

Abstract
Mammalian endothelial cells are deficient in cystathionine β synthetase (CBS) activity, which is responsible for homocysteine (Hcy) clearance. This deficiency makes the endothelium the prime target for Hcy toxicity. Hcy induces integrin shedding in microvascular endothelial cells (MVEC) by increasing matrix metalloproteinase (MMP). Hcy competes with inhibitory neurotransmitter γ aminobutyric acid (GABA)-A receptor. We hypothesized that Hcy transduces MVEC remodeling by increasing metalloproteinase activity and shedding β-1 integrin by inactivating the GABA-A/B receptors, thus behaving as an excitatory neurotransmitter. MVEC were isolated from mouse brain. The presence of GABA-A receptor was determined by immunolabeling. It was induced by muscimol, an agonist of GABA-A receptor as measured by Western blot analysis. Hcy induced MMP-2 activity in a dose- and time-dependent manner, measured by zymography. GABA-A/B receptors ameliorated the Hcy-mediated MMP-2 activation. Hcy selectively increased the levels of tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-3 but decreased the levels of TIMP-4. Treatment with muscimol decreased the levels of TIMP-1 and TIMP-3 and increased the levels of TIMP-4 to control. Hcy caused a robust increase in the levels of a disintegrin and metalloproteinase (ADAM)-12. In the medium of MVEC treated with Hcy, the levels of β-1 integrin were significantly increased. Treatment with muscimol or baclofen (GABA-B receptor agonist) ameliorated the levels of β-1 integrin in the medium. These results suggested that Hcy induced ADAM-12. Significantly, Hcy facilitated the β-1 integrin shedding. Treatment of MVEC with muscimol or baclofen during Hcy administration ameliorated the expression of metalloproteinase, integrin-shedding, and constrictive collagen remodeling, suggesting a role of Hcy in GABA receptor-mediated cerebrovascular remodeling.
Index Entries: MMP, TIMP, ADAM, β-1 integrin, neurotransmitter
INTRODUCTION
Homocysteine (Hcy) is an important risk factor for the development of cardiovascular diseases and stroke (1,2). Epidemiological studies have indicated that hyperhomocysteinemia (HHcy) is an important contributory factor for the development of atherosclerotic lesions and arterial hypertension (3). Numerous studies have demonstrated the pathophysiologic role of Hcy in large conduct arterial endothelial cells in vivo and in vitro (4–6). However, little is known about the role of Hcy in microvessel endothelial cells (MVEC). This is especially true with regard to the role of Hcy in MVEC remodeling. Hcy induces endocardial microvascular endothelial dysfunction (7,8) and microvascular impairment (9). Mammalian endothelial cells are deficient in cystathionine β synthetase (CBS) activity (10,11), which is responsible for Hcy clearance. In addition, Hcy instigates structural (12) and functional microvascular impairment by generating superoxide in the brain (13). Therefore, it is highly likely that endothelial cells are prime targets for Hcy toxicity. Hcy activates endothelial cells and increases matrix metalloproteinases (MMPs) activity (14). Tissue inhibitor of metalloproteinases (TIMPs) modulates MMP activity (15). Hcy also alters the expression of TIMP, leading to increase MMP activity (16). A disintegrin and metalloproteinase (ADAM) plays significant role in disintegrin and shedding the cell surface receptor (17). The cell–cell and cell–matrix interactions are mediated by integrin receptors (18). The shedding of integrin by Hcy-mediated activated metalloproteinase transduces MVEC.
γ aminobutyric acid (GABA) receptors are inhibitory neurotransmitter receptors (19–21) and play a very important role in regulation of arterial blood pressure (22,23). The GABA-A, GABA-B, and GABA-C receptors are present in the central nervous system (19–23), heart (24), and the endothelium (25). The chronic activation of GABA-A receptor decreases arterial blood pressure through a mechanism associated with increased endothelial nitric oxide (26,27). Interestingly, this finding is consistent with a moderate increase in vessel diameter (approx 5%) after acute treatment with GABA in isolated rat cerebral parenchymal arterioles (28). Hcy decreases the bioavailability of endothelial nitric oxide (29) and displaces competitively muscimol, a GABA-A receptor agonist (30). This result suggests that Hcy may attenuate inhibitory GABA-A receptor function in endothelial cells. Interestingly, antagonists to N-methyl-D-aspartate (NMDA) receptors also protect Hcy-mediated toxic effects in rats (31); hence, homocysteic acid or Hcy may behave as the excitatory neurotransmitter (32).
Hcy induces cell adhesive molecules in leukocytes and platelets (33). The inhibition of a disintegrin and metalloproteinase ameliorates Hcy-mediated constrictive vascular remodeling by endothelial cells (34). In addition, Hcy instigates integrin shedding in MVEC by increasing MMP-9 activity (35), but it is unclear whether Hcy induces ADAM and collagen constrictive remodeling in MVEC. We show that Hcy-mediated constrictive collagen remodeling is alleviated by GABA-A/B receptor agonists, muscimol, and baclofen (36). Here, we extend the notation that GABA receptors ameliorate Hcy-mediated integrin shedding and constrictive collagen remodeling in MVEC.
MATERIALS AND METHODS
Cell Culture
Mouse brain MVEC were isolated from the gray matter of the cerebral cortex as described (35,37). In brief, cerebral gray matter was extracted from the mouse brain and minced into pieces in minimal essential medium (MEM) at pH 7.4. Tissue suspension was incubated with dispase (0.3% w/v) for 3 h at 37°C, and was centrifuged at 5800g for 10 min. The pellet was resuspended in 13% (w/v) dextran (average MW 70,000) and centrifuged at 5800g for 10 min. Pellet was suspended in MEM and incubated with collagenase/dispase (1 mg/mL) for 2 h at 37°C then centrifuged at 1000g for 10 min. The pellets were layered on 50% Percoll gradient and this mixture was centrifuged 1400g for 10 min, which resulted in two bands. The red-thick band below the white fatty band was recovered for MVEC. The band was washed by centrifugation for 10 min at 1000g and the pellet was suspended in MEM. MVEC were directly plated in culture medium containing (50% v/v) MEM, 50% (w/v) F-12 nutrient mixture (Ham), 11% (v/v) plasma derived equine serum, 50 mg/mL heparin, 100 g streptomycin, and 100 g penicillin-G. MVEC were then incubated at 37°C with 95% (v/v) CO2 in air. After formation of confluent monolayers (10–14 d), experiments were performed. To characterize the endothelial cells, MVEC were labeled with CD-31 (35).
Characterization of GABA-A Receptor in Primary Cultures
MVEC were stained with GABA-A receptor beta chain antibody to identify GABA-A receptor. In brief, MVEC monolayers were fixed at room temperature for 10 min in 95% ethanol and 5% glacial acetic acid. Cells were then incubated with 1:100 dilution of mouse anti-GABA-A receptor beta chain monoclonal antibody (Chemicon, Corp., CA) for 3 h. Secondary anti-mouse immunoglobulin (Ig)G-fluorescein conjugated was used to detect the fluorescence. For negative controls, the cells were incubated without mouse anti-GABA-A receptor monoclonal antibody; however, secondary antibody detection was kept the same. MVEC were viewed with an inverted microscope (Leica) equipped for transmission and fluorescence.
To determine the purity of MVEC, cells stained with CD-31 (an endothelial cell-specific marker) and GABA-A receptor beta chain antibody were quantitated by fluorescence. MVEC monolayers were washed with 0.1 M phosphate-buffered saline (PBS) and blocked for 20 min at 37°C with 20% (v/v) horse serum in 0.1 M PBS. Cells were subsequently incubated with 1:100 dilution of mouse anti-CD-31-fluorescein conjugated monoclonal antibody overnight at 37°C. Cells were then washed with 0.1 M PBS. For GABA-A receptor, cells were incubated with anti-GABA-A receptor antibody, and then incubated with anti-mouse IgG (Fc-specific) FITC conjugate (1:200 dilution) at 37°C overnight. For control/background, cells were incubated only with anti-mouse IgG (Fc specific) FITC conjugate (1:200 dilution) overnight at 37°C. Cells were detached with 0.25% trypsin. Trypsinized cell suspensions were used for fluorescence measurements with a spectrophotometer (Spex-Fluorolog-2). FITC fluorescence was measured at 518 nm with band-slit of 2.5 mm by exciting at 494 nm with 1.25 band-slit.
GABA-A and -B receptors were isolated from MVEC homogenates incubated with their respective antibodies and immunoprecipated with IgG-agarose beads. Antibody-antigen complexes were dissociated with 0.1% sodium dodecyl sulfate (SDS). To determine whether Hcy competes with muscimol, fluorescamine-homocysteine (F-Hcy) is prepared (38). Not only are the chemical yields low, but the reactants and products are small molecules that are very unlikely to be separated on Sephadex G-50. The collected fractions were separated based on absorbance at 380 nm and fluorescence at 480 nm when excited at 380 nm (38). We observed two peaks: one with absorption maximum at 380 nm, and other with fluorescence maximum at 480 nm. Because free-fluorescamine is not fluorescent, we use fluorescence measure to determine the separation and purity of F-Hcy.
GABA-A and GABA-B receptors (10 μg) were incubated at increasing concentrations of F-Hcy. Because GABA-receptor protein fluorescence exhibits a fairly hydrophobic emission, the GABA-receptor protein emission was measured at 334 nm (2.5 mm band-slit) when excited at 295 nm (1.25 mm band-slit). The protein fluorescence was quenched by F-Hcy (0, 5, 10, 15, and 20 μM). To displace F-Hcy, 20 μM muscimol or baclofen were added. Fluorescence intensity depends on the polarity and the environment of the fluorophore. This is extensively proven for Trp fluorescence in proteins in which intensity will go up in a hydrophobic environment. To minimize this effect, we created Trp fluorescence intensity standard curves to estimate the bound antibody and the concomitant fluorescence intensity.
Treatment of MVEC With Hcy and MMP Activity by Zymography
The media containing serum is essential for normal cell growth and proliferation. Media contains all the necessary nutrients and amino acids for cell metabolism. The serum contains many growth factors. Previous studies from our laboratory demonstrated that serum induces growth promoters in endothelial cells (39). It is known that after the cells reach confluency, their growth is steady and needs only MEM. Therefore, to determine the direct effect of Hcy, independent of serum response, we treated confluent cells with Hcy in serum-free MEM. Confluent MVEC were washed with PBS at pH 7.4 and attenuated in serum-free media for 24 h at 37°C. Cells were incubated with Hcy at various concentrations (6, 12, and 20 μM) for 24 h. To determine the optimum time for MMP activity, the cells were incubated for 0, 6, 12, and 18 h with 12 μM Hcy. The culture medium was dialyzed in PBS overnight and concentrated with static concentrator Minicon B-15 (Millipore). MMP activity was measured with 1% gelatin SDS-polyacrylamide gel electrophoresis (PAGE) (40). The gels were stained with Coomassie blue and lytic activity was scanned by a densitometer (Odessey).
Western Blot Analysis of TIMP-1, -3, and -4, β-1 Integrin, ADAM-12, and GABA-A Receptor
Confluent MVEC were washed with PBS and attenuated in serum-free media for 24 h at 37°C. MVEC were incubated with 20 μM Hcy and 40 μM of Muscimol. Other MVEC were incubated with 20 μM Hcy and 40 μM of muscimol. The culture medium was collected and concentrated. MVEC membrane fraction protein was solubilized at 4°C for 2 h in buffer composed of 1% (w/v) Triton X-100, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 140 mM NaCl, 10 mM Tris-HCl pH 7.5, and protease inhibitors cocktail (leupeptin 1 mg/mL, pepstatin 0.7 mg/mL, trypsin inhibitors 0.1 mg/mL, phenylmethylsulfonyl fluoride 1 mM). Membrane fractions were also concentrated. Identical amounts of total protein were loaded onto 10% SDS-PAGE under reducing conditions. After SDS-PAGE, protein was transferred on to nitro-cellulose membranes by a Bio-Rad transblotter. TIMP-3 and -4 were measured by anti-TIMP-3 or -4 antibodies (1:1000 Sigma). Secondary antibody, anti-rabbit IgG alkaline phosphatase conjugate (1:000, Sigma) was used as the detection system. For GABA-A receptor blot, secondary antibody anti-mouse IgG alkaline phosphatase conjugate was used. TIMP-1 was measured by using anti-TIMP-1 monoclonal mouse antibody (1:10000, Calbiochem). Secondary anti-mouse horseradish peroxidase conjugate (1:5000, Chemicon) was used. ECL plus (Amersham Biosciences) was used as detection for horseradish peroxidase conjugate.
ADAM-12 and β-1 integrin in membrane fraction and β-1 in culture media were measured by anti-ADAM-12 or anti-β-1 integrin antibodies (1:1000, Chemicon). Actin was blotted using anti-actin antibody (Sigma) in cell extracts. Bands in Western blots were scanned by a densitometer.
Collagen Gel Constriction Assay
To determine the effect of Hcy on collagen constriction, native type 1 collagen gels were incubated with 106 MVEC. To prepare collagen gels, a stock solution of 3.4 mg/mL of native type1 collagen (Sigma) was dissolved in 12 mM HCl overnight at room temperature (16). Confluent MVEC were incubated in serum-free medium for 24 h at 37°C with 5% (v/v) CO2. Confluent cells were then suspended in 10X PBS (0.1 M Na2HPO4, 1.5 M NaCl). To achieve 1 mL of polymerizing collagen gel, 0.1 mL10X PBS containing MVEC were mixed with 0.08 mL 0.1 M NaOH and 0.82 mL collagen solution. One milliliter of aliquots was cast in each well of 24-well plates and incubated for 60 min at 37°C for gelation. Endothelial cells tend to aggregate in collagen gel, causing asymmetric constriction (34). For treatment with Hcy and GABA A/B agonists, muscimol, or baclofen were added to a final volume of 1 mL serum-free medium, and layered in collagen gel. Gel diameters were measured with Bio-Rad Quantity One software.
Effects of Muscimol, Baclofen, and Antibodies on Hcy-Mediated Collagen Gel Constriction by MVEC
Collagen gels containing 106 MVEC were incubated with or without Hcy at 37°C. Gel diameters were measured at 24 h. Effects of muscimol (GABA-A agonist) and baclofen (GABA-B agonist) on Hcy-mediated MVEC-induced collagen gel constriction were measured by incubating 40 μM of agonists with 20 μM Hcy. The antibody (1:100 dilution), GABA-A, B receptors, MMP-2, or ADAM-12 were added with 20 μM Hcy in 1-mL cell-free medium to collagen gels. Gels were incubated at 37°C for 24 h.
Statistical Analysis
Data were presented as average ±SD from six different experimental groups. We evaluated differences between groups using a two-way analysis of variance, followed by the Bonferroni post hoc test (41), focusing on effects of Hcy (E vs E+H) and treatment (Hcy treatment vs Hcy plus muscimol or baclofen treatments). Significance was indicated by p < 0.05.
RESULTS
GABA-A and B Receptors in MVEC
To determine whether MVEC were positive for GABA-A receptor, in situ immunolabeling was performed using GABA-A receptor β-chain antibody. Primary MVEC were positive for pericellular GABA-A receptor (Fig. 1A). To determine the number of MVEC that were positive for GABA-A receptor, we performed fluorescence analysis in MVEC labeled with CD-31-FITC and GABA-A receptor. The results suggested that 73 ± 9% of CD-31 positive MVEC were also positive for GABA-A receptor (Fig. 1C). Although Hcy seemed to have higher affinity for GABA-A than -B receptor (Fig. 1D,E), both muscimol and baclofen displaced the F-Hcy-bound GABA receptor. In addition, the strong leveling off in GABA-A receptor Trp fluorescence quenching by F-Hcy suggests buried and partly inaccessible Trp groups in GABA-A receptor (Fig. 1D).
Fig. 1.

Representative micrograph of GABA-A receptor labeling in microvessel endothelial cells (MVEC). Cells were grown onto cover slips. The adherent cells were labeled with anti-GABA-A receptor antibody (A). Control demonstrates cells labeled without primary antibody to GABA-A receptor (B). The magnification for both panels was ×40. The arrows indicate pericellular labeling. (C) Quantitative fluorescence analysis of MVEC positive for CD-31 and GABA-A receptor: FITC relative fluorescence intensity unit (FIU), scale 0–1 FIU, was measured in MVEC suspensions. Background represents negative control, with secondary FITC labeled antibody, suggesting no binding. Identical numbers of MVEC were used in each sample. (D) Fluorescence quenching of GABA-A receptor titrated with F-Hcy in the presence and absence of muscimol. (E) Fluorescence quenching of GABA-B receptor titrated with F-Hcy in the presence and absence of baclofen.
Hcy-Mediated MMP Activation and Suppression of the GABA Receptor
Hcy increased MMP-2 activity in a dose- and time-dependent manner (Fig. 2). We performed experiments with 12 and 20 μM Hcy for different times. Although at 12 μM Hcy, MMP activity leveled off at 18 h, at 20 μM, it leveled off at about 12 h (Fig. 2). These levels were associated with expression of MMP-2 and not with ex vivo MMP activation, because ex vivo incubation of MMP-2 with Hcy did not alter the MMP activity (data not shown).
Fig. 2.
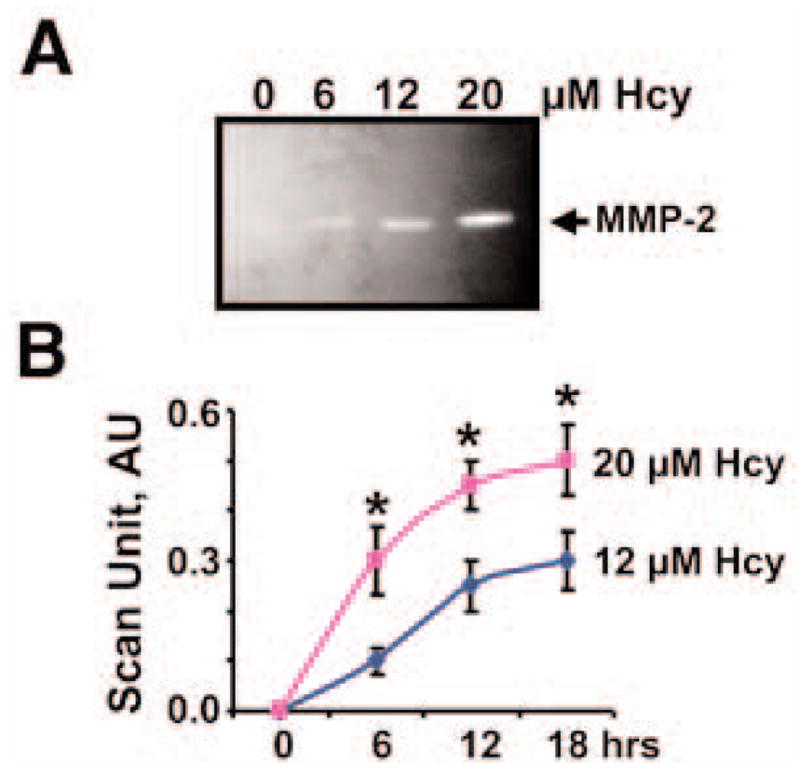
Hcy dose and Hcy time dependence of MMP-2 activity. (A) Representative zymographic analysis of medium from confluent MVEC cultured in serum-free MEM in the presence of 0, 6, 12, and 20 μM Hcy. (B) Line graphs of Hcy time-dependent MMP-2 activity, measured by zymography and scanned with a densitometer, at 0, 6, 12, and 18 h at 12 and 20 μM Hcy. *Significant difference (p < 0.01) when compared with 0 Hcy at 0 h of incubation.
To determine whether muscimol, anti-GABA-A receptor, and baclofen ameliorated the Hcy-induced MMP activation, Hcy-treated cells were cultured in the presence and absence of muscimol, anti-GABA-A receptor or baclofen. Muscimol, anti-GABA-A receptor, and baclofen decreased the Hcy-mediated MMP-2 activity significantly (p < 0.01) (Fig. 3). The results suggested that affinity of muscimol is higher than baclofen (Fig. 1D,E). The treatment with baclofen ameliorated the Hcy-induced MMP-2 to a lesser extent than muscimol. To determine whether muscimol induced GABA-A receptor in MVEC, Western blot analysis of cell extracts prepared from MVEC treated with Hcy, and with and without muscimol, suggested that Hcy decreased, whereas muscimol induced GABA-A receptor in MVEC (Fig. 3). The combination of Hcy and muscimol did not show any difference when compared to muscimol alone. This result suggested that Hcy has less impact in the presence of muscimol.
Fig. 3.

(A) Zymographic analysis of MMP-2 activity in the presence and absence of muscimol: 106 MVEC were cultured alone (E); with 40 μM muscimol (M); with 20 μM Hcy (H); and with Hcy and muscimol or anti-GABA-A antibody (AGA) or baclofen (B). After 24 h cell media were analyzed for MMP-2 activity. Western blot analysis of GABA-A receptor in MVEC. Cells were homogenized. Homogenates were loaded onto reducing SDS-PAGE and blotted using GABA-A receptor antibody. Corresponding actin bands are shown. (B) The bands in zymographic gels and Western blots were scanned and normalized with actin. *p < 0.01 compared with E alone. **p < 0.01 compared with E+H.
Differential Expression of TIMP-1, -3, and -4
To determine whether Hcy-induced TIMPs and muscimol attenuated this induction, MVEC were treated with Hcy and cultured in the presence or absence of muscimol. There was a selective increase in the levels of TIMP-1 and -3 by Hcy and a decrease in the level of TIMP-4. The treatment with muscimol decreased the Hcy-mediated TIMP-1 and -3, and increased Hcy-mediated TIMP-4 (Fig. 4). Interestingly, muscimol alone increased the levels of TIMP-4. The combination of muscimol and Hcy restored normal TIMP-4 levels.
Fig. 4.

Western blots analysis of TIMP-1, -3 and -4. (A) 106 MVEC were cultured alone (E); with 40 μM muscimol (M); with 20 μM Hcy (H); and with Hcy and muscimol. After 24 h, cell homogenates were prepared and analyzed for TIMP-1, -3, and -4 by Western blots. (B) The bands in Western blots were scanned and normalized with actin. *p < 0.01 compared with E alone. **p < 0.01 compared with E+H.
Hcy-Mediated β1-Integrin Shedding
To determine whether Hcy induced β-1-integrin and ADAM, MVEC were treated with Hcy. There was a robust increase in ADAM-12 by Hcy. Although slight, there was a significant (p < 0.01) decrease in β-1 integrin in MVEC and a robust increase in the medium of MVEC treated with Hcy, suggesting integrin-shedding by ADAM-12 (Fig. 5). Cotreatment of MVEC with Hcy and muscimol, anti-GABA-A receptor, or baclofen ameliorated the induction of ADAM.
Fig. 5.

Western blots analysis of membrane (mem) β-1 integrin, ADAM-12, and soluble (sol) β-1 integrin in the medium. (A) 106 MVEC cultured alone (E); with 40 μM muscimol (M); with 20 μM Hcy (H); and with Hcy and muscimol or anti-GABA-A antibody (AGA) or baclofen (B). After 24 h, cell homogenates were prepared and analyzed for β1 integrin (mem) and ADAM-12. Cell medium was collected and analyzed for β1 integrin (sol). (B) Bands in Western blots were scanned and normalized with actin. *p < 0.01 compared with E alone. **p < 0.01 compared with E+H.
To determine whether changes in ADAM in MVEC were associated with integrin shedding, we measured the levels of β-1 integrin in the medium of MVEC treated with Hcy in the presence and absence of muscimol, anti-GABA-A receptor, or baclofen. There was a robust increase in the shed β-1 integrin in the medium of MVEC treated with Hcy. This shedding was inhibited by cotreatment of the MVEC with muscimol, anti-GABA-A receptor or baclofen (Fig. 5).
To determine whether the integrin shedding is associated with collagen remodeling, the collagen gel constriction was measured. Hcy-induced collagen gel constriction was ameliorated by anti-GABA-A receptor, anti-GABA-B receptor, anti-MMP-2, and anti-ADAM-12 antibodies (Fig. 6). Hcy decreased the levels of intact β-1 integrin and at the same time increased the levels of soluble β-1 integrin in the cultured medium. This is consistent with the notion that ADAM-12 shed the integrin. Treatment with GABA receptor agonists ameliorated the Hcy-mediated integrin shedding.
Fig. 6.

(A) Hcy-mediated collagen gel constriction: E, 0 h, MVEC in collagen gel for 0 h; E, 24 h, MVEC in collagen gel for 24 h; E+Hcy, MVEC in collagen gel in the presence of 20 μM Hcy at 0 and 24 h. (B) Inhibition of Hcy (H)-mediated collagen gel constriction by anti-GABA-A antibody (AGA); by anti-GABA-B antibody (AGB); by anti-MMP-2 antibody (AM2), and by anti-ADAM-12 antibody (AAD). MVEC incorporated into collagen gels, and incubated with antibodies (1:100 dilutions) plus Hcy (20 μM) for 24 h. C, Collagen gel; C+E, Collagen+MVEC; C+E+H, Collagen+MVEC + Hcy. *p < 0.05 compared with C+E. **p < 0.05 compared with C+E+H.
DISCUSSION
The results of this study suggest a role of Hcy as an excitatory neurotransmitter in MVEC remodeling. Hcy mediated the transduction in MVEC by shedding the β1 integrin. This was, in part, the result of the increased levels of MMP-2 and ADAM-12 activity. The treatment with GABA-A receptor agonist (muscimol) and GABA-B receptor agonist (baclofen) of inhibitory neurotransmitter receptors ameliorated the MVEC transduction by inhibiting the integrin shedding and metalloproteinase activity.
We have demonstrated for the first time that MVEC contain GABA-A/B receptors, which were suppressed by Hcy (Fig. 1). The Ki’s reported for the interaction of muscimol and synaptic membrane extract were in millimolar range (30). Here, we reported the apparent Kds in micromolar range for the interaction of muscimol with immunopurified GABA receptors, that was deduced from Fig. 1D,E. The muscimol treatment induced the GABA-A receptor in MVEC and ameliorated the Hcy-mediated MMP activation (Figs. 2 and 3). Others suggested that treatment with GABA-A receptor antagonist resulted in increased levels of MMP-9 activity (42). This finding may suggest that Hcy behaves as an antagonist of GABA-A receptor. Elevated levels of Hcy, or hyperhomocysteinemia, attenuated and impaired the inhibitory neurotransmitter mechanism, preventing to decrease in blood pressure (26). It is known that the GABA-A receptor agonist, muscimol, decreases arterial blood pressure (43–45). There may also be a synergism between the increased levels of Hcy and blood pressure, causing microvascular dysfunction (7,8). The present study suggests that Hcy behaved as an antagonist of inhibitory neurotransmitter and synergized the microvascular dysfunction.
Remodeling, by its very nature, implies synthesis and degradation of connective tissue matrix. A critical balance between MMP and TIMP is associated with constitutive vascular remodeling. However, an imbalance leads to pathological remodeling. The role of differential regulation of TIMPs by Hcy suggested for the first time that Hcy increased TIMP-1 expression in MVEC. TIMP-1 induces mitogenesis in endothelial cells (46), and Hcy induces TIMP-1 (47). Our study suggests that muscimol attenuates Hcy-mediated increased TIMP-1 levels in MVEC. TIMP-3 levels were increased by Hcy treatment in MVEC (Fig. 4). Others have suggested increased TIMP-3 in smooth muscle cell apoptosis, leading to vascular remodeling (48). Our results may suggest that Hcy increases TIMP-3 levels, and that these levels are associated with increased vascular remodeling/apoptosis. The GABA-A receptor ameliorated the Hcy-mediated TIMP-3 induction and may decrease vascular apoptosis and remodeling. TIMP-4 levels were decreased by Hcy and reverted to control levels after treatment with muscimol (Fig. 4). This may suggest that the increased metalloproteinase activity was probably from the decreased levels of TIMP-4 during HHcy. However, the activation of an inhibitory neurotransmitter mechanism may ameliorate the elevated levels of metalloproteinase in HHcy.
The β-1 integrin deficiency caused a delay in the expression of a major neurotransmitter like GABA (49). Our results suggest that Hcy increased metalloproteinase and ADAM-12 and decreased β-1 integrin in MVEC. There was a robust increase in soluble β-1 integrin levels in the medium of MVEC cultured in the presence of Hcy. The treatment with muscimol or baclofen reverted ADAM-12, and β-1 levels to normal in the MVEC and β1 in the medium of MVEC. These results suggest that Hcy induced ADAM-12 and metalloproteinase, leading to β-1 integrin shedding (Fig. 5). The activation of inhibitory neurotransmitter in MVEC, ameliorated the transduction in MVEC and decreased the metalloproteinase activity and constrictive collagen remodeling (Fig. 6).
In summary, the anti-GABA-A antibody and agonist to GABA-B receptor (baclofen) ameliorated Hcy-mediated MMP activation. This suggests that Hcy-mediated toxic effects are neutralized by induction of GABA-A and B receptors. Our study also showed for the first time the role of GABA receptors in TIMPs expression and induction. The results on Hcy-mediated integrin shedding, and MMP activation suggest the role of Hcy in constrictive collagen remodeling. Induction of GABA receptors ameliorated the constrictive collagen remodeling. This mechanism may have significance in amelioration of cerebrovascular remodeling in hyperhomocysteinemia.
Acknowledgments
A part of this study was presented at the Annual Meeting of Experimental Biology 2003, San Diego, CA. This work was supported in part by National Institutes of Health grants HL-71010 and HL-74185.
References
- 1.Horstmann S, Kalb P, Kozoil J, Gardner H, Wagner S. Profiles of MMPs, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke. 2003;34:2165–2170. doi: 10.1161/01.STR.0000088062.86084.F2. [DOI] [PubMed] [Google Scholar]
- 2.Li Z, Sun L, Zhang H, et al. Elevated plasma Hcy was associated with hemorrhagic and ischemic stroke, but methylenetetrahydrofolate reductase gene C677T polymorphism was a risk factor for thrombotic stroke. Stroke. 2003;34:2085–2090. doi: 10.1161/01.STR.0000086753.00555.0D. [DOI] [PubMed] [Google Scholar]
- 3.Sutton-Tyrrell K, Bostom A, Selhub J, Zeigler-Johnson C. High homocysteine levels are independently related to isolated systolic hypertension in older adults. Circulation. 1997;96:1745–1749. doi: 10.1161/01.cir.96.6.1745. [DOI] [PubMed] [Google Scholar]
- 4.Lentz SR, Erger RA, Dayal S, Maeda N, Malinow MR, Heistad DD, et al. Folate dependence of HHcy and vascular dysfunction in CBS mice. Am J Physiol. 2000;279:H970–H975. doi: 10.1152/ajpheart.2000.279.3.H970. [DOI] [PubMed] [Google Scholar]
- 5.Upchurch GR, Jr, Welch GN, Fabian AJ, et al. Homocysteine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J Biol Chem. 1997;272:17012–17017. doi: 10.1074/jbc.272.27.17012. [DOI] [PubMed] [Google Scholar]
- 6.Upchurch GR, Jr, Welch GN, Fabian AJ, Pigazzi A, Keaney JF, Jr, Loscalzo J. Stimulation of endothelial nitric oxide production by homocysteine. Atherosclerosis. 1997;132:177–185. doi: 10.1016/s0021-9150(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 7.Miller A, Mujumdar V, Palmer L, Bower JD, Tyagi SC. Reversal of endocardial endothelial dysfunction by folic acid in homocystinemic hypertensive rats. Am J Hyperten. 2002;15:157–163. doi: 10.1016/s0895-7061(01)02286-5. [DOI] [PubMed] [Google Scholar]
- 8.Miller A, Mujumdar V, Shek E, Guillot J, Angelo M, Palmer L, et al. Hyperhomocyst(e)inemia induces multiorgan damage. Heart & Vessels. 2000;15:135–143. doi: 10.1007/s003800070030. [DOI] [PubMed] [Google Scholar]
- 9.Ungvari Z, Pacher P, Rischak K, Szollar L, Koller A. Dysfunction of NO mediation in isolated rat arterioles with methionine-induced HHcy. Arterioscler Thromb Vasc Biol. 1999;19:1899–1904. doi: 10.1161/01.atv.19.8.1899. [DOI] [PubMed] [Google Scholar]
- 10.Finkelstein JD. The metabolism of Hcy: pathways and regulation. Eur J Pediatr. 1998;157:S40–S44. doi: 10.1007/pl00014300. [DOI] [PubMed] [Google Scholar]
- 11.Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 12.Baumbach GL, Sigmund CD, Bottiglieri T, Lentz SR. Structure of cerebral arterioles in cystathionase beta synthase-deficient mice. Circ Res. 2002;91:931–937. doi: 10.1161/01.res.0000041408.64867.1d. [DOI] [PubMed] [Google Scholar]
- 13.Zhang JW, Deb S, Gottschall PE. Regional and differential expression of gelatinases in rat brain after systemic kainic acid or bicuculline administration. Eur J Neurosci. 1998;10:3358–3368. doi: 10.1046/j.1460-9568.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 14.Hunt MJ, Tyagi SC. Peroxisome proliferators compete and ameliorate homocysteine-mediated endocardial endothelial cells activation. Am J Physiol. 2002;283:C1073–C1079. doi: 10.1152/ajpcell.00152.2002. [DOI] [PubMed] [Google Scholar]
- 15.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 16.Mujumdar VS, Tyagi SC. Temporal regulation of extracellular matrix components in transition from compensatory hypertrophy to decompensatory heart failure. J Hypertens. 1999;2:261–270. doi: 10.1097/00004872-199917020-00011. [DOI] [PubMed] [Google Scholar]
- 17.Tyagi SC, Hoit BD. Metalloproteinase in myocardial adapatation and maladaptation. J Cardiovasc Pharmacol Ther. 2002;7:241–246. doi: 10.1177/107424840200700407. [DOI] [PubMed] [Google Scholar]
- 18.Lampugnani MG, Resnati M, Dejana E, Marchisio PC. The role of integrins in the maintenance of endothelial monolayer integrity. J Cell Biol. 1991;112:479–90. doi: 10.1083/jcb.112.3.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnard EA, Skolnick P, Olsen RW, et al. International union of pharmacology. XV Subtypes of GABA-a receptors: classification on the basis of subunit structure and receptor function. Pharmacol Rev. 1998;50:291–312. [PubMed] [Google Scholar]
- 20.Bowery NG, Bettler B, Froestl W, et al. International union of pharmacology: XXXIII. Mammalian GABA-β receptors: structure and function. Pharmacol Rev. 2002;54:247–263. doi: 10.1124/pr.54.2.247. [DOI] [PubMed] [Google Scholar]
- 21.Chebib M, Johnston GA. The ABC of GABA receptors. Clin Exp Pharmacol Physiol. 1999;26:937–940. doi: 10.1046/j.1440-1681.1999.03151.x. [DOI] [PubMed] [Google Scholar]
- 22.Krantis A. GABA in the mammalian enteric nervous system. News Physiol Sci. 2000;15:284–289. doi: 10.1152/physiologyonline.2000.15.6.284. [DOI] [PubMed] [Google Scholar]
- 23.Mifflin SW. What does the brain know about blood pressure. News Physiol Sci. 2001;16:266–271. doi: 10.1152/physiologyonline.2001.16.6.266. [DOI] [PubMed] [Google Scholar]
- 24.Zhang HY, McPherson BC, Liu H, Maman TS, Rock P, Yao Z. H2O2 opens mitochondrial K-ATP channels and inhibits GABA receptors via PKC-epsilon in cardiomyocytes. Am J Physiol. 2002;282:H1395–H1403. doi: 10.1152/ajpheart.00683.2001. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Liu G. A novel method to determine the localization of high and low affinity GABA transporters to the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Res Brain Res Protoc. 1999;4:288–294. doi: 10.1016/s1385-299x(99)00031-8. [DOI] [PubMed] [Google Scholar]
- 26.Kishi T, Hirooka Y, Sakai K, Shigematsu H, Shimokawa H, Takeshita A. Overexpression of eNOS in the RVLM causes hypotension and bradycardiac via GABA release. Hypertension. 2001;38:896–901. [PubMed] [Google Scholar]
- 27.Li YF, Patel KP. Paraventricular nucleus of the hypothalamus and elevated sympathetic activity in heart failure: the altered inhibitory mechanisms. Act Physiol Scand. 2003;177:17–26. doi: 10.1046/j.1365-201X.2003.01043.x. [DOI] [PubMed] [Google Scholar]
- 28.Takayasu M, Dacey RG. Effects of inhibitory and excitatory amino acid neurotransmitters on isolated cerebral parenchymal arterioles. Brain Res. 1989;482:393–396. doi: 10.1016/0006-8993(89)91207-9. [DOI] [PubMed] [Google Scholar]
- 29.Mujumdar VS, Aru GM, Tyagi SC. Induction of oxidative stress by homocyst(e)ine impairs endothelial function. J Cell Biochem. 2001;82:491–500. doi: 10.1002/jcb.1175. [DOI] [PubMed] [Google Scholar]
- 30.Griffiths R, Williams DC, O’Neill C, Dewhurst IC, Ekuwem CE, Sinclair CD. Synergistic inhibition of muscimol binding to calf-brain synaptic membrances in the presense of L-homocysteine and pyridoxal 5′-phosphate. Eur J Biochem. 1983;137:467–478. doi: 10.1111/j.1432-1033.1983.tb07850.x. [DOI] [PubMed] [Google Scholar]
- 31.Folbergrova J. NMDA and not non-NMDA receptor antagonists are protective against seizures induced by homocysteine in neonatal rats. Exp Neurol. 1994;130:344–350. doi: 10.1006/exnr.1994.1213. [DOI] [PubMed] [Google Scholar]
- 32.Olney JW, Price MT, Salles S, et al. L-homocysteic acid: an endogenous excitotoxic ligand of the NMDA receptor. Brain Res Bull. 1987;19:597–602. doi: 10.1016/0361-9230(87)90077-3. [DOI] [PubMed] [Google Scholar]
- 33.Dardik R, Varon D, Tamarin I, Zivelin A, Salomon O, Shenkman B. Hcy and ox-LDL enhanced platelet adhesion to endothelial cells under flow conditions: distinct mechanisms of thrombogenic modulation. Thromb Haemost. 2000;83:338–344. [PubMed] [Google Scholar]
- 34.Mujumdar VS, Tummalapalli CM, Aru GM, Tyagi SC. Mechanism of constrictive vascular remodeling: a role of PPAR. Am J Physiol. 2002;282:C1009–C1015. doi: 10.1152/ajpcell.00353.2001. [DOI] [PubMed] [Google Scholar]
- 35.Shastry S, Tyagi SC. Homocysteine induces metalloproteinase and shedding of β-1 integrin in microvessel endothelial cells. J Cell Biochem. 2004;93:207–213. doi: 10.1002/jcb.20137. [DOI] [PubMed] [Google Scholar]
- 36.Shastry S, Moning L, Tyagi N, Steed M, Tyagi SC. GABA receptors and nitric oxide ameliorate constrictive collagen remodeling in hyperhomocysteinemia. J Cell Physiol. 2005;205:422–427. doi: 10.1002/jcp.20416. [DOI] [PubMed] [Google Scholar]
- 37.Audus KL, Borchardt RT. Bovine brain microvessel endothelial cell monolayers as a model system for the blood-brain barrier. Ann N Y Acad Sci. 1987;507:9–18. doi: 10.1111/j.1749-6632.1987.tb45787.x. [DOI] [PubMed] [Google Scholar]
- 38.Tyagi SC. Homocysteine redox receptor and regulation of extracellular matrix components in vascular cells. Am J Physiol. 1998;274:C396–C405. doi: 10.1152/ajpcell.1998.274.2.C396. [DOI] [PubMed] [Google Scholar]
- 39.Tyagi SC, Kumar SG, Glover G. Induction of tissue inhibitor and matrix metalloproteinase by serum in human heart-derived fibroblast and endomyocardial endothelial cells. J Cell Biochem. 1995;58:360–371. doi: 10.1002/jcb.240580309. [DOI] [PubMed] [Google Scholar]
- 40.Tyagi SC, Matsubara L, Weber KT. Direct extraction and estimation of collagenase(s) activity by zymography in microquantities of rat myocardium and uterus. Clin Biochem. 1993;26:191–198. doi: 10.1016/0009-9120(93)90025-2. [DOI] [PubMed] [Google Scholar]
- 41.Tarone RE. A modified Bonferroni method for discrete data. Biometrics. 1990;46:515. [PubMed] [Google Scholar]
- 42.Zhang F, Slungaard A, Vercellotti GM, Iadecola C. Superoxide-dependent cerebrovascular effects of homocysteine. Am J Physiol. 1998;274:R1704–R1711. doi: 10.1152/ajpregu.1998.274.6.R1704. [DOI] [PubMed] [Google Scholar]
- 43.Baum T, Becker FT. Hypotensive and postural effects of the GABA agonist muscimol and of clonidine. J Cardiovasc Phamacol. 1982;4:165–169. doi: 10.1097/00005344-198203000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Sweet CS, Wenger HC, Gross DM. Central antihypertensive properties of muscimol and related GABA agonists and the interaction of muscimol with baroceptor reflexes. Can J Physiol Pharmacol. 1979;57:600–605. doi: 10.1139/y79-092. [DOI] [PubMed] [Google Scholar]
- 45.Unger T, Becker H, Dietz R, et al. Antihypertensive effect of the GABA receptor agonists muscimol in SHR. Role of the sympathoadrenalaxis. Circ Res. 1984;54:30–37. doi: 10.1161/01.res.54.1.30. [DOI] [PubMed] [Google Scholar]
- 46.Tyagi SC, Meyer L, Kumar SG, Schmaltz RA, Reddy HK, Voelker DJ. Induction of tissue inhibitor of metalloproteinase and its mitogenic response to endothelial cells in human atherosclerotic and restenotic lesions. Can J Cardiol. 1996;12:353–362. [PubMed] [Google Scholar]
- 47.Yang Z, Zou AP. Homocysteine enchances TIMP-1 expression and cell proliferation associated with NADPH oxidase in rat mesangial cells. Kidney Int. 2003;63:1012–1020. doi: 10.1046/j.1523-1755.2003.00825.x. [DOI] [PubMed] [Google Scholar]
- 48.Baker AH, Zaltsman AB, George SJ, Newby AC. Divergent effects of tissue inhibitor of metalloproteinase-1, -2, or -3 overexpression on rat vascular smooth muscle cell invasion, proliferation, and death in vitro. TIMP-3 promotes apoptosis. J Clin Invest. 1998;101:1478–1487. doi: 10.1172/JCI1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andressen C, Arnhold S, Puschmann M, et al. β1 integrin deficiency impairs migration and proliferation of mouse embryonic stem cell derived neurons. Neurosci Lett. 1998;251:165–168. doi: 10.1016/s0304-3940(98)00535-7. [DOI] [PubMed] [Google Scholar]