

Abstract
A variety of molecular modeling, molecular docking, and first-principles electronic structure calculations were performed to study how the α4β2 nicotinic acetylcholine receptor (nAChR) binds with different species of two typical agonists, S-(−)-nicotine and R-(−)-deschloroepibatidine, each of which are distinguished by different free bases and protonation states. Based on these results, predictions were made regarding the corresponding microscopic binding free energies. Hydrogen bonding and cation-π interactions between the receptor and the respective ligands were found to be the dominant factors differentiating the binding strengths of different microscopic binding species. The calculated results and analyses demonstrate that for each agonist, all the species are interchangeable and can quickly achieve a thermodynamic equilibration in solution and at the nAChR binding site. This allows quantitation of the equilibrium concentration distributions of the free ligand species and the corresponding microscopic ligand-receptor binding species, their pH-dependence, and their contributions to the phenomenological binding affinity. The predicted equilibrium concentration distributions, pKa values, absolute phenomenological binding affinities and their pH-dependence are all in good agreement with available experimental data, suggesting that the computational strategy from the microscopic binding species and affinities to the phenomenological binding affinity is reliable for studying α4β2 nAChR-ligand binding. This should provide valuable information for future rational design of drugs targeting nAChRs. The general strategy of the “from-microscopic-to-phenomenological” approach for studying interactions of α4β2 nAChR with S-(−)-nicotine and R-(−)-deschloroepibatidine may also be useful in studying other types of ligand-protein interactions involving multiple molecular species of a ligand and in associated rational drug design.
Introduction
The alkaloid nicotine, initially found in tobacco leaves, is the addictive compound that maintains tobacco smoking behavior. 1, 2, 3 More than any other abused psychostimulant, nicotine addiction is the number one cause of preventable mortality and is responsible for over 4 million smoking-related deaths each year.4,5 Nicotine produces its effects on the central nervous system (CNS) by interacting with nicotinic acetylcholine receptors (nAChRs) that are essential for synaptic transmission. Neuronal nAChRs are members of a superfamily of ligand-gated ion channels, which modulate the function of many major neurotransmitter systems and thereby influence a broad range of brain functions, such as cognition, learning and memory.5, 6, 7, 8, 9 Selective nAChR agonists/antagonists have therapeutic potential in the treatment of Alzheimer’s disease, Parkinson’s disease, dyskinesias, Tourette’s syndrome, schizophrenia, attention deficit disorder, anxiety, and pain, as well as tobacco-use cessation agents.5,9,10,11,12,13,14,15,16,17
Each nAChR structure consists of five protein subunits which transverse the neuronal cell membrane. A total of 12 different subunits (α2 to α10 and β2 to β4) have been identified for neuronal nAChRs, and all of the nAChR protein subunits characterized to date possess a high degree of sequence homology.18 Homo-oligomeric nAChRs can be formed by α7, α8, or α9 subunits, but only α7 is widely distributed in the mammalian CNS. Most nAChR subtypes are composed of two or more different types of subunits, combinations of α and β subunits, the most common subtype being α4β2* which accounts for over 90% of the high-affinity nicotine binding sites in brain.19 Activation of α4*-containing nAChRs is sufficient for nicotine-induced reward, tolerance, and sensitization effects,4 and the β2 subunit has been identified immunocytochemically a a in all dopaminergic neurons in the ventral tegmental area in midbrain. Thus, α4β2 nAChRs have been recognized as a major target mediating the pathology of several kinds of CNS pathologies and diseases4,5,9 and a variety of α4β2 agonists and antagonists have been discovered. Therefore, it is particularly important to understand how the α4β2 nAChR interacts with nicotine and other ligands (i.e. agonists and/or antagonists).
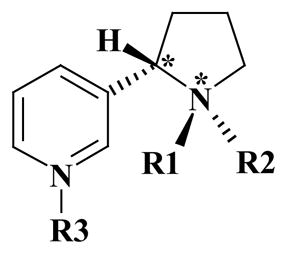
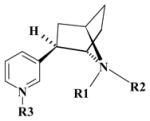
On the other hand, a great challenge is to achieve a full understanding of the binding of ligand molecules to nAChRs. Other than acetylcholine, most nAChR agonists, including nicotine, and nAChR antagonists contain a protonatable amine moieties (see Table 1). In many cases, such a functional group allows for the co-existence of multiple species of the ligand in solution and, possibly, at the nAChR binding site through the dynamic interchange and equilibration between the different protonated forms of the ligand (see below). Herein, different molecular species of a given ligand refer to either different protonation states or different diastereomeric species that might be interchangeable in solution. 20 For example, S-(−)-nicotine has a total of six stable molecular species and R-(−)-deschloroepibatidine has a total of four stable molecular species in solution, as shown in Table 1.
Table 1.
Nomenclature of the different molecular species for S-(−)-nicotine and R-(−)-deschloroepibatidine and available experimental pKa values.
 | ||||||
|---|---|---|---|---|---|---|
| Name | Config. | R1 | R2 | R3 | Charge (e) | pKa (ref. 1) |
| SR | (S, R) | CH3 | Lone pair | Lone pair | 0 | |
| SRH | (S, R) | CH3 | H | Lone pair | 1.0 | ~8.0 |
| SRHH | (S, R) | CH3 | H | H | 2.0 | ~3.0 |
| SS | (S, S) | Lone pair | CH3 | Lone pair | 0 | |
| SSH | (S, S) | H | CH3 | Lone pair | 1.0 | |
| SSHH | (S, S) | H | CH3 | H | 2.0 | |
| ||||||
| DCEH | R | H | H | Lone pair | 1.0 | |
| DCEH2 | R | H | H | H | 2.0 | |
| DCEa | R | H | Lone pair | Lone pair | 0.0 | |
| DCEb | R | Lone pair | H | Lone pair | 0.0 | |
To understand the binding between a receptor and a ligand, first the 3D structure of the receptor and especially the topography of the ligand binding domain (LBD) needs to be known. Although the latest refined structure of the Torpedo (α)2(βγδ) nAChR at 4A˚ resolution has provided fundamental insights into the mechanisms of channel function, it is basically a modeled structure of the resting state of the receptor.21 The binding site in this structure is distorted by inter- and intra-subunit interactions, and thus, is not suitable for studying binding with ligands. Nevertheless, the reported X-ray crystal structure of the homologous acetylcholine-binding protein (AChBP)22, 23 can serve as a suitable template for homology modeling of the LBD of the nAChR. The first X-ray crystal structure of the AChBP22 incorporates an N-2-hydroxyethylpiperazine-N-9-(2-ethanesulphonic acid) (HEPES) molecule at its binding site. The HEPES-bound AChBP was considered to approximate the ligand-free state of this protein, but the protein conformation is different from the latest X-ray crystal structure of the nicotine-bound complex.23 Earlier 3D models of α4β2 nAChRs built on the HEPES-bound AChBP structure did not consider such conformational rearrangements.24,25 Thus, the detailed atomic contacts between nicotine and the receptor are not correct.23 Currently, the nicotine-bound AChBP is the most satisfactory and appropriate template to model the LBD of nAChRs for the purpose of studying ligand-receptor binding. The combined use of this latest X-ray crystal structure of the AChBP complex and the computational chemistry approach enables a reasonable 3D LBD structure of α4β2 nAChR to be built.
Once a 3D LBD structure of the α4β2 nAChR is built, the important questions to be answered will include the following: how does each possible molecular species (with a given protonation state of each ligand) of the S-(−)-nicotine ligand bind with the receptor, and are the six molecular species (i.e. the different combinations of free base and protonation states) of S-(−)-nicotine all interchangeable in solution and/or at the nAChR binding site? The answers to these questions would also suggest that different molecular species of other ligands may or may not be interchangeable in solution at the nAChR binding site. Other important questions include: 1) What is the dominant molecular species for a given ligand binding with the nAChR, when more than one species of the ligand can bind? 2) Can the binding free energies for all the possible molecular species of a ligand binding with the nAChR be determined accurately? 3) Can the distributions of the possible molecular species of a ligand in solution and at the nAChR binding site be determined? 4) Is the dominant molecular species of a ligand in its binding with the nAChR necessarily also the dominant molecular species of the ligand in solution? 5) Is the distribution of molecular species of a ligand at the nAChR binding site dependent on the pH of the solution? 6) Can a pH change alter the dominant molecular species of a ligand at the nAChR binding site? In this respect, the experimentally measurable ligand-receptor binding affinity, called the “phenomenological binding affinity” herein for convenience, could include significant contributions from multiple microscopic binding species. However, such experimental data usually do not provide information about the microscopic binding species. Thus, a further question to be answered is: Is it possible to develop a general strategy to evaluate the phenomenological binding affinity of a ligand with a receptor by quantitatively accounting for the multiple microscopic ligand-receptor binding species?
The present study aims at developing and testing a reliable computational strategy by answering the fundamental questions elaborated above. For this purpose, the 3D structure of the LBD of the human α4β2 nAChR was modeled, focusing on its binding with two representative agonists, S-(−)-nicotine, which has a phenomenological binding affinity of Kd = 1.0 to 2.3 nM,10e,10f and R-(−)-deschloroepibatidine, which has a higher phenomenological binding affinity of Kd = 0.020±0.001 nM.10b All of the six molecular species of S-(−)-nicotine and the four molecular species of R-(−)-deschloroepibatidine were docked into the binding site of the α4β2 nAChR and the microscopic binding free energies were calculated. Meanwhile, reaction coordinate calculations, based on first-principles electronic structure theory, were carried out to examine a key transition state involved in the interchange between the different molecular species of S-(−)-nicotine. The calculated activation free energy and the corresponding rate constant reveal how fast the interchange between the different molecular species occurs in solution. The first-principles electronic structure calculations accounting for solvent effects were also performed to determine the relative Gibbs free energies of the molecular species of these ligands in solution. Further statistical analyses of all the energetic results obtained lead to quantitative predictions of the distributions of all the molecular species in solution and at the nAChR binding site. These results enable us to quantitatively link the microscopic binding affinities with the phenomenological binding affinity of a given ligand binding with the receptor. These quantitative predictions will aid in the better understanding the phenomenological binding affinities of the S-(−)-nicotine and R-(−)-deschloroepibatidine with α4β2 nAChRs and will help predict how the distributions of the microscopic binding species and the phenomenological binding affinities are dependent on the pH of the environment. The calculated results are in good agreement with available experimental observations.1,5,6,8,9,22,23 New insights into the microscopic and phenomenological binding and the general computational strategy developed in this study provide a basis to guide future rational design of more potent and selective nAChR agonists/antagonists for α4β2 and for other nAChR subtypes.
Computational methods
Homology modeling for the LBD domain of α4β2 nAChR
In order to study S-(−)-nicotine and R-(−)-deschloroepibatidine binding with the α4β2 nAChR in atomic detail, a homology model of the LBD of human α4β2 nAChR was built on the latest X-ray crystal structure of the AChBP (PDB entry of 1UW6 at 2.2 Å resolution)23 by using the Homology module of InsightII (version 2000, Accelrys, Inc., San Diego, CA). AChBP is not an ion channel (it is a soluble protein that lacks the transmembrane/intracellular parts compared to nAChRs), but importantly displays many nAChR properties, including binding of nAChR ligands and a conformational change in response to agonist binding Interestingly, the highest percentage of identity (26.5%) has been found with the ligand binding domain of the α7 neuronal nAChR subtype. The identity percentage increases dramatically when considering only the loops forming the agonist binding pocket (40 – 60%), as expected from the functional homology. The sequence alignment was generated by ClusterW with the Blosum scoring function.26,27 The sequence identity is 22% for 4 subunit and 20% for β 2 subunit, showing the medium homology with the template. Several recent studies by an array of advanced techniques, such as solid-state NMR, Lysine scanning, fluorescence measurements, and theoretical simulations, have also demonstrated structural determinants of agonist binding for AChBP, Torpedo nAChR, and human muscle nAChR, and provided undisputable structural basis for modeling of other members of the nAChR superfamily.28,29,30,31 All the five subunits were modeled simultaneously in order to maintain the complementarity between these subunits at the interface. The best alignment was selected according to both the alignment score and the reciprocal positions of the conserved residues, especially those in or close to the nicotine-binding sites of the template.23 These include the conserved GSWT sequence (in which Trp147 of the α4 subunit corresponds to Trp143 of the AChBP) and the C loop (containing Cys191 and Cys192) between the β-strands 9 and 10 of the α4 subunit, whereas there is a gap of three residues at this loop in the β2 subunit. The coordinates of the conserved regions of each subunit were transformed directly from the template structure (i.e. the X-ray crystal structure of AChBP23), whereas the non-equivalent residues were mutated from the template to the corresponding ones in the α4β2 nAChR. The side chains of these non-conserved residues were relaxed by using the Homology module of InsightII program, in order to remove possible steric overlap (or hinderance) with the neighboring conserved residues. In composing the whole LBD domain of the (α4β2α4β2β2) nAChR, the structural arrangement at the two interfaces between each α4 and β2 subunits, particularly at the binding sites, was kept as similar as possible to that of the template, whereas small adjustments were made for the interfaces that are not involved in the binding sites.
Ionizable residues that are not in the binding site were set to the standard protonated or deprotonated states. A careful check of the structural model allowed the proton be assigned to the Nδ1 atoms for His59, His102, His107, and His160 of the α4 subunits and for His8, His44, His84, and His134 of the β2 subunits under the physiological conditions (pH = ~7.4). For the constructed structural model, the protonation states of ionizable residues may also be titrated by solving Poisson-Boltzmann equation as did in previous electrostatic calculations.32 However, the possible binding pocket of α4β2 nAChR (as described later) does not involve any usually ionizable residue except Tyr; the standard pKa of Tyr is 9.6.32,33 When we only consider the pH 5.0 to 9.0, a Tyr residue is usually expected to be unionized. The initial 3D model was energy-minimized by using the Sander module of the Amber 7 program suite34 with a non-bonded cutoff of 10 Å and a conjugate gradient minimization method. The energy minimization was performed first for 1000 steps with the backbone atoms fixed, while the side chain atoms were relaxed in the gas phase, and then for another 600 steps with the side chain atoms constrained in order to relax the backbone. After each of these stages was finished, the whole receptor was visually checked to make sure that there was no any significant distortion during the energy minimization processes. After 3 rounds of these partial energy-minimization runs, the convergence criterion of 0.001 kcal mol−1 Å−1 was quickly achieved in a full energy minimization. We carefully checked the fully energy-minimized structure to make sure that the overall structure of the final model was not significantly different from that of the initial model and the template. Finally, the modeled structure was validated by using PROCHECK and WHATIF programs.35,36
First-principles electronic structure methods and pKa calculations
All geometries of the S-(−)-nicotine and R-(−)-deschloroepibatidine species involved in this study were fully optimized by employing density functional theory (DFT) using Becke’s three parameter hybrid exchange functional and the Lee-Yang-Parr correlation functional37 (B3LYP) with the 6-31+G(d) basis set. Vibrational frequency calculations were carried out to confirm the optimized stable molecular structures and transition state, and to perform zero-point vibration thermal corrections to the Gibbs free energies. Intrinsic reaction coordinate (IRC)38 calculations were performed to verify the expected connection of the first-order saddle point with the two local minima found on the potential energy surface. The geometries optimized at the B3LYP/6-31+G(d) level were used to carry out second-order Møller-Plesset (MP2) single-point energy calculations with the 6-31+G(d) basis set. All these electronic structure calculations in the gas phase were performed by using the Gaussian03 program.39
Self-consistent reaction field (SCRF) calculations were performed to calculate solvent shifts of the Gibbs free energies by using the geometries optimized at the B3LYP/6-31+G(d) level in the gas phase. The free energy of a molecular species in aqueous solution was taken as a sum of the free energy calculated at the MP2/6-31+G(d)//B3LYP/6-31+G(d) level in the gas phase and the corresponding solvent shift determined by the SCRF calculation at the HF/6-31+G(d) level. The SCRF method used in the calculations is our recently developed GAMESS implementation40 of the surface and volume polarization for electrostatic interactions (SVPE).41 The SVPE model is also known as the fully polarizable continuum model (FPCM),42,43 because it fully accounts for both surface and volume polarization effects in the SCRF calculation. In other SCRF implementations, volume polarization effects are ignored or approximately modeled by modifying the surface polarization charge distribution through a simulation and/or charge renormalization,44,45,46,47,48,49,50,51,52 or the solute charge distribution is simply represented by a set of point charges at the solute nuclei.53,54
Since the solute cavity surface is defined as a solute electron charge isodensity contour determined self-consistently during the SVPE iteration process, the SVPE results, converged to the exact solution of Poisson’s equation with a given numerical tolerance, depend only on the contour value at a given dielectric constant and a certain quantum chemical calculation level.41a This single parameter value has been determined to be 0.001 au based on an extensive calibration study.41b Accordingly, the default 0.001 au contour was used in this study.
Concerning the calculation of absolute pKa characterizing the thermodynamic equilibration between the different protonation states of the ligand(s), there are two kinds of protonated states for S-(−)-nicotine and R-(−)-deschloroepibatidine. One state is the protonation of the nitrogen atom (N1) of the saturated azaheterocyclic ring (the first protonation state), and the other state is the protonation of nitrogen atom (N2) in the pyridine ring (the second protonation state). The pKa is determined by the free energy change (ΔGa) of the protonation process:
| (1) |
| (2) |
in which L represents a neutral structure of ligand. LH+ and LHH2+ refer to the first and second protonation states, respectively. Prediction of the free energy change of a protonation process requires knowing the absolute free energy of the proton (H+) in aqueous solution, ΔGhyd298(H+), in addition to the free energies calculated for all of the molecular species mentioned above. Due to the inherent difficulty of measuring absolute solvation free energy of an ion, the reported “experimental” ΔGhyd298(H+) values have a wide range from −252.6 to −264.1 kcal/mol.55 We recently calculated ΔGhyd298(H+) by using a high-level, ab initio method of incorporating a hybrid supermolecule-continuum approach 56, 57, 58, 59 based on the same SVPE procedure used in the present study. ΔGhyd298(H+) was predicted to be −262.4 kcal/mol.56 Accordingly, this ΔGhyd298(H+) value was used in the present study, and we have:
| (3) |
The first-principles electronic structure approach described above has been used previously to solve a variety of chemical and biochemical problems in solution, and the predicted activation free energies for chemical reactions, pKa values, and thermodynamic properties, etc. are all in good agreement with the available experimental data.45,46
Molecular docking and microscopic binding free energy calculations
After the 3D LBD model of the α4β2 nAChR was obtained by homology modeling, the AutoDock 3.0.5 program60 was used to dock each of the molecular species of S-(−)-nicotine and R-(−)-deschloroepibatidine into the α4β2 nAChR binding sites. There are two equivalent ligand binding sites between two pairs of α4 and β2 subunits. We only need to account for one of these two equivalent sites binding with the ligand because these two equivalent binding sites is largely separated so that the ligand binding in one site should have no significant effect on the ligand binding in the other binding site. The atomic charges used for the docking with all the ligand species were the restrained electrostatic potential (RESP) charges. These RESP charges were determined by performing single-point ab initio electrostatic potential calculations at the HF/6-31G* level on the corresponding geometries optimized at the B3LYP/6-31+G* level, followed by fitting with the standard RESP procedure implemented in the Antechamber module of the Amber 7 program.34
During the docking process, conformational search was performed using the Solis and Wets local search method,61 and the Lamarkian genetic algorithm (LGA)60 was applied to the conformational search for the ligand-receptor binding structure. Among a series of docking parameters, the grid size was enlarged to be 120 × 120 × 120, which is large enough for the long-range electrostatic interaction calculation, and the used grid space was the default value of 0.375A˚. The interacting energy resulting from probing of the LBD of the α4β2 nAChR with the ligand was assessed by the empirical binding free energy and corrected by hydrogen bonding energy, if it exists, according to the distance from the donor to the acceptor.
The docked ligand-receptor complex structures were selected according to the criteria for interacting energy combined with geometric matching quality. These complexes were used as starting structures for further energy minimizations using the Sander module of the Amber 7 program before the final binding structures were achieved. The minimization process was similar to that used for modeling the free α4β2 nAChR structure, i.e. first fixing the backbone atoms of the receptor and the whole ligand structure for 1500 steps, in order to relax the side chains, especially those in the binding site. Then the energy minimization was performed by constraining the side chain atoms and also the ligand structure for another 500 steps. After several rounds of these partial energy-minimization runs, the convergence criterion of 0.001 kcal mol−1 Å−1 was quickly achieved in a full energy minimization. We carefully checked the fully energy-minimized structures to make sure that the overall structures of the receptor in the ligand-receptor complexes were not significantly different from that of the free receptor.
Finally, the microscopic binding free energies (i.e. the binding free energies corresponding to the individual microscopic binding species) were estimated by using the standard AutoDock scoring functional form, including a general hydrogen bonding energy (HBE) equation implemented in the AutoDock 3.0.5 program. As one can see below, the N–H···O type of ligand-receptor hydrogen bond exists in some of the docked structures. Based on the general HBE equation, we have HBE(r) ≈ 5εr012/r12 – 6εr010/r10, in which r is the H···O distance in the considered hydrogen bond, and r0 is the minimum value of the H···O distance for which the HBE equation can be used. We used r0 = 1.60 Å, because it is the shortest H···O distance found in our energy minimizations. The ε value was determined by using the condition that HBE(r) = −5.0 kcal/mol when r = 1.90 Å (the default parameter values of the AutoDock 3.0.5 program).
Most of the computations in the above studies were performed on the supercomputers at University of Kentucky Center for Computational Sciences and at Pacific Northwest National Laboratory. Some computations were carried out on SGI Fuel workstations and a 34-processors IBM x335 Linux cluster available in our own laboratory.
Results and discussion
(1) Ligand-receptor binding
A careful inspection of the optimized 3D model for the LBD of the α4β2 nAChR reveals that all the structural features of the model are consistent with the available structural data for both the AChBP and the α4β2 nAChR.6,8,9,21,22,23 The α4β2 nAChR consists of five subunits of two α4 and three β2 along a five-fold axis (only two of the five subunits are depicted in Figure 1), which is similar to the structural arrangement found in the X-ray crystal structure of AChBP.22,23 The root mean square deviation (RMSD) of the modeled structure from the template structure was found to be 0.32 Å for the Cα atoms and 0.43 Å for the backbone atoms. Further, the nicotine-binding site of the modeled α4β2 nAChR is very close to that of AChBP. One particular region rich in conserved aromatic residues is found at the interface between the α4 and β2 subunits. This region corresponds to the generally named “aromatic cage” in other nAChRs,6,8,9 and is expected to serve as a capture area for ligand binding, in light of previous experimental and modeling studies of other nAChR subtypes.5,6,9,62,63,64
Figure 1.


SRH of S-(−)-nicotine and DCEH of R-(−)-deschloroepibatidine binding to the α4β2 nAChR. (A) Viewing the SRH structure in the complex perpendicular to the fivefold axis. Only two subunits of the receptor are shown, along with the ligand in CPK mode. (B) Similar view for DCEH in the complex with the receptor. (C) Viewing the SRH structure (in stick) in the binding site oriented toward the α4 subunit. The pocket is represented in molecular surface format, colored with electrostatic potential in which blue represents positive charge and red represents negative charge; the front part of the pocket has been removed for clarity. (D) A similar view of DCEH at the α4β2 nAChR binding site.
Depicted in Figure 1 are some representative binding complex structures for both S-(−)-nicotine (SRH, Figure 1A) and R-(−)-deschloroepibatidine (DCEH in Figure 1B) binding. As expected, both SRH and DCEH are situated at the interface between subunits α4 and β2, and they are completely buried by the pocket-forming residues of the receptor. However, their relative orientations in the receptor binding site differ somewhat, mostly involving the pyridine ring (Figure 1C and Figure 1D). The principal (+) binding side is composed of more conservative residues from loops A, B, and C of the α4 subunit (according to the notations used in the X-ray crystal structure of AChBP), while the complementary (−) binding side contains more variable modulating residues from loops D and E of the β2 subunit. Both ligands, acting as cations due to the protonated saturated azaheterocyclic nitrogen, are wedged deeply in the bottom of the aromatic cage at the binding site.
As shown in Figure 2, the most important interactions between the receptor and ligands come from hydrogen bonding of the N–H···O type, and from charge-charge interactions between the protonated nitrogen on saturated azaheterocyclic ring of the ligands and the carbonyl group at the backbone of αTrp147. The protonated nitrogen atom in the saturated azaheterocyclic ring has a formal charge of +1, affording a strong attraction to the carbonyl oxygen of αTrp147. This protonated nitrogen is also stabilized by the aromatic moiety through cation-π interactions. Based on the general hydrogen bonding energy (HBE) equation implemented in the AutoDock 3.0.5 program, an N–H···O hydrogen bond with an H···O distance of 1.90 A˚, as shown in the modeled SRH-receptor complex (Figure 2A), contributes −5.0 kcal/mol to the mutual interaction energy at T = 298.15 K.60 Based on the critical distances shown in Figure 2 and listed in Table 2, the difference in HBE between SRH and DCEH is obvious. The hydrogen bond of αTrp147 with DCEH (Figure 2B) should be significantly stronger than that with SRH (Figure 2A), since the H···O distance with DCEH is ~0.2 A˚ shorter than that with SRH. DCEH also has stronger cation-π interactions with αTrp147 than does SRH, because not only does DCEH have one more hydrogen atom make contact with the aromatic side chain of αTrp147, but also the distance from each hydrogen atom to the center of the aromatic side chain of αTrp147 is significantly shorter than that in the SRH interaction (Table 2).
Figure 2.

Molecular interactions at the atomic level. (A) Interactions between SRH of S-(−)-nicotine and the α4β2 nAChR in which residues from the receptor are labeled and shown in stick; the ligand is shown in ball-and-stick. A dashed line represents the hydrogen bonding (HB) interactions between the proton at the nitrogen of pyrrolidine ring of SRH and the carbonyl oxygen of the αTrp147 backbone. Another dashed line represents the cation-π interaction between the protonated nitrogen of SRH and the aromatic side chain of αTrp147, and shows the distance to the center (a pseudo-atom in ball) of the same side chain. (B) Similar view of interactions between DCEH of R-(−)-deschloroepibatidine and the receptor. (C) Superimposed binding conformations of all six molecular species of S-(−)-nicotine extracted from the complex structures by molecular docking. (D) Superimposed binding conformations of all four molecular species of R-(−)-deschloroepibatidine extracted directly from the molecular docking complexes.
Table 2.
Key intermolecular distances involved in the hydrogen-bond (HB) and cation-π interactions in the microscopic binding complexes of several molecular species of S-(−)-nicotine and R-(−)-deschloroepibatidine with the α4β2 nAChR (R).
| HB distance (Å) | Cation-π distance (Å) | |
|---|---|---|
| SRH-R | 1.90 | 3.30 |
| SRHH-R | 1.82 | 3.39 |
| DCEH-R | 1.70 | 2.82, 2.72 |
| DCEH2-R | 1.84 | 2.69, 2.96 |
| DCEa-R | 1.91 | 2.69* |
no cation-π interaction.
In addition to hydrogen bonding and cation-π interactions, some aromatic and hydrophobic interactions also significantly contribute to the receptor binding of both SRH and DCEH. At the principal (+) side, the ligands make contact with side chains of residues in the aromatic cage, which includes Trp147, Tyr188, and Tyr195 of the α4 subunit and Trp53 of the β2 subunit. αTyr91 interacts closely with the ligands through its phenolic group and helps to form the aromatic cage. The vicinal αCys190 and αCys191 residues enhance the ligand-receptor interactions, mainly through the disulfide moiety via hydrophobic and long-range electrostatic interactions, since the sulfur atoms possess partial negative charges. αThr148 is located within a 5 A˚ distance around the ligands and its side chain weakly interacts with the ligands. On the complementary (−) side, the Try53 side chain of the β2 subunit is an important component of the aromatic cage, and interacts with the ligands through the aromatic moieties. βVal109, βPhe117, and βLeu119 significantly contribute to the hydrophobic interactions with both of the ligands, whereas β Asn107 and βAla108 only interact with DCEH.
The above described mode of interactions of SRH with the modeled α4β2 nAChR is quite similar to the nicotine binding with AChBP demonstrated in the crystal structure.23 The similarity exists not only in the general orientation of the SRH in the binding pocket, but also in many key interactions including typical hydrogen bonding and cation-π interactions. Although the composition of the principal (+) binding side in the present SRH—α4β2 nAChR complex is almost the same as that in the crystal structure,23 the details of interactions are somewhat different. For example, the distance between the saturated azaheterocyclic nitrogen of SRH and the carbonyl oxygen at the backbone of the conserved αTrp147 is 2.72 A˚ and the length of hydrogen bond is 1.90 A˚ in our modeled SRH—α4β2 nAChR complex, whereas the corresponding distance between the saturated azaheterocyclic nitrogen of nicotine and the carbonyl oxygen of Trp143 in the crystal structure of AChBP is 2.54 Å. Some significant difference exists on the complementary (−) side of the binding pocket. In the crystal structure,23 residues on the complementary (−) side contains either the short branched residue Leu112, or positively charged residue Arg104, having weaker interactions with the pyridine ring of nicotine, since the pyridine ring prefers contacts with aromatic or hydrophobic groups to highly polar atoms. As replacement at the same side in the α4β2 nAChR, these positions are filled with more hydrophobic and aromatic residues βVal109, βPhe117, and βLeu119. Those branched side chains make better contacts with the pyridine ring of nicotine and, therefore, enhance the intermolecular packing of SRH with α4β2 nAChR (Figure 2A). These non-conserved residues are at least partially responsible for the higher binding affinity of SRH with α4β2 nAChR (Table 3) compared to nicotine binding with AChBP.23 Such enhanced interactions are more apparent in R-(−)-deschloroepibatidine binding, as R-(−)-deschloroepibatidine is more bulky than SRH and can pack more tightly with residues at the complementary side (Figure 2B).
Table 3.
The calculated microscopic binding free energies (ΔG), microscopic binding affinities (Kd), and concentration fractions (at pH 7.4) of different molecular species of the ligands S-(−)-nicotine and R-(−)-deschloroepibatidine in solution, and at the α4β2 nAChR binding site. The calculated phenomenological binding affinities ( and ) are also provided.
| Fraction in solution | Fraction in the binding site | ΔG (kcal/mol) | Kd (nM) | ||
|---|---|---|---|---|---|
| S-(−)-nicotine | |||||
|
| |||||
| SR | 2.650×10−2 | 0.320×10−4 | −8.14 | 1080.00 | |
| SRH | 0.939 | 0.999 | −12.17 | 1.10 | |
| SRHH | 0.101×10−4 | 0.210×10−4 | −12.57 | 0.56 | |
| SS | 0.262×10−4 | 0.297×10−7 | −8.11 | 1130.00 | |
| SSH | 3.450×10−2 | 0.104×10−3 | −8.69 | 429.00 | |
| SSHH | 4.760×10−7 | 0.655×10−8 | −9.59 | 94.20 | |
|
| |||||
| phenomenological |
|
||||
|
| |||||
| R-(−)-deschloroepibatidine | |||||
|
| |||||
| DCEH | 0.991 | 0.999 | −13.92 | 0.057 | |
| DCEH2 | 2.290×10−3 | 0.001 | −14.68 | 0.016 | |
| DCEa | 3.160×10−3 | 1.570×10−4 | −13.64 | 0.091 | |
| DCEb | 3.710×10−3 | 6.890×10−8 | −8.96 | 272.0 | |
|
| |||||
| phenomenological |
|
||||
Our modeled receptor-ligand binding structures are consistent with the known binding information derived from the experimental studies, including site-directed mutagenesis, on the agonist-binding sites of AChBP and nAChRs.65,66,67,68,69 For example, early site-directed mutagenesis studies revealed large decreases of agonist affinities following mutations on the aromatic residues at the subunit interface as αTyr93, αTrp149, αTyr190, and αTyr198 (in numbering of the Torpedo nAChR).65,66,67 Recently reported experimental studies using unnatural amino acid mutagenesis revealed that nicotine employs a hydrogen bond to a backbone carbonyl of αTrp147.31 These aromatic residues are found to be at the most conserved positions by sequence alignment with the AChBP for several α subunits including the α4. These essential residues are αTyr91, αTrp147, αTyr188, and αTyr195 in our modeled LBD of α4β2 nAChR and its complexes with SRH and DCEH, as seen in Figures 1, 2A, and 2B. αTrp147 was found to be the most important determinant of binding with the α2, α3 and α4 subunit containing nAChRs for the typical agonist α-neurotoxin.68 It was found that the γTrp55Leu mutation at the αγ interface of the Torpedo nAChR resulted in a 7000-fold decrease of ACh binding affinity,69 demonstrating the importance of the residue γTrp55 in Torpedo nAChR. The corresponding residue βTrp53 in our modeled LBD of α4β2 nAChR is involved in the cation-π interactions with both SRH and DCEH (Figure 2A and 2B). In addition, a solid-state NMR study revealed a disulfide bond between Cys192 and Cys193,28 which is equivalent to the disulfide bond between αCys190 and αCys191 in our modeled LBD of α 4β2 nAChR.
Figure 2C shows the relative positions of the six molecular species (SR, SRH, SRHH, SS, SSH, and SSHH) of S-(−)-nicotine stabilized in the binding site of the α4β2 nAChR. All of the six molecular species bind with the receptor in a very similar way, but some significant differences can be seen in the vicinity of pyridine moiety. Another significant difference exists in the orientation of the proton at the saturated azaheterocyclic nitrogen. The N–H···O hydrogen bonding and cation-π interactions exist only in the complexes of the receptor binding with SRH and SRHH (Table 2). The relative positions of the four R-(−)-deschloroepibatidine species stabilized at the receptor binding site are depicted in Figure 2D, and show no significant difference in the orientation of R-(−)-deschloroepibatidine framework for the four ligand-receptor binding complexes.
(2) Microscopic binding affinity
Based on each of the docked microscopic ligand-receptor binding species mentioned above, the microscopic ligand-receptor binding free energy (ΔG) and the corresponding dissociation constant (Kd) were calculated. The calculated microscopic ΔG and Kd values are summarized in Table 3 for comparison. As seen in Table 3, the binding affinity of the R-(−)-deschloroepibatidine species is generally higher than that of the S-(−)nicotine species, except for DCEb. The calculated microscopic binding free energies are dominated by the hydrogen bonding and cation-π interactions between the receptor and the ligand, based on the numerical information summarized in Tables 2 and 3. The shorter the H···O distance in the N–H···O hydrogen bond, the stronger the hydrogen bond and, therefore, the larger the contribution of the hydrogen bond to the binding affinity. Within the six microscopic binding species of S-(−)-nicotine, only the microscopic binding species SRH and SRHH are involved in ligand-receptor hydrogen bonding. Thus, SRH and SRHH bind with the receptor much more strongly than the other four species of S-(−)-nicotine, as is supported by the calculated binding free energies (see Table 3). SRH is associated with the lowest microscopic binding free energy (−12.57 kcal/mol), i.e. the highest microscopic binding affinity, which is consistent with the shortest H···O distance in the N–H···O hydrogen bond of SRH with the receptor.
Of the four microscopic binding species of R-(−)-deschloroepibatidine, i.e. DCEH, DCEH2, DCEa, and DCEb, only DCEb does not form a hydrogen bond with the receptor and, therefore, its binding affinity with the receptor is much lower than those for the other three species. The highest binding affinity (i.e. the lowest binding free energy) is associated with DCEH2 (−14.68 kcal/mol), due to the strengthened electrostatic interactions of the receptor with the saturated azaheterocyclic nitrogen in DCEH2, as compared to that with the unprotonated pyridine species in DCEH. DCEa has a slightly lower binding affinity with the α4β2 nAChR than DCEH and DCEH2, due to the loss of the cation-π interactions between the saturated azaheterocyclic nitrogen and the αTrp147 side chain.
(3) Distribution of molecular species of ligands in solution
Interchange between different species of S-(−)-nicotine in solution
In order to determine the distribution of all the molecular species of a given ligand, i.e. S-(−)-nicotine or R-(−)-deschloroepibatidine, in solution, we first need to know whether these molecular species are interchangeable and are able to reach a thermodynamic equilibration in solution. In the case of the six molecular species of S-(−)-nicotine, it is clear that a thermodynamic equilibration between SR, SRH, and SRHH, in which the methyl group on the saturated azaheterocyclic nitrogen is trans to the pyridine ring can exist, because the protonation/deprotonation processes are well-recognized as being very fast. For the same reason, a thermodynamic equilibration between SS, SSH, and SSHH, in which the methyl group on the saturated azaheterocyclic nitrogen is cis to the pyridine ring can also exist (see Figure 3 for the structures of these species). An important question to be answered is whether the interchange between the first set of molecular species (SR, SRH, and SRHH) and the second set of molecular species (SS, SSH, and SSHH) is possible, and if the interchange occurs, how fast is this interchange? We propose that it is not likely that a direct interchange between SRH and SSH or between SRHH and SSHH can occur. However, these species are indirectly interchangeable when the direct interchange between SR and SS can occur. For example, the pathway of the change from SRH to SSH is SRH → SR → SS → SSH. The pathway of the change from SRHH to SSHH is SRHH → SRH → SR → SS → SSH → SSHH. Further, the different microscopic binding structures (L-R) are also indirectly interchangeable. For example, the pathway of the change from SRH-R to SSH-R is SRH-R (complex) → SRH (free ligand + free receptor) → SR (free ligand + free receptor) → SS (free ligand + free receptor) → SSH (free ligand + free receptor) → SSH-R (complex).
Figure 3.

Schematic representation of the structural interchange in solution and pKa values of the six molecular species of S-(−)-nicotine. The calculated activation free energies and the corresponding rate constants are also shown.
Is a direct interchange between SR and SS possible? Clearly, in both the SR and SS species, the saturated azaheterocyclic nitrogen utilizes three sp3 hybrid orbitals to form σ-bonds with three C-atoms; the remaining sp3 hybrid orbital is occupied by a lone-pair of electrons. The interchange between SR and SS can only result from a pyramidal inversion of the saturated azaheterocyclic nitrogen, resulting in a change in the orientation of the methyl group (i.e. from cis to trans with the pyridine ring, or vice verse). During the hypothetical nitrogen inversion from SR (or SS) to SS (or SR), an sp3 hybridization of the nitrogen atom in one stable structure will gradually change into a sp2 hybridization when the three C–N bonds are in the same plane, followed by a gradual change into the new sp3 hybridization in the stable inverted structure. Reaction coordinate calculations revealed a transition state (TS) structure between stable structures SR and SS. The geometry optimized for this transition state is depicted in Figure 4, along with the stable SR and SS geometries optimized. The optimized TS geometry reveals that the three C–N bonds are almost planar. In the TS structure, three sp2 hybrid orbitals of the saturated azaheterocyclic nitrogen are used to form three C–N bonds, and the lone-pair becomes a pure p orbital.
Figure 4.
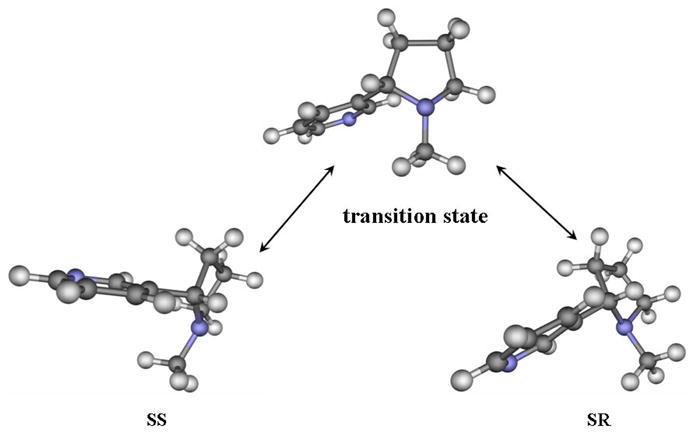
Geometries optimized at the B3LYP/6-31+G* level for the free base species SR and SS of S-(−)-nicotine and the transition state involved in the structural interchange between SR and SS.
The activation free energy, ΔGav(SR→SS), for the structural change from SR to SS is the free energy change from SR to TS, whereas the activation free energy, ΔGav(SS→SR), for the structural change from SS to SR is the free energy change from SS to TS. Based on our first-principles electronic structure calculations accounting for the solvent effects, ΔGav(SR→SS) = 6.78 kcal/mol and ΔGav(SS→SR) = 2.64 kcal/mol, when T = 298.15 K. Based on the calculated activation free energies, the corresponding rate constants can be evaluated by using the conventional transition state theory (CTST),70 i.e.
| (4) |
where kB is the Boltzmann constant, T is the absolute temperature, h is Planck’s constant, and ΔGav is the activation free energy. Thus, our calculations predict that k(SR→SS) = ~7 × 107 s−1 and k(SR→SS) = ~8 × 1010 s−1. Such large rate constants for the structural interchange between SR and SS reveal that a thermodynamic equilibration of the six nicotine species can be achieved very quickly in solution. This allows us to calculate the Boltzmann distribution of the concentrations of the six S-(−)-nicotine species in solution by using the calculated relative Gibbs free energies.
In a similar manner, the four molecular species of R-(−)-deschloroepibatidine can also be expected to quickly reach a thermodynamic equilibration in solution. The interchange between structures DCEa and DCEb of R-(−)-deschloroepibatidine involves two pathways. One pathway is associated with a nitrogen pyramidal inversion, similar to the pathway for the interchange between species SR and SS of S-(−)-nicotine. The other pathway is through protonation and deprotonation, using the protonated structure DCEH as an intermediate between structures DCEa and DCEb, as shown in Figure 5.
Figure 5.

Schematic representation of the structural interchange in solution and pKa values of different molecular species of R-(−)-deschloroepibatidine.
Distribution of the molecular species of S-(−)-nicotine
Thermodynamic equilibration between a molecular species and its deprotonated state is characterized by the pKa associated with the deprotonation process. The pKa values calculated for the SRH, SSH, SRHH, and SSHH species of S-(−)-nicotine, by using our first-principles electronic structure approach, are given in Figure 3. Thermodynamic equilibration between a pair of enantiomers, i.e. SR/SS, SRH/SSH, or SRHH/SSHH, is determined by the relative Gibbs free energies. Based on our first-principles electronic structure calculations, the free energy of SR is 4.14 kcal/mol lower than that of SS, when T = 298.15 K. Remarkably, the free energy difference between the two enantiomers becomes much smaller after they are protonated; the calculated free energy of SRH is only 1.96 kcal/mol lower than that of SSH. Further protonation does not decrease this free energy difference; i.e. the calculated free energy of SRHH is 2.11 kcal/mol lower than that of SSHH. The calculated free energy differences and pKa values together indicate that SRH should be the primary molecular species of S-(−)-nicotine in solution under physiologic conditions (pH 7.4), but other molecular species, particularly SSH and SR, should also be present in significant concentrations.
We can compare the calculated results with available experimental data to see how good the calculations are. Whidby and Seeman’s NMR spectroscope studies 71 demonstrated that the protonated S-(−)-nicotine species with the methyl group trans to the pyridine ring predominates in solution to the extent of more than 90%. The latter observations indicate that the free energy of SRH should be lower than that of SSH by at least 1.37 kcal/mol, when T = 298.15 K. The experimentally-derived free energy difference of > 1.37 kcal/mol is in good agreement with our calculated free energy difference of 1.96 kcal/mol between SRH and SSH. In addition, the pKa values 8.95 and 2.43 calculated for SRH and SRHH, respectively, are also reasonably close to the corresponding experimental pKa values of ~8.0 and ~3.0.1 The experimental pKa values were determined by analyzing of the thermodynamic data available for different protonation states in both the gas phase and solution (including the heat of vaporization).1 We note that the experimental pKa values are phenomenological, whereas our calculated pKa values are only associated with the microscopic structures SR, SRH, and SRHH. In principle, the microscopic pKa values are not exactly equal to the corresponding phenomenological pKa values unless only SR, SRH, and SRHH of nicotine exist in solution. Nevertheless, in light of our calculated relative free energies discussed above, the concentrations of SR, SRH, and SRHH are much higher than the corresponding concentrations of SS, SSH, and SSHH. Therefore, the microscopic pKa values associated with SR, SRH, and SRHH should be very close to the corresponding phenomenological pKa values determined at the same level of theory.
The calculated free energy differences and pKa values can also be used to quantitatively evaluate the concentrations of all the molecular species in solution as a function of pH. Assuming that the concentration of SRH is C0 at a given pH, the concentrations of other related species can be evaluated using the well-known Boltzmann distribution as:
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
In these equations, ΔG(SR → SS) is the free energy change from SR to SS and represents the pKa of species X (X = SRH, SRHH, SSH, or SSHH). The total concentration ( ) of S-(−)-nicotine in solution is a sum of the concentrations of all these species:
| (10) |
Based on Eqs.(5) to (10), the concentration fraction, , for each species total can be evaluated. Depicted in Figure 6 are the calculated concentration fractions from pH 0 to 14, when T = 298.15 K. As seen in Figure 6, the species in which the methyl group is trans to the pyridine ring are dominant in solution over the whole pH range. The cationic form (SRH) is dominant around physiologic pH, but the free base form (SR) is also present as a significant fraction. At very high pH, SR is present as the highest fraction, whereas at a very low pH, the dication (SRHH) is present as the highest fraction, and decreases sharply as the pH increases.
Figure 6.

pH-dependence of the calculated equilibrium distribution (molar fraction) of different molecular species of S-(−)-nicotine in solution. The labeled curves (i.e. the curves labeled with a circle, triangle, star, or pentagon) refer to the results evaluated by using the calculated pKa values of SRH and SRHH, whereas the unlabeled curves in the same colors represent the corresponding results evaluated by using the experimental pKa values for SRH and SRHH. When the experimental pKa values were used for SRH and SRHH, the pKa value used for SSH was the calculated pKa value of SSH minus 0.95, since the same computational approach overestimated the pKa of SRH by ~0.95; the pKa value used for SSHH was the calculated pKa value of SSHH plus 0.57, since the same computational approach underestimated the pKa of SRHH by ~0.57.
Distribution of the molecular species of R-(−)-deschloroepibatidine in solution
The same approach used to calculate the thermodynamic distribution of the S-(−)-nicotine species in solution can also be utilized to evaluate the thermodynamic distribution of the four molecular species of R-(−)-deschloroepibatidine in solution. Assuming the concentration of DCEH to be C1, the concentrations of the other three species can be calculated as follows:
| (11) |
| (12) |
| (13) |
In these equations, is the pKa of DCEH2 whose conjugated base is DCEH, is the pKa of DCEH when its conjugated base is considered to be DCEa, and is the pKa of DCEH when its conjugated base is considered to be DCEb. The total concentration ( ) of R-(−)-deschloroepibatidine in solution is calculated by
| (14) |
Figure 7 illustrates the pH-dependence of the concentration fractions for the four species of R-(−)-deschloroepibatidine. As seen in Figure 7, the monocationic form (DCEH) is dominant around physiologic pH. At very high pH, the two free base forms (DCEa and DCEb) are present in the highest fractions, whereas at very low pH, the dication (DCEH2) is present in the highest fraction.
Figure 7.

pH-dependence of the calculated equilibrium distribution (molar fraction) of different molecular species of R-(−)-deschloroepibatidine in solution.
(4) Distribution of microscopic binding species
Herein, we relate a microscopic binding species to a ligand-receptor binding structure, in which the receptor binds with a specific molecular species of a given ligand. As a given ligand (i.e. S-(−)-nicotine or R-(−)-deschloroepibatidine) exists as multiple molecular species in solution, these multiple molecular species can also exist at the nAChR binding site and, therefore, one has to consider multiple microscopic binding species for a ligand binding with the receptor. Thermodynamic distribution of the microscopic binding species of a given ligand is determined by both the thermodynamic distribution of the molecular species of the free ligand in solution and the relative microscopic binding free energies. Considering a microscopic ligand-receptor binding species (LR) formed from a specific molecular species (L) of the ligand and the α4β2 nAChR (R), we have
| (15) |
| (16) |
in which ΔGbind is the microscopic binding free energy between L and R, and Kd is the dissociation constant of the microscopic binding complex LR.
For S-(−)-nicotine, a microscopic binding species LR can be SR-R, SRH-R, SRHH-R, SS-R, SSH-R or SSHH-R. Similarly for R-(−)-deschloroepibatidine, LR can be DCEa-R, DCEb-R, DCEH-R, or DCEH2-R. The total concentration ( ) of all the microscopic S-(−)-nicotine-receptor binding species is a sum of the concentrations of all of the microscopic binding species:
| (17) |
Similarly, we can also evaluate the total concentration ( ) of all the microscopic total R-(−)-deschloroepibatidine-receptor binding species:
| (18) |
Equations (16) to (18) reveal that the concentration fractions of the microscopic binding species are also pH-dependent, since the concentration fractions of the molecular species of the free ligand are dependent on the pH of the solution. However, the concentration fractions of the microscopic binding species should not be the same as the concentration fractions of the corresponding species of the free ligand in solution, unless the ΔGbind values for different microscopic binding species are exactly the same. Figures 8 and 9 illustrate the calculated pH-dependence of the concentration fractions of the microscopic binding species for α4β2 nAChR binding with S-(−)-nicotine and R-(−)-deschloroepibatidine, respectively. As seen in Figure 8, the concentration fractions of SS-R, SSH-R, and SSHH-R are overwhelmed by those of SR-R, SRH-R, and SRHH-R for S-(−)-nicotine binding with the receptor. The overall fraction of the S-(−)-nicotine species (i.e. SS, SSH, and SSHH) with the methyl group cis to the pyridine ring in the microscopic S-(−)-nicotine-receptor binding (Figure 8) is even smaller than that of the same free ligand species in solution (Figure 6).
Figure 8.

pH-dependence of the calculated equilibrium distribution (molar fraction) of different microscopic binding complexes between S-(−)-nicotine and the α4β2 nAChR. The labeled curves (i.e. the curves labeled with a circle, triangle, star, or pentagon) refer to the results evaluated by using the calculated pKa values of SRH and SRHH, whereas the unlabeled curves in the same color represent the corresponding results evaluated by using the experimental pKa values for SRH and SRHH. When the experimental pKa values were used for SRH and SRHH, the pKa value used for SSH was the calculated pKa value of SSH minus 0.95, since the same computational approach overestimated the pKa of SRH by ~0.95; the pKa value used for SSHH was the calculated pKa value of SSHH plus 0.57, since the same computational approach underestimated the pKa of SRHH by ~0.57.
Figure 9.

pH-dependence of the calculated equilibrium distribution (molar fraction) of different microscopic binding complexes between R-(−)-deschloroepibatidine and the α 4β2 nAChR.
α4β2 nAChR binding of the four molecular species of R-(−)-deschloroepibatidine is illustrated in Figure 9. The overall concentration fractions of the receptor binding with the dication (DCEH2), monocation (DCEH), and the free base species (DCEa and DCEb) are very similar to the corresponding overall concentration fractions of the three species in solution (Figure 7). A significant difference exists in the relative fractions associated with the two free base species, DCEa and DCEb. The concentration fractions of species DCEa and DCEb are very close to each other in solution, whereas the concentration fractions of the binding species DCEa-R and DCEb-R are quite different. The fraction of DCEb-R is negligible compared to that of DCEa-R; also, the microscopic binding species DCEa-R is dominant at high pH. This is because the calculated DCEb-R binding free energy (−8.96 kcal/mol) is significantly higher than the calculated DCEa-R binding free energy (−13.64 kcal/mol).
(5) Phenomenological binding affinity
Generally speaking, the experimentally measured ligand-receptor binding affinity is the phenomenological binding affinity which includes contributions from all of the possible microscopic binding species. As discussed above, the total concentration of the molecular species of the ligand in solution can be calculated by using Eq. (10) or (14) and the total concentration of the microscopic binding species can be calculated by using Eq.(17) or (18). Based on the total of the concentrations of the microscopic binding species ( or ), the structures of the free ligand ( or ), and the free receptor ([R]) in solution, the “phenomenological dissociation constant” ( ) can be evaluated using the following equation:
| (19) |
The superscript “ligand” in Eq.(19) is “nicotine” for S-(−)-nicotine or “DCEH” for R-(−)-deschloroepibatidine. The corresponding “phenomenological binding free energy” can be evaluated by using the phenomenological dissociation constant :
| (20) |
In terms of the technical detail of the calculation process, we take S-(−)-nicotine as an example. One can simply use two arbitrarily given concentrations, one is [SRH] and the other is [SRH-R]. Then, the concentrations of the other five molecular species and the other five microscopic binding structures can be evaulated at any pH by using the relative free energies of the free molecular species and the determined microscopic binding constants. Then, one can easily evaluate , [R], and required to calculate through Eq.(19). The calculated overall binding constant is independent of the arbitrarily chosen [SRH] and [SRH-R] values.
Both the calculated phenomenological dissociation constant and the phenomenological binding free energy are pH-dependent for S-(−)-nicotine and R-(−)-deschloroepibatidine (Figures 10 and 11). This is because the thermodynamic distributions of the microscopic species both in solution and at the receptor binding site are dependent on the pH of the solution, so that and are also pH-dependent. The predicted pH-dependence of the phenomenological binding of S-(−)nicotine with the nAChR is qualitatively consistent with a recent experimental observation with voltage-clamp electrophysiology which demonstrated that the efficacy of nicotine as an agonist at the α4β2 nAChR decreased from ~90% at pH 6.5 to ~10% at pH 9.0; the observed nicotine efficacy at pH 9.0 is only a ninth of that at pH 6.5.72 As seen in Figure 10, when the experimental pKa values are used, our calculated value for S-(−)-nicotine binding with the α4β2 nAChR is 1.32 nM at pH 6.5 and 13.34 nM at pH 9.0. This calculated value increase of ~10-fold is in excellent agreement with the efficacy decrease of ~9-fold observed experimentally. When the calculated pKa values are used, the same values of 1.32 nM and 13.34 nM for S-(−)-nicotine binding with the α4β2 nAChR are obtained at pH 7.45 and pH 9.95, respectively. Predicting the same values at the higher pH values is consistent with the fact that our calculations overestimated the pKa of SRH by 0.95 (Figure 3).
Figure 10.
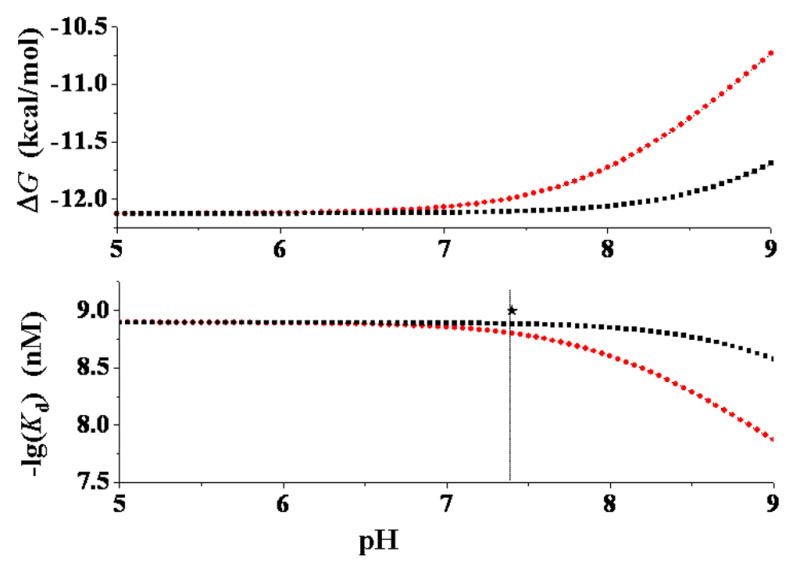
pH-dependence of the calculated phenomenological binding free energy (ΔG) and binding affinity (Kd) of S-(−)-nicotine with the α4β2 nAChR. The black curves refer to the results evaluated by using the calculated pKa values of SRH and SRHH, whereas the red curves represent the corresponding results evaluated by using the experimental pKa values for SRH and SRHH. The experimentally determined −log(Kd) value (9.0) is3 also indicated as black star. When the experimental pKa values were used for SRH and SRHH, the pKa value used for SSH was the calculated pKa value of SSH minus 0.95, since the same computational approach overestimated the pKa of SRH by ~0.95; the pKa value used for SSHH was the calculated pKa value of SSHH plus 0.57, since the same computational approach underestimated the pKa of SRHH by ~0.57.
Figure 11.

pH-dependence of the calculated phenomenological binding free energy (ΔG) and binding affinity (Kd) of R-(−)-deschloroepibatidine at the α4β2 nAChR. The experimentally determined −log(Kd) value (10.7) is also indicated as black star.
Figures 10 and 11 reveal a qualitative trend that decreasing the pH of the solution generally favors the phenomenological binding affinity. However, the and values are not sensitive to changes in the pH 5 to 7 range for S-(−)-nicotine (Figure 10) and in pH 6 to 9 range for R-(−)-deschloroepibatidine (Figure 11). The and values calculated for S-(−)-nicotine under physiologic conditions (pH 7.4) are 1.3 nM and −12.10 kcal/mol, respectively, which is in excellent agreement with the reported experimental range of the Ki values (1.0 to 2.3 nM).10e,10f The and values calculated for R-(−)-deschloroepibatidine under the physiologic conditions (pH 7.4) are 0.064 nM and −13.89 kcal/mol, respectively, which is also in excellent agreement with the reported experimental Ki value of 0.020 ± 0.001 nM10b or 0.031 nM.11a Thus, the calculated absolute phenomenological binding affinity data are also consistent with the available experimental data.
The general computational strategy for studying interactions of the α4β2 nAChR with S-(−)-nicotine and R-(−)-deschloroepibatidine from the microscopic binding species and affinities to the phenomenological binding affinities might also be useful for studying other types of ligand-protein interactions involving multiple molecular species of a specific ligand. As is well known, a receptor is activated or inhibited by a ligand through a specific interaction with the binding site. The (microscopic) binding information at the atomic level is usually provided by X-ray crystallography or NMR spectroscopy,22,23,73,74,75 whereas the (phenomenological) binding affinity is measured through reaction kinetics and/or thermodynamics studies.5,76,77 When these two aspects are complementary to each other, one can achieve a better understanding of these biological processes.73,74,75 The computational modeling approach provides a unique way of predicting microscopic binding events and for quantitatively linking microscopic binding to phenomenological binding events. In view of this “from-microscopic-to-phenomenological” computational strategy, the dominant species of a ligand binding with a receptor is not necessarily the dominant species of the ligand in solution, particularly when the dominant structural form of the ligand in solution has a lower binding affinity at that receptor.
Conclusion
Molecular modeling, molecular docking, and first-principles electronic structure calculations carried out in this study have demonstrated how the α4β2 nAChR binds with different species of two typical agonist molecules, i.e. six species of S-(−)-nicotine and four species of R-(−)-deschloroepibatidine, and have provided a prediction of the corresponding microscopic binding free energies. In both cases, of these two molecules hydrogen bonding and cation-π interactions between the receptor and the ligand were found to be the dominant factors that differentiate the binding strengths of the different microscopic binding species.
Our reaction coordinate calculations using first-principles electronic structure theory revealed a transition state (TS) for the structural interchange between two diastereomeric protonated species (SR and SS) of the free base form of S-(−)-nicotine in solution. In light of the calculated low activation free energies (6.78 kcal/mol for SR → SS and 2.64 kcal/mol for SR → SS) and very high rate constants (~7 × 107 s−1 for SR → SS and ~8 × 1010 s−1 for SR → SS) for the structural interchange of these species in solution, as well as some further analysis, we conclude that all the molecular species (distinguished by the free base and the different protonation states) of the ligand, i.e. S-(−)-nicotine or R-(−)-deschloroepibatidine, can quickly achieve a thermodynamic equilibration in solution and at the receptor binding site. This allows us to evaluate the equilibrium concentration distributions of the free ligand species and the corresponding microscopic ligand-receptor binding species. The calculated equilibrium concentration distributions of the ligand species clearly show their pH-dependence and provide the microscopic information required for further determination of the phenomenological binding affinity of the ligand with the α4β2 nAChR.
The predicted equilibrium concentration distributions, pKa values, absolute phenomenological binding affinities of the ligand species and their pH-dependence are all in good agreement with available experimental data. This suggests that the computational strategy of studying interactions of ligands with receptors from their microscopic binding species and affinities to the phenomenological binding affinity is reliable for studying ligand-α4β2 nAChR binding, and thus, should be a valuable approach for future rational design of drugs targeting the α4β2 nAChR.
The general strategy of the “from-microscopic-to-phenomenological” approach used in the present work could also be useful in future studies of other types of ligand-protein interactions involving multiple molecular species of a ligand, and in any related rational drug design endeavors. Such a strategy enables us to quantitatively account for the thermodynamic distribution of all the possible microscopic ligand-protein binding species for a given ligand, their pH-dependence, and their contributions to the phenomenological binding affinity.
Supplementary Material
Acknowledgments
This research was supported in part by funds from the College of Pharmacy and the Center for Computational Sciences (CCS) at University of Kentucky, and by NIH grant U19DA017548. We also utilized the EMSL Molecular Sciences Computing Facility (a DOE’s national user facility operated by Pacific Northwest National Laboratory) under Grand Challenge grants GC3565 and GC9598.
Footnotes
Supporting Information Available. Stereo version of Figures 2A and 2B; complete citations of references 10(a), 10(e), 34, 39, and 40. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gorrod JW, Jacob P III, editors. Analytic Determination of Nicotine and Related Compounds and Their Metabolites. Elsevier; New York: 1999. [Google Scholar]
- 2.Quikk M. Trends Neurosci. 2004;27:561–568. doi: 10.1016/j.tins.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Hogg RC, Bertrand D. Science. 2004;306:983–985. doi: 10.1126/science.1106030. [DOI] [PubMed] [Google Scholar]
- 4.Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- 5.Wonnacott S, Sidhpura N, Balfour DJK. Curr Opin Pharm. 2005;5:53–59. doi: 10.1016/j.coph.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Karlin A. Nature Rev Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- 7.Miyazawa A, Fujiyoshi Y, Unwin N. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 8.Karlin A. Neuron. 2004;41:841–842. doi: 10.1016/s0896-6273(04)00151-5. [DOI] [PubMed] [Google Scholar]
- 9.Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA. Trends Neurosci. 2004;27:329–336. doi: 10.1016/j.tins.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 10.(a) Briggs CA, et al. Pharm Biochem Behav. 1997;57:231–241. doi: 10.1016/s0091-3057(96)00354-1. [DOI] [PubMed] [Google Scholar]; (b) Carroll FI, Liang F, Navarro HA, Brieaddy LE, Abraham P, Damaj MI, Martin BR. J Med Chem. 2001;44:2229–2237. doi: 10.1021/jm0100178. [DOI] [PubMed] [Google Scholar]; (c) Efange SMN, Tu Z, Hohenberg K, Francesconi L, Howell RC, Rampersad MV, Todaro LJ, Papke RL, Kung MP. J Med Chem. 2001;44:4704–4715. doi: 10.1021/jm010129z. [DOI] [PubMed] [Google Scholar]; (d) Sharples CGV, Karig G, Simpson GL, Spencer JA, Wright E, Millar NS, Wonnacott S, Gallagher T. J Med Chem. 2002;45:3235–3245. doi: 10.1021/jm020814l. [DOI] [PubMed] [Google Scholar]; (e) Sullivan JP, et al. J Tharm Exp Therap. 1997;283:235. [PubMed] [Google Scholar]; (f) Wilkins LH, Jr, Grinevich VP, Ayers JT, Crooks PA, Dwoskin LP. J Tharm Exp Therap. 2003;304:400. doi: 10.1124/jpet.102.043349. [DOI] [PubMed] [Google Scholar]
- 11.(a) Tonder JE, Olesen PH. Curr Med Chem. 2001;8:651–674. doi: 10.2174/0929867013373165. [DOI] [PubMed] [Google Scholar]; (b) Bunnelle WH, Dart MJ, Schrimpf MR. Curr Topics Med Chem. 2004;4:299–334. doi: 10.2174/1568026043451438. [DOI] [PubMed] [Google Scholar]
- 12.Wilkins LH, Jr, Grinevich VP, Ayers JT, Crooks PA, Dwoskin LP. J Pharm Exp Ther. 2003;304:400–410. doi: 10.1124/jpet.102.043349. [DOI] [PubMed] [Google Scholar]
- 13.Dwoskin LP, Sumithran SP, Zhu J, Deaciuc AG, Ayers JT, Crooks PA. Bioorg Med Chem Lett. 2004;14:1863–1867. doi: 10.1016/j.bmcl.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 14.Crooks PA, Ayers JT, Xu R, Sumithran SP, Grinevich VP, Wilkins LH, Deaciuc AG, Allen DD, Dwoskin LP. Bioor Med Chem Lett. 2004;14:1869–1874. doi: 10.1016/j.bmcl.2003.10.074. [DOI] [PubMed] [Google Scholar]
- 15.Miller DK, Crooks PA, Zheng G, Grinevich VP, norrholm SD, Dwoskin LP. J Pharm Exp Ther. 2004;310:1035–1045. doi: 10.1124/jpet.104.068098. [DOI] [PubMed] [Google Scholar]
- 16.Pallavicini M, Moroni B, Bolchi C, Clementi F, Fumagalli L, Gotti C, Vailati S, Valoti E, Villa L. Bioor Med Chem Lett. 2004;14:5827–5830. doi: 10.1016/j.bmcl.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 17.Wei ZL, Petukhov PA, Xiao Y, Tuckmantel W, George C, Kellar KJ, Kozikowski AP. J Med Chem. 2003;46:921. doi: 10.1021/jm025613w. [DOI] [PubMed] [Google Scholar]
- 18.Elliott KJ, Ellis SB, Berckhan KJ, Urrutia A, Chavez-Noriega LE, Johnson EC, Velicelebi G, Harpold MM. J Mol Neurosci. 1996;7:217–28. doi: 10.1007/BF02736842. [DOI] [PubMed] [Google Scholar]
- 19.(a) Flores CM, Rogers SW, Pobreza LA, Wolfe BB, Kellar KJ. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]; (b) Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, Novere NL, Vincent P, Pich EM, Brulet P, Changeux JP. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]; (c) Marubio LM, Arroyo-Jimenez MD, Cordero-Erausquin M, Lena C, Novere NL, d’Exaerde AD, Huchet M, Damaj MI, Changeux JP. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]; (d) Paradiso KG, Steinbach JH. J Physiol. 2003;553:857–871. doi: 10.1113/jphysiol.2003.053447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crooks PA. In: Analytic Determination of Nicotine and Related Compounds and Their Metabolites. Gorrod JW, Jacob P III, editors. Elsevier; New York: 1999. p. 69. [Google Scholar]
- 21.Unwin N. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 22.Brejc K, Dijk WJ, Klaassen RV, Schuurmans M, Oost J, Smit AB, Sixma TK. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 23.Celie PHN, Rossum-Fikkert SE, Dijk WJ, Brejc K, Smit AB, Sixma TK. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 24.Le Novere N, Grutter T, Changeux J. Proc Natl Acad Sci. 2002;99:3210–3215. doi: 10.1073/pnas.042699699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schapira M, Abagyan R, Totrov M. BMC Struct Biol. 2002;2:1–8. doi: 10.1186/1472-6807-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henikoff S, Henikoff JG. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williamson PTF, Watts TA, Addona GH, Miller KW, Watts A. Proc Natl Acad Sci USA. 2001;98:2346. doi: 10.1073/pnas.031361698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sine SM, Wang HL, Bren N. J Biol Chem. 2002;277:29210. doi: 10.1074/jbc.M203396200. [DOI] [PubMed] [Google Scholar]
- 30.Gao F, Bren N, Burghard TP, Hansen S, Henchman RH, Taylor P, McCammon JA, Sine SM. J Biol Chem. 2005;280:8443. doi: 10.1074/jbc.M412389200. [DOI] [PubMed] [Google Scholar]
- 31.Cashin AL, Petersson EJ, Lester HA, Dougherty DA. J Am Chem Soc. 2005;127:350. doi: 10.1021/ja0461771. [DOI] [PubMed] [Google Scholar]
- 32.Ullmann GM, Knapp EW. Eur Biophys J. 1999;28:533–551. doi: 10.1007/s002490050236. [DOI] [PubMed] [Google Scholar]
- 33.Nozaki Y, Tanford C. Methods Enzymol. 1967;11:715–734. [Google Scholar]
- 34.Case DA, et al. AMBER. Vol. 7. University of California; San Francisco: 2002. [Google Scholar]
- 35.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 36.Vriend G. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 37.(a) Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (c) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623. [Google Scholar]
- 38.(a) Gonzalez C, Schlegel HB. J Chem Phys. 1989;90:2154. [Google Scholar]; (b) Gonzalez C, Schlegel HB. J Phys Chem. 1990;94:5523. [Google Scholar]
- 39.Frisch MJ, et al. Gaussian 03, Revision A.1. Gaussian, Inc; Pittsburgh, PA: 2003. [Google Scholar]
- 40.Schmidt MW, et al. J Comput Chem. 1993;14:1347. [Google Scholar]
- 41.(a) Zhan CG, Bentley J, Chipman DM. J Chem Phys. 1998;108:177. [Google Scholar]; (b) Zhan CG, Chipman DM. J Chem Phys. 1998;109:10543. [Google Scholar]; (c) Zhan CG, Chipman DM. J Chem Phys. 1999;110:1611. [Google Scholar]
- 42.(a) Zhan C-G, Norberto de Souza O, Rittenhouse R, Ornstein RL. J Am Chem Soc. 1999;121:7279. [Google Scholar]; (b) Zhan CG, Zheng F. J Am Chem Soc. 2001;123:2835. doi: 10.1021/ja005529a. [DOI] [PubMed] [Google Scholar]; (c) Zheng F, Zhan CG, Ornstein RL. J Phys Chem B. 2002;106:717. [Google Scholar]; (d) Zhan CG, Dixon DA, Sabri MI, Kim MS, Spencer PS. J Am Chem Soc. 2002;124:2744. doi: 10.1021/ja0113394. [DOI] [PubMed] [Google Scholar]; (e) Dixon DA, Feller D, Zhan CG, Francisco SF. J Phys Chem A. 2002;106:3191. [Google Scholar]; (f) Dixon DA, Feller D, Zhan CG, Francisco SF. Int J Mass Spectrom. 2003;227:421. [Google Scholar]; (g) Zhan CG, Dixon DA, Spencer PS. J Phys Chem B. 2003;107:2853. [Google Scholar]
- 43.(a) Chen X, Zhan CG. J Phys Chem A. 2004;108:3789. [Google Scholar]; (b) Chen X, Zhan CG. J Phys Chem A. 2004;108:6407. [Google Scholar]; (c) Zhan CG, Dixon DA, Spencer PS. J Phys Chem B. 2004;108:6098. [Google Scholar]
- 44.Tomasi J, Persico M. Chem Rev. 1994;94:2027. [Google Scholar]
- 45.Mejias JA, Lago S. J Chem Phys. 2000;113:7306. [Google Scholar]
- 46.Cramer CJ, Truhlar DG. In: Solvent Effects and Chemical Reactions. Tapia O, Bertran J, editors. Kluwer; Dordrecht: 1996. p. 1. [Google Scholar]
- 47.Chipman DM. J Chem Phys. 2000;112:5558. [Google Scholar]
- 48.Barone V, Cossi M, Tomasi J. J Chem Phys. 1997;107:3210. [Google Scholar]
- 49.Tomasi J, Mennucci B, Cances E. J Mol Struct (Theochem) 1999;464:211. [Google Scholar]
- 50.Cancès E, Mennucci B. J Chem Phys. 2001;114:4744. [Google Scholar]
- 51.Cossi M, Rega N, Scalmani G, Barone V. J Chem Phys. 2001;114:5691. [Google Scholar]
- 52.Chipman DM. J Chem Phys. 2002;116:10129. [Google Scholar]
- 53.Tawa GJ, Topol IA, Burt SK, Caldwell RA, Rashin AA. J Chem Phys. 1998;109:4852. [Google Scholar]
- 54.Topol IA, Tawa GJ, Burt SK, Rashin AA. J Chem Phys. 1999;111:10998. [Google Scholar]
- 55.Mejias JA, Lago S. J Chem Phys. 2000;113:7306. [Google Scholar]
- 56.Zhan CG, Dixon DA. J Phys Chem A. 2001;105:11534–11540. [Google Scholar]
- 57.Zhan CG, Dixon DA. J Phys Chem A. 2002;106:9737–9744. [Google Scholar]
- 58.Zhan CG, Dixon DA. J Phys Chem B. 2003;107:4403–4417. [Google Scholar]
- 59.Zhan CG, Dixon DA. J Phys Chem A. 2004;108:2020–2029. [Google Scholar]
- 60.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 61.Solis FJ, Wets RJB. Maths Opera Res. 1981;6:19–30. [Google Scholar]
- 62.Williamson PTF, Watts TA, Addona GH, Miller KW, Watts A. Proc Natl Acad Sci USA. 2001;98:2346–2351. doi: 10.1073/pnas.031361698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sine SM, Wang HL, Bren N. J Biol Chem. 2002;277:29210–29223. doi: 10.1074/jbc.M203396200. [DOI] [PubMed] [Google Scholar]
- 64.Gao F, Bren N, Burghardt TP, Hansen S, Henchman RH, Taylor P, McCammon JA, Sine SM. J Biol Chem. 2005;280:8443–8451. doi: 10.1074/jbc.M412389200. [DOI] [PubMed] [Google Scholar]
- 65.Galzi JL, Bertrand D, Devillers-Thiery A, Revah F, Bertrand S, Changeux JP. FEBS Lett. 1991;294:198. doi: 10.1016/0014-5793(91)80668-s. [DOI] [PubMed] [Google Scholar]
- 66.O’Leary ME, White MM. J Biol Chem. 1992;267:8360. [PubMed] [Google Scholar]
- 67.Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. J Biol Chem. 1994;269:8808. [PubMed] [Google Scholar]
- 68.Malany S, Osaka H, Sine SM, Taylor P. Biochem. 2000;39:15388. doi: 10.1021/bi001825o. [DOI] [PubMed] [Google Scholar]
- 69.Xie Y, Cohen JB. J Biol Chem. 2001;276:2417. doi: 10.1074/jbc.M009085200. [DOI] [PubMed] [Google Scholar]
- 70.Alvarez-Idaboy JR, Galano A, Bravo-Pérez G, Ruíz ME. J Am Chem Soc. 2001;123:8387. doi: 10.1021/ja010693z. [DOI] [PubMed] [Google Scholar]
- 71.Whidby JF, Seeman JI. J Org Chem. 1976;41:1585. doi: 10.1021/jo00871a022. [DOI] [PubMed] [Google Scholar]
- 72.Petersson Ej, Choi A, Dahan DS, Lester HA, Dougherty DA. J Am Chem Soc. 2002;124:12662–12663. doi: 10.1021/ja028206i. [DOI] [PubMed] [Google Scholar]
- 73.Ridge KD, Abdulaev NG, Sousa M, Palczewski K. Trends Biochem Sci. 2003;28:479–487. doi: 10.1016/S0968-0004(03)00172-5. [DOI] [PubMed] [Google Scholar]
- 74.Vrielink A, Sampson N. Curr Opin Struct Biol. 2003;13:709–715. doi: 10.1016/j.sbi.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 75.Lefkowitz RJ. Trends Pharm Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 76.Leavitt S, Freire E. Curr Opin Struct Biol. 2001;11:560–6. doi: 10.1016/s0959-440x(00)00248-7. [DOI] [PubMed] [Google Scholar]
- 77.Cashin AL, Petersson EJ, Lester HA, Dougherty DA. J Am Chem Soc. 2005;127:350–356. doi: 10.1021/ja0461771. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.