naive CD4+CD62L+CD44- population using FACSAria II (BD, NJ). Cells were activated by plate-bound anti-CD3/CD28 (both 10 μg/ml; eBioscience, CA) in media for 3-4 days either under neutral conditions or with IL-6 (20 ng/ml) plus human TGF-β1 (2.5 ng/ml), anti-IFN-γ (10 μg/ml, BioXCell), anti-IL-4 (10 μg/ml, BD pharmingen) and anti-mouse IL-2 (mIL-2) (10 μg/ml, BioXCell). Human IL-2 (hIL-2) was used at 100 IU/ml unless indicated otherwise.
Intracellular staining and flow cytometry
The following antibodies were used: For cell sorting: anti-CD4-PerCPCy5.5, anti-CD62L-APC and anti-CD44-PE (all BD, NJ). For intracellular staining: anti-IL-17A-PE is from BD (NJ). Anti-IL-17F-Alexa Fluor® 647, and anti-FOXP3-Alexa Fluor® 647 were purchased from eBioscience (CA). In brief, cells were stimulated for 2 hours with PMA and ionomycin with the addition of brefeldin A (GolgiPlug; BD, NJ). Afterwards, cells were fixed with 4% formyl saline, permeabilized with 0.1% saponin buffer and stained with fluorescent antibodies before analyzing on a FACS Calibur (BD, NJ). Events were collected and analyzed with Flow Jo software (Tree Star, Ashland, OR).
Retroviral transduction
Naïve CD4+ T cells were activated with plate-bound anti-CD3, anti-CD28 and anti-IFN-γ for 24 hours. The activated cells were transduced with supernatants containing hNGFR retrovirus, hNGFR-caSTAT5 retrovirus, GFP retrovirus or GFP-RORγt virus in the presence of polybrene (8ug/ml) by centrifuge at 2500 rpm for 90 minutes at 30 degree. The same procedure of transduction was repeated 24 hours later and differentiation cytokines were added for 2 days.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as described previously9. Naïve CD4+ T cells were activated and polarized for 3-4 days followed by cross-linking for 8 min with 1% formaldehyde. The cells were harvested and lysed by sonication. After pre-clearing with protein A agarose beads (Upstate, VA), cell lysates were immunoprecipitated with anti-H3K4m3 (ab8580, Abcam), anti-STAT5A/STAT5B antibody (PA-ST5A and PA-ST5B, R&D Systems), anti-STAT3 antibody (14-6727, ebioscience, CA), anti-H3Ac (06-599, Millipore), anti-H3K27m3 (07-449, Millipore), anti-p300 (C-20, Santa Cruz, CA) or anti-NCoR2 (ab24551, Abcam) overnight at 4°C. After washing and elution, crosslinks were reversed at 65°C for 4 hr. The eluted DNA was purified, samples analyzed by quantitative-PCR with customer designed primers and probes (Supplemental Table 1) using a 7500 real time PCR system (both Applied Biosystems, CA). The Ct value for each sample was normalized to corresponding input value.
Chromatin immunoprecipitation and massive parallel sequencing (ChIP-seq)
ChIP-seq experiments were performed as described previously41. Briefly, CD4+ T cells were activated with TCR stimulation and polarized with IL-6 and TGF-β1 for 3 days. Before harvesting, cells were restimulated with IL-6 for STAT3 precipitation or IL-2 for STAT5 precipitation for 30 minutes. Cross-linked T cells were treated with MNase to generate approximately 80% mononucleosomes and 20% dinucleosomes. Chromatin from 2 × 107 cells was used for each ChIP experiment, which yielded approximately 100 ng of DNA. Antibodies against anti-STAT5A/STAT5B antibody (PA-ST5A and PA-ST5B, R&D Systems), and anti-STAT3 (14-6727, ebiosciences, CA) were used. The ChIP DNA fragments were blunt-ended, ligated to the Illumina adaptors, and sequenced using the Illumina 1G Genome Analyzer. Sequenced reads of 25 bp were obtained using the Illumina Analysis Pipeline (all Illumina, CA). All reads were mapped to the mouse genome (mm9) and only uniquely matching reads were retained. Those unique tags were mapped into non-overlapping 200 bps windows of genome for STAT3 binding sample. Significant peaks (islands) were identified based on window tag-count threshold determined from a P-value of 0.05 (defined by Poisson background model).
Histopathology
Mouse tissues were fixed in 10% formyl-saline followed by embedding in paraffin blocks. Tissue sections were stained in hematoxylin and eosin and reported by a pathologist in a blinded manner.
Isolation of lamina propria lymphocytes
Large intestines were removed, cleared from mesentery, fat and Peyer’s patches, cut into pieces and washed in HBSS w/o Ca2+/Mg2+. After incubation in HBSS with EDTA, epithelial layer cells were removed and the remaining tissue was digested with collagenase and DNAse I (both Roche, IN) at 37°C. Lamina propria lymphocytes were recovered from the supernatant, purified over a 40%, 80% percol gradient and the interface layer washed twice in media before stimulation with phorbol myristate acetate (PMA), ionomycin and brefeldin A.
Real-time PCR
Total RNA was isolated with the use of mirVana™miRNA kit (Ambion, TX); cDNA was synthesized with Taqman reverse transcription Kit (Applied Biosystems, CA) according to the manufacture’s instructions. Quantitative PCR was performed with an ABI PRISM 7700 sequence detection system with site-specific primers and probes (Applied Biosystems, CA). The comparative threshold cycle method and an internal control (β-actin) were used to normalize the expression of the target genes. The primers and probes from Applied Biosystems: mouse ACTB 4352341E and mouse SOCS3 (Mm00545913).
Immunoloblot analysis
T cell protein extracts were separated by 4-12% SDS-PAGE and were transferred to nitrocellular membranes (invitrogen). Memberanes were probed with anti-STAT5 (Tyr694) (9351, Cell signaling technology), anti-STAT3 (Tyr705) (9145, Cell signaling technology) and anti-actin (Millipore).
Supplementary Material
Acknowledgments
We thank J. Simone and J. Lay (Flow Cytometry Section, NIAMS) and the NIAMS LACU staff for their excellent technical support. We thank Drs. S. Kuchen and F.C. Eberle comments on the manuscript. The Intramural Research programs of NIAMS and NIAID have supported this work.
Footnotes
Author contributions X.Y designed and performed experiments, analyzed and wrote the manuscript; K.G. designed and performed experiments and helped to write the manuscript; S.S.T helped to analyze gut lymphocytes; J.R.C examined the histopathology and J.Z, K.H and J.G provided mice and helped perform experiments; Y.K. performed the ChIP-seq experiments and the data analyzed by L.W, H.W.S, Y.K and G.V; J.O’S designed experiments, analyzed all acquired data and helped write the manuscript. A.L designed, performed or interpreted all the experiments and wrote the manuscript.
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Weaver CT, Hatton RD. Interplay between the TH17 and TReg cell lineages: a (co-)evolutionary perspective. Nat Rev Immunol. 2009;9:883–889. doi: 10.1038/nri2660. [DOI] [PubMed] [Google Scholar]
- 2.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buonocore S, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;464:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu HC, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 5.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 2007;19:652–657. doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 8.Ghoreschi K, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durant L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang XO, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 12.Mathur AN, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–4907. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 13.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Harris TJ, et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol. 2007;179:4313–4317. doi: 10.4049/jimmunol.179.7.4313. [DOI] [PubMed] [Google Scholar]
- 15.Milner JD, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cenit MC, et al. STAT3 locus in inflammatory bowel disease and multiple sclerosis susceptibility. Genes Immun. 2010;11:264–268. doi: 10.1038/gene.2010.10. [DOI] [PubMed] [Google Scholar]
- 17.Stumhofer JS, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 18.Zhou L, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elias KM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mucida D, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 21.Moriggl R, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10:249–259. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- 22.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 23.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 24.Yao Z, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diveu C, et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol. 2009;182:5748–5756. doi: 10.4049/jimmunol.0801162. [DOI] [PubMed] [Google Scholar]
- 26.Wong PK, et al. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest. 2006;116:1571–1581. doi: 10.1172/JCI25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukasa R, et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity. 2010;32:616–627. doi: 10.1016/j.immuni.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem. 2007;282:5969–5972. doi: 10.1074/jbc.C600322200. [DOI] [PubMed] [Google Scholar]
- 29.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 31.Wei G, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakajima H, Brindle PK, Handa M, Ihle JN. Functional interaction of STAT5 and nuclear receptor co-repressor SMRT: implications in negative regulation of STAT5-dependent transcription. Embo J. 2001;20:6836–6844. doi: 10.1093/emboj/20.23.6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadlack B, et al. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 34.Sadlack B, et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995;25:3053–3059. doi: 10.1002/eji.1830251111. [DOI] [PubMed] [Google Scholar]
- 35.Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc Natl Acad Sci U S A. 1997;94:3168–3171. doi: 10.1073/pnas.94.7.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen AC, et al. Cutting edge: Decreased accumulation and regulatory function of CD4+ CD25(high) T cells in human STAT5b deficiency. J Immunol. 2006;177:2770–2774. doi: 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- 37.Hafler DA, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 38.Lowe CE, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39:1074–1082. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 39.Hoyer KK, Kuswanto WF, Gallo E, Abbas AK. Distinct roles of helper T-cell subsets in a systemic autoimmune disease. Blood. 2009;113:389–395. doi: 10.1182/blood-2008-04-153346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park JH, et al. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxiclineage T cells. Nat Immunol. 2010;11:257–264. doi: 10.1038/ni.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei L, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32:840–851. doi: 10.1016/j.immuni.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esashi E, et al. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity. 2008;28:509–520. doi: 10.1016/j.immuni.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu M, et al. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol Cell. 2009;36:682–695. doi: 10.1016/j.molcel.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walker SR, et al. Reciprocal effects of STAT5 and STAT3 in breast cancer. Mol Cancer Res. 2009;7:966–976. doi: 10.1158/1541-7786.MCR-08-0238. [DOI] [PubMed] [Google Scholar]
- 45.Rayman JB, et al. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ. Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol. 2000;20:5797–5807. doi: 10.1128/mcb.20.16.5797-5807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- 48.Saccani S, Pantano S, Natoli G. Modulation of NF-kappaB activity by exchange of dimers. Mol Cell. 2003;11:1563–1574. doi: 10.1016/s1097-2765(03)00227-2. [DOI] [PubMed] [Google Scholar]
- 49.Lee CK, et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17:63–72. doi: 10.1016/s1074-7613(02)00336-9. [DOI] [PubMed] [Google Scholar]
- 50.Yang XO, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.