

Abstract
This Letter describes the synthesis and SAR, developed through an iterative analogue library approach, of an mGluR4 positive allosteric modulator lead based on a pyrazolo[3,4-d]pyrimidine scaffold. Despite tremendous therapeutic potential, Compound 7, VU0080421, and related congeners represent only a handful of mGluR4 positive allosteric modulators ever described.
Glutamate is the major excitatory neurotransmitter in the central nervous system, exerting its effects through both ionotropic and metabotropic glutamate receptors. The metabotropic glutamate receptors (mGluRs) are members of the GPCR family C, characterized by a large extracellular amino-terminal agonist binding domain. To date, eight mGluRs have been cloned, sequenced and assigned to three groups (Group I: mGluR1 and mGluR5; Group II: mGluR2 and mGluR3; Group III: mGluRs 4,6,7,8) based on their sequence homology, pharmacology and coupling to effector mechanisms.1 The Group III mGluRs are the least explored and characterized of the mGluRs, but despite this fact, mGluR4 has garnered a great deal of attention as a therapeutic target for multiple indications.2
The reason for the slower pace of development within Group III mGluRs concerns the availability of ligands.2,3 Most pharmacological studies employ prototypical Group III agonists such as L-(+)-2-amino-4-phosphonobutryic acid, L-AP4, 1 or functionalized carboxyphenylglycines 2, which have limited CNS penetration (Fig. 1).4 A major breakthrough in the field occurred when Maj and co-workers reported on the discovery of (−)-PHCCC 3, the first mGluR4 positive allosteric modulator (PAM), derived from the mGluR1 negative allosteric modulator (NAM) (−)-CPCCOEt 4.5 (−)-PHCCC possesses an EC50 of 4.1 µM, with a 5.5-fold leftward shift of the glutamate response curve and selectivity versus mGluRs 2, 3, 5, 6, 7, 8.5,6 However, (−)-PHCCC is a partial antagonist (30%) of mGluR1.
Figure 1.

Chemical structures of orthosteric mGluR4 agonists L-AP4 (1), functionalized phenylglycines (2) and the mGluR4 PAM (−)-PHCCC (3) which was derived from the mGluR1 NAM (−)-CPCCOEt (4).
SAR for (−)-PHCCC is very ‘flat’, with virtually any chemical modifications resulting in a complete loss of mGluR4 PAM activity (Fig. 2), a finding common with several series of mGluR PAMs.7–10 Despite this, (−)-PHCCC has been a very important proof of concept (POC) compound demonstrating a therapeutic role for selective mGluR4 activation in Parkinson’s disease,6,11 anxiety,12 depression,13 neuroprotection14 and oncology.15
Figure 2.

Chemical modifications and the resulting ‘flat’ SAR for (−)-PHCCC.
In all of these pioneering POC experiments, PHCCC was either administered through intracerebroventricular injection (icv) or by employing toxic 50% DMSO vehicles which disrupt the blood-brain barrier, as PHCCC possesses poor physiochemical properties and limited brain penetration.6,11–15 In order to advance this field, new mGluR4 PAMs are required with improved efficacy, physiochemical properties and novel molecular architectures. In this Letter, we describe the discovery and SAR of a novel mGluR4 PAM, based on a pyrazolo[3,4-b]pyrimidine scaffold, derived from an HTS campaign.
Our mGluR4 PAM HTS identified three pyrazolo[3,4-d]pyrimidines that afforded a concentration-dependent potentiation of an EC20 of glutamate in mGluR4/Gqi5 CHO cells (Fig. 3).7 When HTS stocks were evaluated with full concentration-response curves, 7, 1-(2,4-diphenyl)-4-(3-methylpiperdin-1-yl)-1H-pyrazolo[3,4-d]pyrimidine, was a stand-out compound with an EC50 for potentiation of 4.6 µM while having no effect on mGluR4 in the absence of glutamate (Fig. 4).16
Figure 3.

Concentration-dependent potentiation of glutamate in mGluR4/Gqi5 CHO cells by pyrazolo[3,4-d]pyrimidine compounds 5, 6 and 7 in the high throughput screening campaign (HTS).
Figure 4.

Compound 7, potentiates mGluR4 activation by glutamate. In the absence of glutamate, 7 does not activate mGluR4. In the presence of an EC20 concentration of glutamate, 7 caused a concentration-dependent potentiation of mGluR4 with an EC50 for potentiation of 4.6 µM, equivalent to PHCCC, EC50 ~ 4.1 µM.5,6 Data represents the average of at least three independent determinations.
Additionally, 7 was superior to PHCCC in terms of fold-shift of the glutamate concentration-response curve. As shown in Figure 5, PHCCC induces a 5.5-fold leftward shift of the glutamate response curve with an elevation in the glutamate max, whereas 7 elicits a 27.2(±8.5)-fold shift with no increase in the glutamate max. Compound 7 was selective versus mGluR5, but was a full antagonist of mGluR1 (IC50 = 2.6 µM). This finding was surprising since PHCCC was derived from a series of mGluR1 antagonists (the (−)-CPCCOEt 4 series), it was not surprising that 3 was a 30% partial antagonist of mGluR1, but 7 is structurally unrelated. It is possible that PHCCC and 7 share a common allosteric binding site on mGluR4 that is also conserved in mGluR1, but in the absence of radioligands for mGluR4, this issue will have to be addressed at a later time.
Figure 5.

Both PHCCC (3) and 7 shift the glutamate agonist response curves to the left 5.5- and 27.2 (±8.5)-fold, respectively at 30 µM (EC50 shifts from 7.5 ± 1.6 µM to 317 ± 100 nM). Interestingly, PHCCC increases maximal response whereas 7 affords no increase in the glutamate max, akin to other mGluR5 PAMs reported from our labs.7
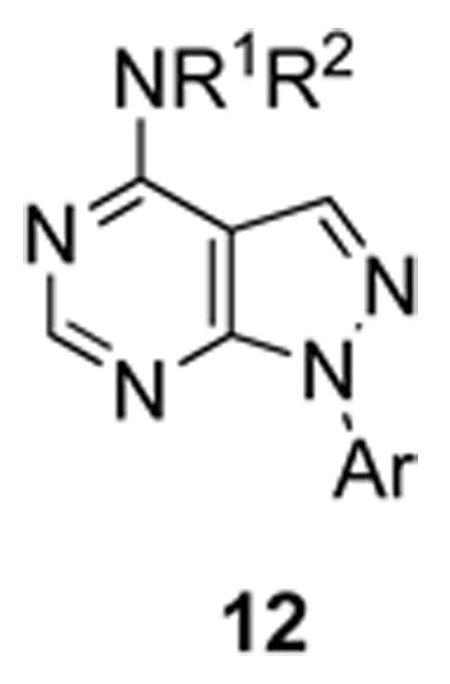
With a bona fide novel mGluR4 PAM lead in hand, we employed an iterative analogue library synthesis approach to rapidly evaluate SAR for 7. Despite HTS stocks of 7 confirming structure and purity, re-synthesis of 7 (VU0080241) resulted in a compound with slightly lower potency and a less robust fold shift (EC50 ~ 5 uM, fold shift 11.8±3.3, Table 1); however, VU0080241 still represents an improvement over PHCCC. In light of the ‘flat’ SAR observed by ourselves and others in the field with PHCCC,5–7 we initially synthesized multidimensional diversity libraries to determine the scope and breadth of the SAR. However, classical synthetic approaches to pyrazolo[3,4-d]pyrimidines yields were typically moderate (<50%) with prolonged reaction times at high temperatures (multiple steps requiring >48 h at reflux).17 As recently reported, we have developed a rapid, high yielding and general microwave-assisted approach to access diverse pyrazolo[3,4-d]pyrimidines, and this expedited protocol was employed to produce analogues of VU0080421 (Scheme 1).18 The synthesis began by reacting 2-(ethoxymethylene)malononitrile 8 with seven arylhydrazines under microwave irradiation to deliver 5-amino-1-aryl-1H-pyrazole-4-carbonitriles 9. The nitrile analogues 9 were then hydrolyzed with H2SO4 to produce the corresponding 5-amino-1-aryl-1H-pyrazole-4-carboxamides 10. A microwave-assisted condensation employing carboxamides 10 in neat formamide smoothly afforded the corresponding analogous 1-aryl-1H-pyrazolo[3,4-d]pyrimidines 11. MAOS conditions (POCl3, DMF, 120 °C, 45 min, mw) were used for the conversion to the chloro congeners, which were then subjected to a microwave-assisted SNAr reaction to provide the final analogues 12 based on HTS lead 7, VU0080421.19 In short order, over 126 analogues 12 (7 arylhydrazines × 18 amines) were prepared and evaluated as mGluR4 PAMs (Fig. 6).
Table 1.
Glutamate fold shifts by VU0080421 analogues 12.
 | |||
|---|---|---|---|
| Compound | NR1R2 | Ar | Fold shifta |
| (−)-PHCCC, 3 | 5.5 | ||
|
7 VU0080241 |
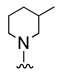 |
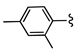 |
27.2 11.8 |
| 12a | 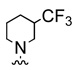 |
9.9 | |
| 12b | 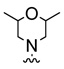 |
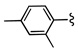 |
3.5 |
| 12c | 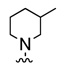 |
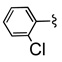 |
2.7 |
| 12d | 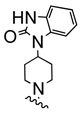 |
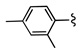 |
2.4 |
Fold shifts represent the average of at least three independent experiments performed in quadruplicate at 30 µM.
Scheme 1.

Reagents and conditions: (a) ArNHNH2, EtOH, mw, 105 °C, 10 min, 54–77%; (b) i) H2SO4, 0°C, ii) H2NCHO, mw, 200 °C, 20 min, 48–91%; (c) i) POCl3, DMF, mw, 120 °C, 45 min, ii) HNR1R2, DMF, PS-DIEA, mw, 90 °C, 20 min, 84–99%.18,19
Figure 6.

Monomers employed in the diversity library of pyrazolo[3,4-d]pyrimidines 12 (7 arylhydrazines × 18 amines) based on HTS lead 7, VU0080421. Monomers in boxes indicate that they produced analogues 12 that potentiated an EC20 of glutamate in mGluR4/Gqi5 CHO cells.
As in the case of PHCCC, analogues 12 of VU0080241 were uniformly inactive on mGluR4 with only the parent HTS lead 7, the re-synthesized VU0080421 and four other analogues (12a–12d) showing any activity as mGluR4 PAMs. SAR for this series was ‘flat’ with only a 3.9% active rate. With one exception (12c, containing a 2-chlorophenyl group), the 2,4-dimethylphenyl moiety (6, 7, 12a, 12b, 12d) was required for activity, and little diversity was tolerated with respect to the nature of the NR1R2 moiety. As shown in Table 1, analogues 12 lost efficacy, EC50s ≥ 10 µM, but provided robust leftward shifts of the glutamate response curve 2.4- to 9.9-fold. One explanation for the ‘flat’ SAR is that the allosteric binding sites which VU0080421 and (−)-PHCCC occupy are very shallow, similar to the second, non-MPEP, allosteric binding site on mGluR5 that CPPHA occupies.8,9 In addition, in vitro DMPK studies identified stability issues with VU0080241. Importantly, VU0080241 was found to be unstable in fortified liver microsome preparations, with only 9% of the parent compound remaining after 90 minutes.
Despite the disappointing SAR and microsomal instability, VU0080421 (7) represents a significant advance in the mGluR4 PAM field. VU0080421 (7) possesses a large 11.8- to 27.2-fold shift, the largest we have observed for an mGluR PAM, and it does not contain the oxime or amide NH moieties that are speculated to contribute to the observed lack of brain penetration for PHCCC in vehicles other than DMSO. Moreover, VU0080421 represents a novel chemotype for a PAM of mGluRs and is one of only a handful of reported PAMs of mGluR4. Further refinements to VU0080241 and related series of mGluR4 PAMs are in progress and will be reported in due course.
Acknowledgement
The authors thank C. David Weaver, Emily L. Days, Tasha Nalywajko, Cheryl A. Austin and Michael Baxter Williams for their critical contributions to the HTS portion of the project as well as the National Institute of Mental Health, the Michael J. Fox Foundation, the Vanderbilt Department of Pharmacology and the Vanderbilt Institute of Chemical Biology for support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.a) Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]; b) Conn PJ, Pin J-P. Annu. Rev. Pharmacol. Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2.a) Marino MJ, Hess JF, Liverton N. Curr. Top. Med. Chem. 2005;5:885–895. doi: 10.2174/1568026054750263. [DOI] [PubMed] [Google Scholar]; b) Marino MJ, Conn PJ. Curr. Opin. Pharm. 2006;8:98–102. doi: 10.1016/j.coph.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 3.a) Yang Z-Q. Curr. Top. Med. Chem. 2005;5:913–918. doi: 10.2174/1568026054750272. [DOI] [PubMed] [Google Scholar]; b) Niswender CM, Jones JK, Conn PJ. Curr. Top. Med. Chem. 2005;5:847–857. doi: 10.2174/1568026054750254. [DOI] [PubMed] [Google Scholar]
- 4.a) Thomsen C, Kristensen P, Mulvihill E, Haldeman B, Suzdak PD. Eur. J. Pharmacol. 1992;227:361–362. doi: 10.1016/0922-4106(92)90018-q. [DOI] [PubMed] [Google Scholar]; b) Gasparini F, Bruno V, Battaglia G, Lukic S, Leonhardt T, Inderbitzen W, Laurie D, Sommer B, Varney MA, Hess SD, Johnson EC, Kuhn R, Urwyler S, Sauer D, Poret C, Schmutz M, Nicoletti F, Flor PJ. J. Pharmacol. Exp. Ther. 1999;289:1678–1687. [PubMed] [Google Scholar]
- 5.Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, Stoehr N, Stein T, Gasparini F, Vranesic I, Kuhn R, Nicoletti F, Flor PJ. Neuropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 6.Marino MJ, Willaim DL, Jr, O’Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG, Conn PJ. PNAS. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, Rodriguez AL, Marlo JE, de Paulis T, Thompson AD, Days EL, Nalywajko T, Austin CA, Williams MB, Ayala JE, Williams R, Lindsley CW, Conn PJ. Mol Pharmacol. doi: 10.1124/mol.108.049551. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Z, Wisnoski DD, O’Brien JA, Lemiare W, Williams DL, Jr, Jacobson MA, Wittman M, Ha S, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg. Med. Chem.Lett. 2007;17:1386–1391. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 9.O’Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Sur C, Duggan ME, Pettibone DJ, Conn J, Williams DL. J. Pharmacol. Exp. Therapeut. 2004;309(2):568–577. doi: 10.1124/jpet.103.061747. [DOI] [PubMed] [Google Scholar]
- 10.Lindsley CW, Wisnoski DD, Leister WH, O’Brien JA, Lemiare W, Williams DL, Jr, Burno M, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. J. Med. Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 11.Battaglia G, Busceti CL, Molinaro G, Biagioni F, Traficante A, Nicoletti F, Bruno V. J. Neurosci. 2006;26:7222–7229. doi: 10.1523/JNEUROSCI.1595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stachowicz K, Klak K, Klodzinska A, Chojnacka-Wojcik E, Pilc A. Eur. J. Pharmacol. 2004;498:153–156. doi: 10.1016/j.ejphar.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Klak K, Palucha A, Branski P, Pilc A. Amino Acids. 2007;32:169–172. doi: 10.1007/s00726-006-0316-z. [DOI] [PubMed] [Google Scholar]
- 14.Canudas AM, Di Giorgi-Gerevini V, Iacovelli L, Nano G, D'Onofrio M, Arcella A, Giangaspero F, Busceti C, Ricci-Vitiani L, Battaglia G, Nicoletti F, Melchiorri D. J. Neurosci. 2004;24:10343–10352. doi: 10.1523/JNEUROSCI.3229-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iacovelli L, Arcella A, Battaglia G, Pazzaglia S, Aronica E, Spinsanti P, Caruso A, De Smaele E, Saran A, Gulino A, D'Onofrio M, Giangaspero F, Nicoletti F. J. Neurosci. 2006;26:8388–8397. doi: 10.1523/JNEUROSCI.2285-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Assay details: Cell culture-human mGluR4/ Gqi5/CHO line. Human mGluR4 (hmGluR4)/CHO cells were stably transfected with the chimeric G protein Gqi5 and single clones were selected via hygromycin resistance and screened for mGluR4-mediated calcium mobilization using the method described below. hmGluR4/CHO cells were cultured in 90% Dulbecco’s Modified Eagle Media (DMEM), 10% fetal bovine serum (FBS), 100 units/ml penicillin/streptomycin, 20 mM HEPES (pH 7.3), 1 mM sodium pyruvate, 2 mM glutamine, 400 µg/ml G418 (Mediatech, Inc. Herndon, VA) and 5 nM methotrexate (Calbiochem, EMD Chemicals, Gibbstown, NJ). Culturing conditions for other mGluR cell lines are described below. All cell culture reagents were purchased from Gibco/Invitrogen (Carlsbad, CA). Calcium fluorescence assay. Primary high throughput screening details were described in 7. Briefly, human mGluR4/Gqi5/CHO cells (30,000 cells/20 µl/well) were plated in black-walled, clear-bottomed 384 well plates (Greiner Bio-One, Monroe, NC) in DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 100 units/ml penicillin/streptomycin (Assay Media). The cells were grown overnight at 37 °C in the presence of 5% CO2. The next day, media was removed and the cells were incubated with 20 µL of 1 µM Fluo-4AM (Invitrogen, Carlsbad, CA) prepared as a stock in DMSO and mixed in a 1:1 ratio with pluronic acid F-127 and diluted in Assay Buffer (Hank’ Balanced Salt Solution, 20 mM HEPES and 2.5 mM Probenecid (Sigma-Aldrich, St. Louis, MO) for 45 minutes at 37 °C. Dye was removed and 20 µL of Assay Buffer was added. Test compounds were diluted into Assay Buffer to generate a 20 µM stock. Ca2+ flux was measured using the Functional Drug Screening System 6000 (FDSS6000, Hamamatsu, Japan). Appropriate baseline readings were taken (10 images at 1 Hz, excitation, 470±20 nm emission, 540±30 nm) and test compounds were added at a final concentration 10 µM.) Confirmation/ Selectivity studies for other mGluRs. Rat mGluRs 1 and 5. Compounds were assessed using 10 point concentration response curves starting at a 30 µM final concentration and diluted by 1:3. For fold shift experiments, glutamate concentration-response curves were performed in the presence of a 30 µM final concentration of compound. Calcium fluorescence assays were employed for counterscreening rat mGluR1/Baby Hamster Kidney (mGluR1/BHK) and rat mGluR5/HEK cells using a similar triple-addition protocol employing appropriate EC20 and EC80 glutamate concentration for each receptor, the exceptions being that cells were plated at 20,000 cells/well and 15,000 cells/well in Assay Media, respectively. Maximum calcium fluorescence, compared to control, was calculated for the EC20 and EC80 peaks, respectively, after exporting raw plate data to Microsoft Excel.
- 17.a) Katsuhiko N, Kawano H, Sasaoka S, Ukawa C, Hirama T, Takada A, Cottam HB, Robins RK. J. Het. Chem. 1994;31:239–243. [Google Scholar]; b) Tiberghien N, Lumley J, Reynolds K, Angell RM, Matthews N, Cockerill GS, Barnes MC. WO 047288. 2005 [Google Scholar]
- 18.Daniels RN, Kim K, Lewis JA, Lebois EP, Muchalski H, Lindsley CW. Tetrahedron Lett. 2008;49:305–310. [Google Scholar]
- 19.Experimental for the Synthesis of VU0080241, 7: 2,4-Dimethylhydrazine hydrochloride was partitioned between 2M sodium hydroxide solution and dichloromethane. The organic layer was separated, dried and reduced under vacuum to give the free hydrazine (1.90 g, 14 mmol). The free hydrazine and ethoxymethylenemalononitrile 8 (1.70 g, 14 mmol) in ethanol were irradiated at 105°C for 10 min by microwave. The crude product was recrystallized from ethanol to yield a yellow solid product 9 (2.30g, 77%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.59 (s, 1 H), 7.17 (s, 1 H), 7.13 (s, 1 H) 7.12 (s, 1 H), 4.47 (s, 2 H), 2.38 (s, 3 H), 2.07 (s, 3 H); 13C NMR (CDCl3, 100 MHz) δ (ppm) 150.51, 140.93, 140.59, 136.20, 132.27, 132.20, 127.85, 127.28, 114.34, 74.35, 21.13, 17.13; LCMS, single peak, 2.57 min, m/e, 213.95 (M+1); Compound 9 (4.6g, 21.7mmol) was then treated with concentrated H2SO4 (30ml) at 0°C. The reaction mixture was stirred at room temperature for 1 hour then quenched with ice. The solution was neutralized with aqueous NH4OH and filtered to provide yellow solid product (4.77g, 95%); A suspension of 5-amino-1-(2,4-dimethylphenyl)-1H-pyrazole-4-carboxamide 10 (2.30 g, 10 mmol) in formamide was irradiated at 200°C for 20min by microwave. The cooled solution was diluted with water. The product was filtered, washed with water and dried over in vacuo to afford a gray solid 1-(3,5-dimethylphenyl)-1H-pyrazolo[ 3,4-d]pyrimidin-4-ol 11 (2.18g, 91%). 1H NMR (DMSO, 400 MHz) δ (ppm) 8.27 (s, 1 H), 8.03 (s, 1 H), 7.23 (d, J = 7.6 Hz, 2 H), 7.23 (d, J = 7.6 Hz, 1 H), 2.35 (s, 3 H), 1.99 (s, 3 H); 13C NMR (CDCl3, 100 MHz) δ (ppm) 157.75, 153.10, 148.85, 139.28, 135.85, 135.02, 134.39, 131.73, 127.91, 127.40, 106.42, 21.07, 17.74; LCMS, single peak, 2.36 min, m/e, 242.96 (M+1); A suspension of 1-(2,4-dimethylphenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-ol 11 (2.36 g, 9.82 mmol) and POCl3 (3ml) in dichloroethane (7 mL) was irradiated at 120 °C for 45 min by microwave. The solvent was removed in vacuo to yield gray solid product. To a solution of 4-chloro-1-(3,5-dimethylphenyl)-1H-pyrazolo[3,4-d]pyrimidine (2.32 g, 9 mmol) and 3-methylpiperidine (3.76 g, 27 mmol) in DMF (24 mL) was added triethylamine (3.16 mL, 27 mmol) at room temperature. The reaction mixture was irradiated in microwave at 90 °C for 15min. The cooled solution was treated with water to provide yellow solid VU 0080241 (7), 1-(2,4-dimethylphenyl)-4-(3-methylpiperidin-1-yl)-1H-pyrazolo[ 3,4-d]pyrimidine (2.67 g, 84%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 8.35 (s, 1 H), 8.12 (s, 1 H), 7.24 (d, J = 8.0 Hz, 1 H), 7.17 (s, 1 H), 7.13 (d, J = 8.0 Hz, 1 H), 4.66 (s, 2 H), 3.21 (t, J = 11.6, Hz, 1 H), 2.86 (t, J = 11.6, Hz, 1 H), 2.38 (s, 3 H), 2.17 (s, 3 H), 1.98-1.60 (m, 4 H), 1.35-1.24 (m, 1 H), 1.03 (d, J = 6.4 Hz, 3 H); 13C NMR (CDCl3, 100 MHz) δ (ppm) 155.83, 155.20,153.82, 140.04, 135.09, 133.61, 133.00, 132.08, 127.50, 127.22, 113.81, 33.06, 31.26, 25.18, 21.21, 21.18, 19.12, 17.88; LCMS, single peak, 2.87 min, m/e, 322.12 (M+1)