

Abstract
We recently found that the adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain and leucine zipper motif (APPL)1 is essential for mediating adiponectin signal to induce liver kinase B (LKB)1 cytosloic translocation, an essential step for activation of AMP-activated protein kinase (AMPK) in cells. However, the underlying molecular mechanisms remain unknown. Here, we demonstrate that treating C2C12 myotubes with adiponectin promoted APPL1 interaction with protein phosphatase 2A (PP2A) and protein kinase Cζ (PKCζ), leading to the activation of PP2A and subsequent dephosphorylation and inactivation of PKCζ. The adiponectin-induced inactivation of PKCζ results in dephosphorylation of LKB1 at Ser307 and its subsequent translocation to the cytosol, where it stimulates AMPK activity. Interestingly, we found that metformin also induces LKB1 cytosolic translocation, but the stimulation is independent of APPL1 and the PP2A-PKCζ pathway. Together, our study uncovers a new mechanism underlying adiponectin-stimulated AMPK activation in muscle cells and shed light on potential targets for prevention and treatment of insulin resistance and its associated diseases.
Adiponectin exerts its antidiabetic and antiinflammatory functions partly by binding to its membrane receptors adiponectin receptor 1 and adiponectin receptor 2 (1, 2). Recent evidence indicated that skeletal muscle tissue is one of the primary target sites for adiponectin action (3). Our previous study showed that the binding of adiponectin promotes the recruitment of adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain and leucine zipper motif (APPL)1 to the receptors, which leads to stimulate downstream targets including the AMP-activated protein kinase (AMPK) and various biological events, such as glucose uptake and fatty acid oxidation in muscle cells (4, 5). In addition, we have found that adiponectin sensitizes insulin signaling by suppressing negative effect of p70 S6-kinase on insulin receptor substrate 1 serine phosphorylation (6), and APPL1 is essential for mediating the insulin sensitizer role of adiponectin (4). Accumulating evidence support the role of APPL1 in mediating adiponectin and insulin signaling in endothelial cells, adipocytes, HEK293 cells, zebrafish, as well as in mouse liver (7–12). Most recently, we showed that APPL1, together with its isoform APPL2, function as a “Yin-Yang” regulator of adiponectin signaling (13).
Several upstream kinases have been reported to activate AMPK in muscle cells, including liver kinase B (LKB)1 and Ca2+/calmodulin-dependent kinase kinase II (14–19). LKB1 is a constitutively active serine/threonine protein kinase that is predominately localized in the nucleus under normal physiological condition (20). By forming a heterotrimeric complex with Ste20-related adaptor protein (STRADα/β) and mouse protein 25 (MO25α/β) or associating with a LKB1 interacting protein, LKB1 is translocated to the cytosol, where it activates its substrates (20–26). It has been showed that LKB1 plays a critical role in adiponectin-induced activation of AMPK in muscle cells (22, 26). Our recent study revealed that adiponectin-stimulated AMPK activation in muscle cells is through two distinct mechanisms: APPL1-independent pathway stimulating Ca2+ release that activates Ca2+/calmodulin-dependent kinase kinase II and APPL1-dependent pathway that promotes LKB1 cytosolic translocation (26). APPL1 acts as an anchoring protein to tether LKB1 in cytosol in response to adiponectin stimulation, which leads to subsequent AMPK phosphorylation and activation (26). However, the underlying molecular mechanism by which APPL1 mediates adiponectin signal to stimulate LKB1 cytosolic translocation remains largely unknown.
Metformin is a widely used drug for the treatment of type 2 diabetes (27). Although studies have implicated AMPK activation as a mediator of metformin action, how metformin activates AMPK is poorly understood (28). One proposed mechanism is via inhibiting complex I activity of the respiratory chain and thereby increasing cellular AMP:ATP ratio and potentiating AMPK phosphorylation by the upstream kinase LKB1 (29, 30). Recent studies have shown that LKB1 is essential for metformin-stimulated AMPK activation in vivo, and LKB1 is translocated to cytosol in response to metformin stimulation (26, 31, 32). Despite its wide usage as an antidiabetic drug, the direct target of metformin is yet to be identified.
In the present study, we have demonstrated protein phosphatase 2A (PP2A) and protein kinase Cζ (PKCζ) as two important regulators of adiponectin signaling in C2C12 cells. In addition, we found that metformin-stimulated AMPK activation is independent of APPL1 and the PP2A-PKCζ pathway, suggesting a selective role of APPL1 in mediating adiponectin signaling. Our study reveals a novel molecular mechanism underlying APPL1-mediated adiponectin signaling in regulating LKB1-AMPK activation and function.
Results
Adiponectin stimulation leads to dephosphorylation of LKB1 at Ser307
We recently found that cytosolic localization of LKB1 is essential for adiponectin-stimulated AMPK phosphorylation, and APPL1 plays an important role in this process (26). To elucidate the underlying molecular mechanism, we tested whether phosphorylation plays a role in LKB1 cellular trafficking in response to adiponectin stimulation. In vivo labeling experiments in C2C12 myoblasts revealed that LKB1 is phosphorylated under basal conditions and adiponectin treatment resulted in a decrease of this phosphorylation in a time-dependent manner (Fig. 1A).
Fig. 1.
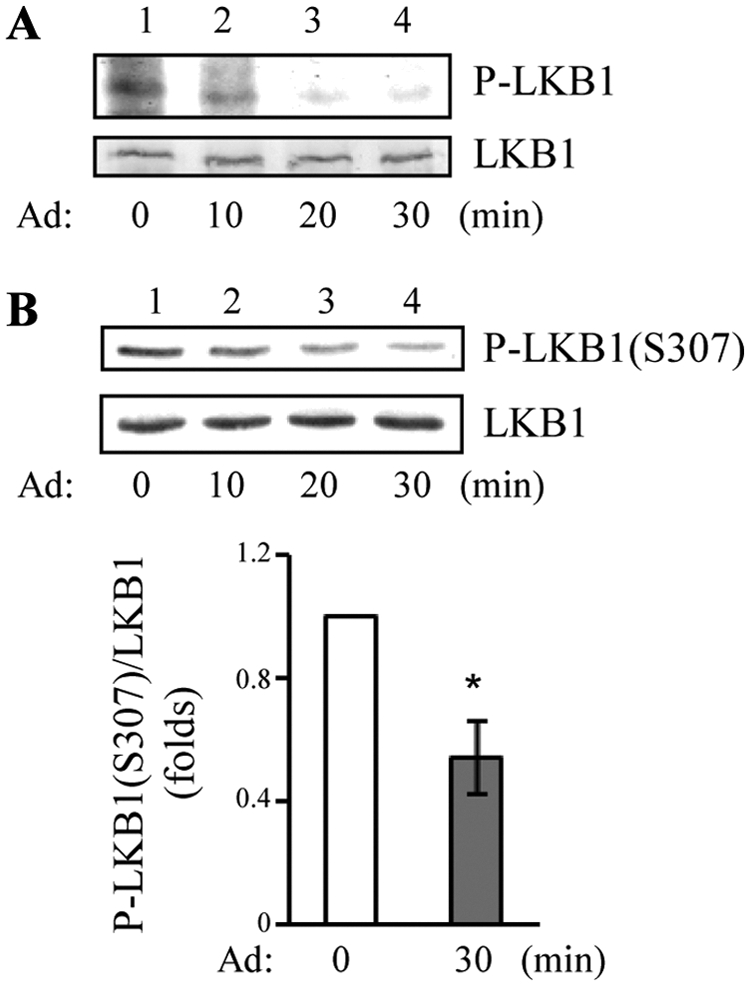
Adiponectin (Ad) induces dephosphorylation of LKB1 at Ser307. A, LKB1 undergoes dephosphorylation in response to adiponectin stimulation. C2C12 myoblasts transiently expressing myc-tagged LKB1 were serum starved, incubated with Krebs-Ringer bicarbonate buffer containing 0.5 mCi of 32P orthophosphate for 4 h, and then treated with or without adiponectin (1 μg/ml) for indicated times. LKB1 was immunoprecipitated with anti-myc monoclonal antibody (second panel), and autoradiography was performed to detect LKB1 phosphorylation (P-LKB1, top panel). B, Adiponectin induces dephosphorylation of LKB1 at Ser307 in cells. After serum starvation, C2C12 myotubes were treated with adiponectin (1 μg/ml) for indicated times. The phosphorylation of endogenous LKB1 at Ser307 and the protein levels were detected by Western blot analysis with antibodies specific to phospho-LKB1-Ser307 [P-LKB1 (S307), top panel] and LKB1 (second panel), respectively. The relative phosphorylation level of LKB1 was shown as graphic representation. The error bars represent mean ± sem from three independent experiments. *, P < 0.05.
By phosphopeptide mapping experiments, we found that LKB1 is phosphorylated exclusively on serine residue(s) in C2C12 myoblasts (Supplemental Fig. 1A, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). In addition, adiponectin treatment diminished serine phosphorylation of LKB1 (Supplemental Fig. 1A, right panel). Sequence analysis revealed that LKB1 contains a putative PKCζ phosphorylation site at Ser307 (Supplemental Fig. 1B), suggesting that PKCζ is a putative upstream kinase of LKB1. Consistent with this, PKCζ could phosphorylate LKB1 in vitro, and the phosphorylation was greatly suppressed in the S307A mutant of LKB1 (Supplemental Fig. 1C, top panel, lane 4 vs. lane 2), and two-dimensional phosphopeptide mapping showed that replacing Ser307 with Ala led to the loss of a major phosphopeptide in LKB1 (Supplemental Fig. 1D). During our study, Xie et al. (33) showed that PKCζ phosphorylated LKB1 at Ser307 under metformin stimulation, further demonstrating that Ser307 of LKB1 is a PKCζ-mediated phosphorylation site in vitro.
To determine whether phosphorylation of LKB1 at Ser307 is regulated by adiponectin, we examined LKB1 phosphorylation in C2C12 myotubes by Western blot analysis using a phospho-specific antibody to Ser307 of LKB1 (Supplemental Fig. 2A). Endogenous LKB1 is phosphorylated at Ser307 under basal conditions (Fig. 1B, top panel, lane 1). Adiponectin treatment attenuated LKB1 phosphorylation at Ser307 in a time-dependent manner (Fig. 1B, top panel, lanes 2–4 vs. lane 1), suggesting a negative regulatory role of adiponectin on LKB1 phosphorylation in cells.
Dephosphorylation of LKB1 at Ser307 promotes LKB1 cytosolic translocation
To determine whether phosphorylation at Ser307 regulates LKB1 subcellular localization, we generated LKB1 mutant, in which Ser307 is mutated to Ala (S307A). Our previous study has shown that adiponectin treatment stimulated cytoplasmic translocation of LKB1 in C2C12 cells at both endogenous and overexpression level (26). Consistent with this finding, adiponectin treatment resulted in a significant increase of LKB1 cytosolic localization in C2C12 myoblasts (Fig. 2A). The LKB1S307A mutant is largely localized in the cytosol at basal condition, and its subcellular localization pattern is very similar to that of the wild-type LKB1 in adiponectin-treated cells (Fig. 2A).
Fig. 2.

Dephosphorylation at Ser307 stimulates cytosolic localization of LKB1. A, Localization of S307A mutant of LKB1 in C2C12 cells. Confocal microscopy images depict the localization of myc-tagged wild-type (WT) and S307A mutant form of LKB1 overexpressed in C2C12 myoblasts treated with or without adiponectin (Ad) (1 μg/ml) for 20 min. The localization of LKB1 (green) was determined by an antibody to the myc-tag. The cell nuclei were stained with DAPI (blue). Scale bar, 20 μm. The cells with cytosolic LKB1 were counted, analyzed, and shown as graphic representation. The error bars represent mean ± sem from three independent experiments. **, P < 0.01 vs. wild-type LKB1 without adiponectin treatment. ns, Nonstatistical significance. B, The effect of S307A mutant of LKB1 on adiponectin-stimulated AMPK activation. C2C12 myoblasts overexpressing myc-tagged wild-type or S307A mutant of LKB1 were treated with or without adiponectin (1 μg/ml) for 20 min. The phosphorylation of AMPK (top panel), AMPK protein level (second panel), and myc-tagged LKB1 (third panel) were detected by Western blot analysis with specific antibodies as indicated. The relative phosphorylation level of AMPK was shown as graphic representation. The error bars represent mean ± sem from three independent experiments. *, **, ***: P < 0.05, P < 0.01, and P < 0.001, respectively, vs. mock transfection without adiponectin treatment. ##, P < 0.01 vs. LKB1 (WT) without adiponectin stimulation. ns, Nonsignificant difference in statistics.
To further elucidate the role of LKB1 phosphorylation in adiponectin-regulated downstream signaling, we examined the effect of S307A mutant of LKB1 on AMPK phosphorylation in C2C12 myoblasts. Overexpression of LKB1 increased basal AMPK phosphorylation that was further enhanced by adiponectin treatment (Fig. 2B, top panel, lanes 3 and 4 vs. lanes 1 and 2). Interestingly, overexpression of LKB1S307A mutant greatly enhanced the basal level of AMPK phosphorylation, which is not further stimulated by adiponectin (Fig. 2B, top panel, lanes 5 and 6 vs. lanes 1 and 2). Together, our data demonstrate that dephosphorylation at Ser307 plays a critical role in LKB1 cytosol translocation and adiponectin-stimulated AMPK activation.
Inactivation of PKCζ promotes LKB1 cytosolic localization and AMPK phosphorylation
Because S307A mutant of LKB1 is located predominantly in the cytosol (Fig. 2A), we investigated whether PKCζ, which is responsible for LKB1 phosphorylation at Ser307 (Supplemental Fig. 1C), plays a role in regulating LKB1 cellular localization. Immunofluorescence studies revealed that under basal conditions, LKB1 is mainly localized in the nucleus in cells coexpressing wild-type PKCζ (Fig. 3A). On the other hand, coexpression of the kinase-dead PKCζ(PKCζ-KD), which acts as a dominant negative mutant of this kinase (34), significantly increased the cytosolic localization of LKB1 under basal conditions. The PKCζ(KD)-induced cytosol localization of LKB1 was not further stimulated by adiponectin treatment (Fig. 3A). These data indicate that the activity of PKCζ positively regulates LKB1 nuclear localization. Consistent with this observation, overexpression of PKCζ-KD led to increase of basal AMPK phosphorylation (Fig. 3B, top panel, lane 5 vs. lane 1). The PKCζ(KD)-induced AMPK phosphorylation was not further enhanced by adiponectin treatment (Fig. 3B, top panel, lane 6 vs. lane 5).
Fig. 3.

Inactivation of PKCζ induces cytosolic translocation of LKB1 and subsequent AMPK activation. A, The kinase activity of PKCζ is inversely related to adiponectin (Ad)-induced LKB1 cytosolic translocation. Localization of hemagglutinin-tagged LKB1 coexpressed with myc-tagged wild type (WT) or kinase inactive mutant (KD) of PKCζ in C2C12 myoblasts with or without adiponectin treatment (1 μg/ml, 20 min) was determined by confocal microscopy images with specific antibodies to the tags as described in Materials and Methods. The cells with cytosolic LKB1 were counted, analyzed, and shown as graphic representation. The error bars represent mean ± sem from three independent experiments. *, P < 0.05 vs. wild-type LKB1 without adiponectin treatment. ns, Nonsignificant difference in statistics. B, The kinase inactive mutant of PKCζ increases basal AMPK phosphorylation. C2C12 myoblasts were transiently transfected with myc-tagged wild-type or kinase inactive mutant (KD) of PKCζ and treated with or without adiponectin (1 μg/ml, 20 min). The phosphorylation of AMPK (top panel), the protein levels of AMPK (second panel) and PKCζ (third panel), and β-tubulin loading control (fourth panel) were detected by Western blot analysis with specific antibodies as indicated. The relative phosphorylation level of AMPK was shown as graphic representation. The error bars represent mean ± sem from three independent experiments. ** and ***, P < 0.01 and P < 0.001, respectively, vs. mock transfection without adiponectin treatment. C, PS of PKCζ blocked adiponectin-induced LKB1 translocation. Localization of myc-tagged LKB1 in C2C12 myoblasts with or without PKCζ-PS pretreatment (10 μm, 1 h) and with or without adiponectin (1 μg/ml, 20 min) stimulation was determined by confocal microscopy images with specific antibodies to the tags as described in Materials and Methods. The cells with cytosolic LKB1 were counted, analyzed, and shown as graphic representation. The error bars represent mean ± sem from three independent experiments. **, P < 0.01 vs. PKCζ-PS negative control without adiponectin treatment. D, Inhibition of PKCζ using PKCζ-PS increases basal AMPK phosphorylation. C2C12 myotubes were pretreated with or without PKCζ-PS (10 μm, 1 h) and treated with or without adiponectin (1 μg/ml, 20 min). LKB1 phosphorylation at Ser307 (first panel), AMPK phosphorylation at Thr172 (third panel), and protein levels of LKB1 (second panel) and AMPK (fourth panel) were determined by Western blot analysis with specific antibodies as indicated. The relative phosphorylation levels of LKB1 and AMPK were shown as graphic representations. The error bars represent mean ± sem from three independent experiments. *, **, and ***, P < 0.05, P < 0.01, and P < 0.001, respectively, vs. PKCζ-PS negative control without adiponectin treatment. E, Suppression of endogenous PKCζ expression enhances basal AMPK phosphorylation. shRNA PKCζ-suppressed or the control C2C12 myotubes were treated with or without adiponectin (1 μg/ml) for 20 min. Thr172 phosphorylation of AMPK (first panel), protein levels of AMPK (second panel), phosphorylation of LKB1 at Ser307 (third panel), protein levels of LKB1 (fourth panel) and PKCζ (fifth panel), and tubulin loading control (bottom panel) were detected by Western blot analysis with specific antibodies as indicated.
To further confirm that the activity of PKCζ negatively regulates LKB1 cytosolic localization, we treated C2C12 cells with a specific inhibitor for PKCζ, PKCζ-pseudosubstrate (PS) (35). Inhibition of PKCζ activity with PKCζ-PS promoted LKB1 translocation from the nucleus to cytosol to a similar extent to that induced by adiponectin treatment (Fig. 3C). Consistent with this observation, inhibition of PKCζ activity by PKCζ-PS decreased basal LKB1 phosphorylation at Ser307 (Fig. 3D, top panel, lane 4 vs. lane 1) and increased basal AMPK phosphorylation (Fig. 3D, third panel, lane 4 vs. lane 1). The inverse correlation of PKCζ activity with LKB1 cytosolic localization was further proved by RNA interference (RNAi) approach. We found that suppression of PKCζ expression by RNAi decreased basal LKB1S307 phosphorylation (Fig. 3E, third panel, lane 3 vs. lane 1) that was further diminished by adiponectin treatment (Fig. 3E, third panel, lane 4 vs. lane 3). In addition, suppression of PKCζ expression greatly enhanced AMPK phosphorylation in C2C12 myotubes (Fig. 3E, top panel, lane 3 vs. lane 1). Together, these results demonstrate that down-regulation of PKCζ activity contributes to LKB1 cytosolic translocation and AMPK activation.
Adiponectin induces APPL1-dependent dephosphorylation of PKCζ
To determine whether PKCζ activity is regulated by adiponectin, we examined the effect of adiponectin on PKCζ phosphorylation at Thr410, an indicator of PKCζ activation in cells (36). Treatment of C2C12 myotubes with adiponectin decreased phosphorylation of endogenous PKCζ at Thr410 in a time-dependent manner (Fig. 4A, top panel). A similar effect of adiponectin on PKCζ phosphorylation was also observed when mouse primary skeletal muscle was treated with adiponectin ex vivo (Fig. 4B, top panel, lane 4 vs. lane 3). Because our previous study has shown that APPL1 plays an essential role in adiponectin-mediated AMPK activation (4, 26), we next tested the role of APPL1 in adiponectin-mediated PKCζ dephosphorylation. As shown in Fig. 4C, the inhibitory effect of adiponectin on PKCζ activity was greatly blocked in APPL1-suppressed C2C12 myotubes compared with the scrambled control cells (Fig. 4C, top panel, lanes 6–8 vs. lanes 2–4), suggesting a potential role of APPL1 in mediating adiponectin-induced down-regulation of PKCζ activity.
Fig. 4.

Adiponectin (Ad) induces inactivation of PKCζ with APPL1-dependent manner. A, The effect of adiponectin on PKCζ activity. After serum starvation, C2C12 myotubes were treated with adiponectin (1 μg/ml) for indicated times. Phosphorylation and protein levels of endogenous PKCζ were detected by Western blot analysis with antibodies specific to phospho-PKCζ-Thr410 (top panel) and PKCζ (second panel), respectively. The relative phosphorylation level of PKCζ was shown as graphic representation. The error bars represent mean ± sem from three independent experiments. ***, P < 0.001. B, Adiponectin induces inactivation of PKCζ in primary skeletal muscle tissue. Mouse skeletal muscle tissue was treated without or with adiponectin (2.5 μg/ml) for 30 min, and endogenous PKCζ was immunoprecipitated with PKCζ antibody. Phosphorylation of PKCζ at Thr410 (top panel) and protein expression levels of PKCζ (second and third panels) were determined by Western blot analysis with specific antibodies as indicated. The relative PKCζ phosphorylation was shown as graphic representation. The error bars represent mean ± sem from three independent experiments. **, P < 0.01. C, Adiponectin-induced inactivation of PKCζ is dependent on APPL1. C2C12 myotubes stably expressing APPL1 shRNA or the scrambled shRNA were serum starved and treated with adiponectin (1 μg/ml) for indicated times. Phosphorylation of PKCζ at Thr410 (top panel), protein expression levels of PKCζ (second panel) and APPL1 (third panel), and tubulin loading control (fourth panel) were determined by Western blot analysis with specific antibodies as indicated. The relative PKCζ phosphorylation was shown as graphic representation. The error bars represent mean ± sem from four independent experiments. *** and *, P < 0.001 and P < 0.05, respectively, vs. the scramble control without adiponectin treatment. D, APPL1 interacts with PKCζ via its C terminus in vitro. C2C12 myoblasts overexpressing myc-tagged PKCζ were serum starved and treated with adiponectin (1 μg/ml) as indicated. The myc-tagged PKCζ was pulled down by GST or GST-APPL1(CT) fusion proteins. The bound PKCζ (top panel) and PKCζ expression control (input, second panel) were detected with anti-myc antibody. E, PKCζ interacts with APPL1 in cells. C2C12 myotubes were serum starved and treated with adiponectin (1 μg/ml) for 15 min. Endogenous APPL1 was immunoprecipitated (IP) with negative control immunoglobulin (NIg) or an anti-APPL1 antibody. Immunoprecipitated APPL1 (second panel), coimmunoprecipitated PKCζ (top panel), and its Thr410 phosphorylation (third panel) were detected with the antibodies as indicated. The endogenous protein levels of PKCζ (forth panel) and APPL1 (fifth panel) in cell lysates were determined with specific antibodies as indicated. The relative APPL1 binding with PKCζ was shown as graphic representation. The error bars represent mean ± sem from three independent experiments. *, P < 0.05.
To understand the mechanism underlying APPL1-mediated PKCζ inactivation, we tested whether PKCζ could interact with APPL1. Affinity binding experiments demonstrated that PKCζ associates with APPL1 in vitro, and the C-terminal part of APPL1 is involved in this interaction (Supplemental Fig. 2B). The binding of PKCζ to the bacterially purified C terminus of APPL1 in vitro was not affected by adiponectin stimulation (Fig. 4D, top panel, lanes 4–5 vs. lane 3). On the other hand, the interaction between APPL1 and PKCζ in cells was stimulated by adiponectin (Fig. 4E, top panel, lane 4 vs. lane 3), suggesting that adiponectin-stimulated posttranslational modifications of APPL1 may regulate this interaction. It is also possible that another cellular component might be involved in regulating this interaction in cells.
PKCζ activity is down-regulated by PP2A in response to adiponectin stimulation
To understand the mechanism underlying adiponectin-induced and APPL1-dependent dephosphorylation of PKCζ, we asked whether PP2A, which has previously been shown to interact with PKCζ in adult cardiac myocytes (37), could contribute to adiponectin-induced inactivation of PKCζ. We found that PP2A can directly dephosphorylate PKCζ, as demonstrated by in vitro dephosphorylation assay (Fig. 5A, top panel). The effect of adiponectin on PP2A activity was also assessed by PP2A substrate assay. As shown in Fig. 5B, adiponectin treatment significantly increased PP2A activity in C2C12 myotubes. Interestingly, adiponectin-induced activation of PP2A (Fig. 5B) is coincident with a decrease in PKCζ phosphorylation in cells (Fig. 4, A and B). To investigate the direct involvement of PP2A in adiponectin-induced PKCζ inactivation, we tested the effect of PP2A activity on PKCζ phosphorylation by treatment of the cells with cantharidin, a compound that can specifically inhibit PP2A at lower concentrations (38, 39). Inhibition of PP2A resulted in an increase of PKCζ phosphorylation at basal level (Fig. 5C, top panel, lane 1 vs. lane 4). Adiponectin-induced down-regulation of PKCζ phosphorylation was significantly blocked in the cells pretreated with cantharidin (Fig. 5C, top panel, lanes 5–6 vs. lanes 2–3). Interestingly, the effect of cantharidin on PKCζ phosphorylation was correlated with LKB1 phosphorylation under both basal level and adiponectin treatment (Fig. 5C, top panel vs. third panel), demonstrating the involvement of PP2A-PKCζ-LKB1 pathway in mediating adiponectin signaling in muscle cells. To determine the mechanism by which adiponectin stimulates PP2A activity, we examined whether PP2A could interact with APPL1. As shown in Fig. 5D, endogenous APPL1 interacts with endogenous PP2A in C2C12 myotubes, and this interaction was stimulated by adiponectin (Fig. 5D, top panel, lane 4 vs. lane 3). The regulatory mechanism for adiponectin-mediated APPL1-PP2A interaction is currently unknown. It is possible that adiponectin-stimulated posttranslational modifications in APPL1 or PP2A could play a role in this regulation. In conclusion, adiponectin induces PKCζ dephosphorylation by promoting its interaction with PP2A, which is facilitated by adaptor protein APPL1.
Fig. 5.

PP2A inhibits PKCζ activity in response to adiponectin (Ad) stimulation. A, In vitro dephosphorylation assay. Recombinant PKCζ (50 ng) was incubated with 2 U of purified PP2A for 30 min at 30 C. After stopping the reaction, the phosphorylation at Thr410 (top panel) and the protein (bottom panel) levels of PKCζ were determined by Western blot analysis with specific antibodies as indicated. B, Adiponectin stimulates PP2A activity. C2C12 myotubes were treated with mock control or adiponectin (1 μg/ml) for different times as indicated. PP2A activity was measured as described in Materials and Methods. The PP2A activity in the cells treated with mock control was set as 100% (activity change is 0% as shown); the activity changes with adiponectin treatments were compared with the control. **, P < 0.01. C, Adiponectin-mediated dephosphorylation of LKB1 and PKCζ is inhibited by cantharidin. C2C12 myotubes were pretreated with or without cantharidin (1 μm, 2 h) and treated with or without adiponectin (1 μg/ml) for different times as indicated. PKCζ phosphorylation at Thr410 (first panel), LKB1 phosphorylation at Ser307 (third panel), and protein levels of PKCζ (second panel) and LKB1 (fourth panel) were determined by Western blot analysis with specific antibodies as indicated. The relative phosphorylation levels of PKCζ and LKB1 were shown as graphic representation. The error bars represent mean ± sem from three independent experiments. * and **, P < 0.05 and P < 0.01, respectively, vs. cantharidin treatment without adiponectin. ns, Nonsignificant difference in statistics. D, Adiponectin stimulates interaction of APPL1 with PP2A in cells. After serum starvation, C2C12 myotubes were treated with adiponectin (1 μg/ml) for 15 min. Endogenous APPL1 was immunoprecipitated with an antibody specific to APPL1. Immunoprecipitated APPL1 (second panel) and coimmunoprecipitated endogenous PP2A (top panel), protein expression level of APPL1 (bottom panel) and PP2A (third panel) in the cell lysates were detected with specific antibodies as indicated. The relative APPL1 binding was shown as graphic representation. The error bars represent mean ± sem from three independent experiments. **, P < 0.01.
Metformin-induced cytosolic translocation of LKB1 is independent of APPL1-PP2A-PKCζ pathway
We and others have previously shown that metformin also induces LKB1 cytosolic translocation in C2C12 cells (26, 31). To determine whether metformin and adiponectin share a common mechanism to regulate LKB1 subcellular localization, we investigated metformin-induced LKB1 localization in APPL1-suppressed cells. We found that suppression of APPL1 expression had no significant effect on metformin-induced AMPK phosphorylation in C2C12 myotubes compared with scrambled control cells (Fig. 6A, top panel, lanes 6–8 vs. lanes 2–4), indicating that APPL1 is not required for metformin-stimulated AMPK activation. In addition, metformin stimulation led to a reduction of LKB1 phosphorylation at Ser307 (Fig. 6A, third panel, lanes 2–4), which is similar to adiponectin-induced dephosphorylation and cytosolic translocation of LKB1 (Figs. 1B and 2A). Interestingly, there is no significant change of metformin-induced dephosphorylation of LKB1 in APPL1 RNAi myotubes compared with that in C2C12 scrambled control (Fig. 6A, third panel, lanes 6–8 vs. lanes 2–4), suggesting that metformin-regulated LKB1 dephosphorylation is independent of APPL1.
Fig. 6.

Metformin-induced subcellular translocation of LKB1 is independent of APPL1-PP2A-PKCζ pathway. A, Metformin-mediated AMPK phosphorylation and LKB1 dephosphorylation are independent of APPL1. C2C12 scrambled control and APPL1 RNAi myotubes were serum starved and treated with metformin (500 μm) for indicated times. Phosphorylation levels of AMPK at Thr172 (top panel), LKB1 at Ser307 (third panel), and protein expression levels of AMPK (second panel) and LKB1 (fourth panel) were detected by Western blot analysis with specific antibodies as indicated. The relative phosphorylation levels of AMPK and LKB1 are shown as graphic representation. The error bars represent mean ± sem from three independent experiments. ** and *, P < 0.01 and P < 0.05, respectively, vs. the scramble control without metformin treatment. #, P < 0.05 vs. the scrambled control without metformine treatment. ns, Nonsignificant difference in statistics. B, Metformin treatment has no effect on the phosphorylation levels of PKCζ and PP2A. C2C12 scrambled control and APPL1 RNAi myotubes were serum starved and treated with metformin (500 μm) for indicated times. Phosphorylation levels of PKCζ at Thr410 (top panel), PP2A-Cα/β at Tyr307 (third panel), and protein expression levels of PKCζ (second panel) and PP2A (fourth panel) were detected by Western blot analysis with specific antibodies as indicated. The relative phosphorylation level of PKCζ is shown as graphic representation. The error bars represent mean ± sem from three independent experiments. **, P < 0.01 vs. the scramble control without metformin treatment. C, Metformin has no effect on APPL1-PP2A interaction. C2C12 myotubes were serum starved and treated with metformin (500 μm) for indicated times. Endogenous APPL1 was immunoprecipitated (IP) with negative control immunoglobulin (NIg) or an antibody specific to APPL1. Immunoprecipitated APPL1 (second panel) and coimmunoprecipitated PP2A (first panel) and protein expression levels of APPL1 (bottom panel) and PP2A (third panel) in cell lysates were detected with specific antibodies as indicated. D, Metformin treatment in db/db mice increases AMPK and decreases LKB1 (Ser307) phosphorylation but has no effect on PKCζ and PP2A phosphorylation in the skeletal muscle. Phosphorylation levels of AMPK at Thr172, LKB1 at Ser307, PKCζ at Thr410, PP2A-Cα/β at Tyr307, and protein expression levels of AMPK, LKB1, PKCζ, and PP2A in the skeletal muscle tissue homogenate from db/db mice administered with vehicle (n = 6) or metformin (150 mg/kg) (n = 6) were detected by Western blotting analysis. The relative phosphorylation levels of AMPK, LKB1, PKCζ, and PP2A are shown as graphic representation. The error bars represent mean ± sem. *, P < 0.05.
To further understand the difference of metformin and adiponectin actions on AMPK activation, we tested whether metformin has any effect on the activities of PP2A and PKCζ. Our data indicated that metformin is unable to regulate phosphorylation of PP2A at Tyr307 (a marker of PP2A activity) (Fig. 6B, third panel) or PKCζ at Thr410 (Fig. 6B) (top panel) in either absence or presence of APPL1. In addition, PP2A substrate assays also indicated that metformin treatment has no effect on PP2A activity in C2C12 myotubes (Supplemental Fig. 3). Unlike adiponectin, metformin had no effect on the interaction between APPL1 and PP2A (Fig. 6C, top panel). Furthermore, we found that metformin did not stimulate PP2A and PKCζ activities in skeletal muscle of db/db mice, although it significantly increased AMPK phosphorylation at Thr172 and reduced LKB1 phosphorylation at Ser307 (Fig. 6D), which is associated with increased insulin sensitivity in the db/db mice (Supplemental Fig. 4). These in vivo data further demonstrate that adiponectin and metformin activate LKB1-AMPK signaling via distinct mechanisms.
Discussion
We have recently shown that LKB1 undergoes adiponectin-stimulated and APPL1-dependent cytosolic translocation, a key step for adiponectin-induced activation of AMPK in muscle cells (26). However, the molecular mechanism underlying APPL1-dependent LKB1 cytosolic translocation remains unknown. In the present study, we present evidence showing that PP2A-PKCζ pathway is a missing link that mediates APPL1-dependent LKB1 cytosolic translocation in response to adiponectin stimulation. Under basal condition, PP2A activity is suppressed, which leads to enhancement of PKCζ activity in cytosol and subsequent promotion of LKB1 nuclear localization by phosphorylating LKB1 at Ser307 (Fig. 7A). Adiponectin stimulation results in association of PP2A with APPL1, which induces activation of PP2A and deactivation of PKCζ, a PP2A substrate that also associates with APPL1 in cells. As a consequence of adiponectin-induced inactivation of PKCζ, LKB1 is accumulated in the cytosol due to less phosphorylation at Ser307 (Fig. 7B), which leads to more binding of LKB1 with APPL1 and activation of AMPK (26).
Fig. 7.

A model of adiponectin-induced subcellular translocation of LKB1 in muscle cells. A, Under basal conditions, PP2A is inactive and dissociated from APPL1, leading to activation of PKCζ in cytosol. The activated PKCζ phosphorylates LKB1 at Ser307, which promotes LKB1 transporting into the nuclei. B, Adiponectin stimulation induces recruitment of both PP2A and PKCζ onto APPL1, leading to activation of PP2A and subsequent dephosphorylation of PKCζ by PP2A. The inactivated PKCζ then results in less phosphorylation of LKB1 at Ser307 and an accumulation of LKB1 in the cytosol, which leads to the binding of LKB1 with APPL1 and activation of AMPK. A hypothetical nuclear phosphatase (X) may also contribute to LKB1 cytosolic translocation in response to adiponectin or metformin response. AdipoR, Adiponectin receptor.
We and others have demonstrated that adiponectin-mediated LKB1 cytosolic translocation is a common mechanism employed by different cell types to mediate AMPK phosphorylation (26, 40, 41). Association of LKB1 with MO25α and STRADα is one of the mechanisms that anchors LKB1 in cytoplasm to mediate AMPK phosphorylation (21, 42). Our previous study has demonstrated that adiponectin has no effect on LKB1-STRAD-MO25 complex formation, and APPL1 do not interact with either STRAD or MO25 in the presence or absence of adiponectin, indicating the presence of an unidentified mechanism for LKB1 translocation (26). Here, we showed for the first time that PKCζ-mediated phosphorylation of LKB1 at Ser307 plays a negative role in adiponectin-stimulated AMPK activation, probably by sequestering LKB1 in the nucleus. This finding is consistent with an early report that the Xenopus homolog of LKB1 (XEEK), which has an uncharged residue (Asn) corresponding to Ser307 in human and mouse LKB1 (Supplemental Fig. 1B), is primarily localized in the cytosol (43). Because AMPK is localized mainly in the cytosol (44), nuclear translocation of the AMPK upstream kinase LKB1 would thus provide a mechanism by which PKCζ negatively regulates AMPK activity. Consistent with this, activation of PKCζ by insulin has been shown to inhibit AMPK activity in heart and β-oxidation in L6 myotubes (45, 46). Recently, inhibitory role of PKCζ on AMPK phosphorylation in L6 myotubes was reported that further supports our findings (47). On the other hand, our result is contradictory to a recent finding by Xie et al. (33), who showed that PKCζ-mediated LKB1 phosphorylation at Ser307 is required for metformin-stimulated LKB1 cytosolic transfer in endothelial cells. The reason for this discrepancy remains unknown, but LKB1 cellular localization may be regulated via different mechanisms in different cells. Recently, another study showed that PKCζ is not involved in metformin-induced AMPK activation in H9c2 myocytes and C2C12 myotubes (48). In addition, study by Hawley et al. (49) demonstrated that metformin-mediated activation of AMPK is through inhibition of mitochondrial respiration in HEK293 cells. Taken together, our data demonstrate that AMPK activation mediated by adiponectin and metformin are through two distinct pathways converging at LKB1.
In this study, we have identified PP2A as a critical regulator in adiponectin-induced LKB1 cytosolic localization and subsequent AMPK phosphorylation and activation. PP2A does not directly dephosphorylate LKB1 at Ser307, which is likely mediated by a distinct phosphatase located in the nucleus (Fig. 7B). Instead, PP2A promotes adiponectin signaling by dephosphorylation and inactivation of PKCζ, which catalyzes LKB1 phosphorylation at Ser307. The mechanism of adiponectin-induced LKB1 dephosphorylation and its cytosolic transfer is not currently known and will be part of our future studies. One possibility is that APPL1/PP2A/PKCζ complex will be translocated into the nucleus to dephosphorylate LKB1 in response to adiponectin stimulation. It is also possible that adiponectin could activate a nuclear phosphatase that specifically targets LKB1.
Interestingly, we found that metformin, which also stimulates LKB1 cytosolic translocation and AMPK activation (26), activates AMPK via an APPL1-PP2A-PKCζ-independent mechanism. This conclusion is supported by the following observations. First, metformin and adiponectin induce LKB1 cytosolic translocation with different time courses (45 min vs. 20 min) (26). In addition, metformin treatment had no effect on the activities of PKCζ and PP2A in cells (Fig. 6B and Supplemental Fig. 3) and in vivo (Fig. 6D). Furthermore, suppressing APPL1 has little effect on metformin-induced dephosphorylation of LKB1 at Ser307 or activation of AMPK (Fig. 6A). Although how metformin promotes LKB1 cytosolic translocation remains unknown, one possibility may be that metformin may stimulate a nuclear phosphatase that directly dephosphorylates LKB1 at Ser307. Further studies will be needed to test this possibility.
In conclusion, we have provided evidence showing that PP2A and PKCζ are two novel signaling molecules involved in mediating adiponectin signaling from APPL1 to LKB1-AMPK pathway. In addition, we have demonstrated that the APPL1-PP2A-PKCζ pathway mediates adiponectin-induced dephosphorylation of LKB1 at Ser307, which plays a key role in regulating LKB1 translocation from the nucleus to cytosol. We also show that metformin-induced LKB1 cytosolic translocation and subsequent AMPK activation is mediated by a mechanism independent of APPL1-PP2A-PKCζ pathway. Our study reveals a novel molecular mechanism underlying adiponectin-induced AMPK activation and the role of APPL1 in this regulation. Because activation of AMPK is a key step in mediating the beneficial effects of adiponectin on fatty acid oxidation in muscle cells (27, 50–52), the intervention of the APPL1-PP2A-PKCζ pathway in the current study may provide critical information for designing therapeutic drug for the treatment of insulin resistance and obesity-related metabolic disorders.
Materials and Methods
Plasmids, adiponectin, chemicals, and antibodies
Plasmids encoding full-length human APPL1, various glutathione S-transferase (GST)-APPL1 fusion constructs [GST-APPL1, GST-APPL1(COOH-terminus) (amino acids 455–693), and GST-APPL1(NH2-terminus) (amino acids 1–270)], the globular form of adiponectin, and the anti-APPL1 antibody were described in our previous publications (4, 26, 53). Plasmids encoding mutant of mouse LKB1(S307A) or kinase inactive PKCζ (PKCζ-K273A) were generated by site-directed mutagenesis. Plasmids encoding amino acid 1–427 of LKB1 was cloned by PCR from a mouse cDNA library and subcloned into a mammalian expression vector pBEX1 (54), in-frame at its COOH terminus with a sequence encoding a hemagglutinin-tag, or the pcDNA 3.1 myc/His A plasmid (Invitrogen, Carlsbad, CA), in-frame at its COOH terminus with a sequence encoding a myc-tag. The phospho-specific antibody to Ser307 of LKB1 was raised in rabbits using phosphopeptide corresponding to mouse LKB1 299–314 (Ac-C-Ahx-IRQIRQH-pS307-WFRKKHP-amide) that was synthesized by 21st Century Biochemicals (Marlboro, MA). Monoclonal antibodies to β-tubulin 2.1 (Sigma, St. Louis, MO), myc-tag (produced in house with the myc 1–9E10.2 cell clone from American Type Culture Collection, Manassas, VA), LKB1 (Upstate-Millipore, Bedford, MA) and polyclonal antibody to phospho-PP2A-Cα/β (Tyr307) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were used in the experiments. PKCζ-PS was from Invitrogen and cantharidin was from Biomol International (Plymouth Meeting, PA). All other antibodies were obtained from Cell Signaling Technologies (Danvers, MA). Metformin was purchased from Sigma-Aldrich (St. Louis, MO).
Cell lines and cell culture
Conditions for culturing and differentiation of C2C12 cells, APPL1-suppressed C2C12 cells, and the scramble control C2C12 cells are described in our previous studies (4). PKCζ short hairpin RNA (shRNA) construct or the control cloned into pLKO.1 vector (catalog no. RMM3981-9590281; Open Biosystems, Auburn, AL) was stably expressed in C2C12 myocytes and selected using puromycin as described previously (4).
In vitro and ex vivo binding assays and coimmunoprecipitation experiments
Experimental procedures for in vitro binding and coimmunoprecipitation are essentially the same as described previously (55). Ex vivo incubation of skeletal muscle with adiponectin was performed as described previously (26).
In vitro kinase assay and phosphopeptide mapping of LKB1
Myc-tagged wild-type LKB1 or LKB1(S307A) mutant was transiently expressed in C2C12 myoblasts and immunoprecipitated with an anti-myc antibody. In vitro phosphorylation was initiated with the addition of 30 μl of kinase buffer [50 mm Tris-HCl (pH 7.5), 5 mm magnesium chloride, 1 mm sodium fluoride, 1 mm sodium orthovanadate, and 1 mm phenylmethanesulfonyl fluoride] containing 2 μCi of [γ-32P]ATP and 0.2 μg of recombinant human PKCζ (Upstate, Temecula, CA). After incubation for 30 min at 30 C, the reaction mixture was washed with ice-cold buffer containing 50 mm HEPES (pH 7.4), 150 mm NaCl, and 0.1% Triton X-100. Phosphorylated LKB1 was separated by SDS-PAGE and visualized by autoradiography. 32P-labeled LKB1 or LKB1(S307A) was trypsinized and examined by two-dimensional thin layer chromatography as described previously (56).
Western blot, immunofluorescence studies, and statistical analyses
Expression and phosphorylation levels of proteins were detected by Western blot analysis of cell lysates or immunoprecipitation with specific antibodies. Quantification of the relative increase in protein phosphorylation (expressed as percentage of basal phosphorylation) was performed using Scion Image Alpha 4.0.3.2 program (Scion Corp., Frederick, MD) and normalized for the amount of protein expression in each experiment. For all Western blot quantifications, “folds” represent the ratio of phosphorylated to nonphoshorylated protein. Percentage of cytosolic LKB1 is calculated as P = C/(N + C), where N represents cells in which LKB1 is localized predominantly in the nucleus, and C represents cells with predominantly cytosolic LKB1. Statistical evaluation of the data was done using one-way ANOVA. * or #, P < 0.05; ** or ##, P < 0.01; ***, P < 0.001.
PP2A activity assays
The PP2A activity assay kit was purchased from Millipore (Bedford, MA), and the reaction was performed according to the manufacturer's instruction (Millipore). In brief, PP2A catalytic subunit was immunoprecipitated from C2C12 myotubes lysate (500 μg of total protein) with 4 μg of anti-PP2A C-subunit antibody. The immunoprecipitated PP2A C-subunit was incubated with 750 μm phosphopeptide (K-R-pT-I-R-R) and Ser/Thr Assay buffer at 30 C for 10 min. The supernatant (25 μl) was mixed with 100 μl of Malachite Green phosphate detection solution provided by the manufactory and kept at room temperature for 10 min. The absorbance at 650 nm was measured. The same assay solution without PP2A incubation was used as a negative control.
In vitro dephosphorylation assays
Recombinant PKCζ (0.05 μg) was incubated in phosphatase buffer [50 mm Tris-HCl (pH 7.0), and 100 μm CaCl2] with indicated units of purified PP2A AC dimmer (subunit A and C mixed together and formed functional PP2A) (Upstate) for 30 min at 30 C. The reactions were terminated by adding sodium dodecyl sulfate gel loading buffer and run on 10% sodium dodecyl sulfate-polyacrylamide gels. The protein and phosphorylation levels of PKCζ were detected by Western blot analysis with specific antibody as indicated.
In vivo metformin studies
Thirty db/db mice (11 wk old) were fed chow diet (diet 5008; Ralston Purina, St. Louis, MO) for 2 wk and then grouped to match blood glucose across treatments before dosing. The animals received either vehicle (n = 10), metformin150 mpk (n = 10), or metformin500 mpk (n = 10) by oral gavageonce daily for three consecutive days. The mice were fasted overnight on the third day, and fasted baseline blood glucose was taken by tail vein blood glucometer readings next day morning. Animals then received a fourth dose of their respective treatment and blood glucose measured every hour thereafter for 4 h. After 4 h, animals were scarified by cervical dislocation, skeletal muscle was removed and frozen immediately in liquid nitrogen. From the frozen tissues, protein extraction was done as described previously (26). Animal protocol was approved by the Merck Research Laboratory Animal Care and Use Committee.
Acknowledgments
We thank Derong Hu for her excellent technical assistance and Dr. Victoria Frohlich (Associate Director, Digital Optical Imaging Facility, University of Texas Health Science Centre at San Antonio) for assistance in confocal microscopy studies.
This work was supported by National Institute of Health Grants R01 DK69930, R01 DK080344, and 3R01DK080344-01A2S1 (to L.Q.D.), AG030979 and DK80157 (to N.M.), and R01 DK76902 (to F.L.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AMPK
- AMP-activated protein kinase
- APPL
- adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain and leucine zipper motif
- GST
- glutathione S-transferase
- LKB
- liver kinase B
- MO25α/β
- mouse protein 25
- PKCζ
- protein kinase Cζ
- PP2A
- protein phosphatase 2A
- PS
- pseudosubstrate
- RNAi
- RNA interference
- shRNA
- short hairpin RNA
- STRADα/β
- Ste20-related adaptor protein.
References
- 1. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. 1995. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749 [DOI] [PubMed] [Google Scholar]
- 2. Kadowaki T, Yamauchi T. 2005. Adiponectin and adiponectin receptors. Endocr Rev 26:439–451 [DOI] [PubMed] [Google Scholar]
- 3. Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. 2010. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 464:1313–1319 [DOI] [PubMed] [Google Scholar]
- 4. Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, Liu F, Dong LQ. 2006. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 8:516–523 [DOI] [PubMed] [Google Scholar]
- 5. Deepa SS, Dong LQ. 2009. APPL1: role in adiponectin signaling and beyond. Am J Physiol Endocrinol Metab 296:E22–E36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang C, Mao X, Wang L, Liu M, Wetzel MD, Guan KL, Dong LQ, Liu F. 2007. Adiponectin sensitizes insulin signaling by reducing p70 S6 kinase-mediated serine phosphorylation of IRS-1. J Biol Chem 282:7991–7996 [DOI] [PubMed] [Google Scholar]
- 7. Chandrasekar B, Boylston WH, Venkatachalam K, Webster NJ, Prabhu SD, Valente AJ. 2008. Adiponectin blocks interleukin-18-mediated endothelial cell death via APPL1-dependent AMP-activated protein kinase (AMPK) activation and IKK/NF-κB/PTEN suppression. J Biol Chem 283:24889–24898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng KK, Lam KS, Wang Y, Huang Y, Carling D, Wu D, Wong C, Xu A. 2007. Adiponectin-induced endothelial nitric oxide synthase activation and nitric oxide production are mediated by APPL1 in endothelial cells. Diabetes 56:1387–1394 [DOI] [PubMed] [Google Scholar]
- 9. Cheng KK, Iglesias MA, Lam KS, Wang Y, Sweeney G, Zhu W, Vanhoutte PM, Kraegen EW, Xu A. 2009. APPL1 potentiates insulin-mediated inhibition of hepatic glucose production and alleviates diabetes via Akt activation in mice. Cell Metab 9:417–427 [DOI] [PubMed] [Google Scholar]
- 10. Lee MH, Klein RL, El-Shewy HM, Luttrell DK, Luttrell LM. 2008. The adiponectin receptors AdipoR1 and AdipoR2 activate ERK1/2 through a Src/Ras-dependent pathway and stimulate cell growth. Biochemistry 47:11682–11692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saito T, Jones CC, Huang S, Czech MP, Pilch PF. 2007. The interaction of Akt with APPL1 is required for insulin-stimulated Glut4 translocation. J Biol Chem 282:32280–32287 [DOI] [PubMed] [Google Scholar]
- 12. Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, Habermann B, Brand M, Zerial M. 2008. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 133:486–497 [DOI] [PubMed] [Google Scholar]
- 13. Wang C, Xin X, Xiang R, Ramos FJ, Liu M, Lee HJ, Chen H, Mao X, Kikani CK, Liu F, Dong LQ. 2009. Yin-Yang regulation of adiponectin signaling by APPL isoforms in muscle cells. J Biol Chem 284:31608–31615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. 2005. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J 24:1810–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, Hardie DG. 1995. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem 270:27186–27191 [DOI] [PubMed] [Google Scholar]
- 16. Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. 2007. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403:139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. 2005. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2:9–19 [DOI] [PubMed] [Google Scholar]
- 18. Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. 2005. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 280:29060–29066 [DOI] [PubMed] [Google Scholar]
- 19. Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. 2005. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2:21–33 [DOI] [PubMed] [Google Scholar]
- 20. Smith DP, Spicer J, Smith A, Swift S, Ashworth A. 1999. The mouse Peutz-Jeghers syndrome gene Lkb1 encodes a nuclear protein kinase. Hum Mol Genet 8:1479–1485 [DOI] [PubMed] [Google Scholar]
- 21. Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR. 2003. MO25α/β interact with STRADα/β enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J 22:5102–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Imai K, Inukai K, Ikegami Y, Awata T, Katayama S. 2006. LKB1, an upstream AMPK kinase, regulates glucose and lipid metabolism in cultured liver and muscle cells. Biochem Biophys Res Commun 351:595–601 [DOI] [PubMed] [Google Scholar]
- 23. Smith DP, Rayter SI, Niederlander C, Spicer J, Jones CM, Ashworth A. 2001. LIP1, a cytoplasmic protein functionally linked to the Peutz-Jeghers syndrome kinase LKB1. Hum Mol Genet 10:2869–2877 [DOI] [PubMed] [Google Scholar]
- 24. Tiainen M, Ylikorkala A, Mäkelä TP. 1999. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA 96:9248–9251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tiainen M, Vaahtomeri K, Ylikorkala A, Mäkelä TP. 2002. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1). Hum Mol Genet 11:1497–1504 [DOI] [PubMed] [Google Scholar]
- 26. Zhou L, Deepa SS, Etzler JC, Ryu J, Mao X, Fang Q, Liu DD, Torres JM, Jia W, Lechleiter JD, Liu F, Dong LQ. 2009. Adiponectin activates AMP-activated protein kinase in muscle cells via APPL1/LKB1-dependent and phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathways. J Biol Chem 284:22426–22435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saenz A, Fernandez-Esteban I, Mataix A, Ausejo M, Roque M, Moher D. 2005. Metformin monotherapy for type 2 diabetes mellitus. Cochrane Database Syst Rev:CD002966. [DOI] [PubMed] [Google Scholar]
- 28. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. 2001. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. 2000. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275:223–228 [DOI] [PubMed] [Google Scholar]
- 30. Owen MR, Doran E, Halestrap AP. 2000. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348(Pt 3):607–614 [PMC free article] [PubMed] [Google Scholar]
- 31. Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. 2008. Phosphorylation of LKB1 at serine 428 by protein kinase C-ζ is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 117:952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. 2005. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xie Z, Dong Y, Zhang J, Scholz R, Neumann D, Zou MH. 2009. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol Cell Biol 29:3582–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Diaz-Meco MT, Berra E, Municio MM, Sanz L, Lozano J, Dominguez I, Diaz-Golpe V, Lain de Lera MT, Alcamí J, Payá CV. 1993. A dominant negative protein kinase C ζ subspecies blocks NF-κ B activation. Mol Cell Biol 13:4770–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Standaert ML, Bandyopadhyay G, Sajan MP, Cong L, Quon MJ, Farese RV. 1999. Okadaic acid activates atypical protein kinase C (ζ/λ) in rat and 3T3/L1 adipocytes. An apparent requirement for activation of Glut4 translocation and glucose transport. J Biol Chem 274:14074–14078 [DOI] [PubMed] [Google Scholar]
- 36. Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. 1998. Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr Biol 8:1069–1077 [DOI] [PubMed] [Google Scholar]
- 37. Wu SC, Solaro RJ. 2007. Protein kinase C ζ. A novel regulator of both phosphorylation and de-phosphorylation of cardiac sarcomeric proteins. J Biol Chem 282:30691–30698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Honkanen RE. 1993. Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett 330:283–286 [DOI] [PubMed] [Google Scholar]
- 39. Kim MH, Shim KS, Kim SH. 2010. Inhibitory effect of cantharidin on osteoclast differentiation and bone resorption. Arch Pharm Res 33:457–462 [DOI] [PubMed] [Google Scholar]
- 40. Fang X, Palanivel R, Cresser J, Schram K, Ganguly R, Thong FS, Tuinei J, Xu A, Abel ED, Sweeney G. 2010. An APPL1-AMPK signaling axis mediates beneficial metabolic effects of adiponectin in the heart. Am J Physiol Endocrinol Metab 299:E721–E729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taliaferro-Smith L, Nagalingam A, Zhong D, Zhou W, Saxena NK, Sharma D. 2009. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene 28:2621–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boudeau J, Scott JW, Resta N, Deak M, Kieloch A, Komander D, Hardie DG, Prescott AR, van Aalten DM, Alessi DR. 2004. Analysis of the LKB1-STRAD-MO25 complex. J Cell Sci 117:6365–6375 [DOI] [PubMed] [Google Scholar]
- 43. Su JY, Erikson E, Maller JL. 1996. Cloning and characterization of a novel serine/threonine protein kinase expressed in early Xenopus embryos. J Biol Chem 271:14430–14437 [DOI] [PubMed] [Google Scholar]
- 44. Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. 2007. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor α gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the α2 form of AMP-activated protein kinase. Mol Cell Biol 27:4317–4327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bandyopadhyay G, Standaert ML, Galloway L, Moscat J, Farese RV. 1997. Evidence for involvement of protein kinase C (PKC)-ζ and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubes. Endocrinology 138:4721–4731 [DOI] [PubMed] [Google Scholar]
- 46. Gamble J, Lopaschuk GD. 1997. Insulin inhibition of 5′ adenosine monophosphate-activated protein kinase in the heart results in activation of acetyl coenzyme A carboxylase and inhibition of fatty acid oxidation. Metabolism 46:1270–1274 [DOI] [PubMed] [Google Scholar]
- 47. Wang L, Balas B, Christ-Roberts CY, Kim RY, Ramos FJ, Kikani CK, Li C, Deng C, Reyna S, Musi N, Dong LQ, DeFronzo RA, Liu F. 2007. Peripheral disruption of the Grb10 gene enhances insulin signaling and sensitivity in vivo. Mol Cell Biol 27:6497–6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ussher JR, Jaswal JS, Wagg CS, Armstrong HE, Lopaschuk DG, Keung W, Lopaschuk GD. 2009. Role of the atypical protein kinase Cζ in regulation of 5′-AMP-activated protein kinase in cardiac and skeletal muscle. Am J Physiol 297:E349–E357 [DOI] [PubMed] [Google Scholar]
- 49. Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, Hardie DG. 2010. Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11:554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. 2002. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8:1288–1295 [DOI] [PubMed] [Google Scholar]
- 51. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. 2003. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423:762–769 [DOI] [PubMed] [Google Scholar]
- 52. Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, Kim JB. 2006. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor α. Diabetes 55:2562–2570 [DOI] [PubMed] [Google Scholar]
- 53. Dong LQ, Zhang RB, Langlais P, He H, Clark M, Zhu L, Liu F. 1999. Primary structure, tissue distribution, and expression of mouse phosphoinositide-dependent protein kinase-1, a protein kinase that phosphorylates and activates protein kinase Cζ. J Biol Chem 274:8117–8122 [DOI] [PubMed] [Google Scholar]
- 54. Bram RJ, Hung DT, Martin PK, Schreiber SL, Crabtree GR. 1993. Identification of the immunophilins capable of mediating inhibition of signal transduction by cyclosporin A and FK506: roles of calcineurin binding and cellular location. Mol Cell Biol 13:4760–4769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wick MJ, Wick KR, Chen H, He H, Dong LQ, Quon MJ, Liu F. 2002. Substitution of the autophosphorylation site Thr516 with a negatively charged residue confers constitutive activity to mouse 3-phosphoinositide-dependent protein kinase-1 in cells. J Biol Chem 277:16632–16638 [DOI] [PubMed] [Google Scholar]
- 56. Liu F, Roth RA. 1994. Identification of serines-967/968 in the juxtamembrane region of the insulin receptor as insulin-stimulated phosphorylation sites. Biochem J 298(Pt 2):471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]