

Abstract
The studies of the HFE mutations: H63D and C282Y in North African populations have revealed the extreme rarity or even the absence of the C282Y mutation. We have examined 200 chromosomes (100 Libyan people live in Benghazi) for the presence of the two HFE mutations by PCR-RFLP analysis by using PCR conditions used to amplify both Autosomal and Y chromosomal STRs. We have found that the allele frequencies are, respectively, 17% for the H63D and 0% for the C282Y. These results are consistent with the worldwide spread of the H63D mutation and the north European restriction of the C282Y.
Keywords: HFE gene mutations, H63D, C282Y, haemochromatosis, allele frequency, Benghazi, Libyan population
Introduction
Hereditary hemochromatosis (HH) is an autosomal recessive blood disorder that is characterized by an increase in iron absorption, which, if untreated, can lead to excessive iron deposits in organs and eventual organ failure [1]. HH is caused by a mutation of the HFE gene that regulates iron absorption. Since the disease is recessive, a person must have two mutated genes in order for the disorder to be expressed (termed homozygous). The two main mutations that can occur are C282Y mutation (accounting for 90-95% of cases) and H63D mutation. H63D is associated with a less severe form of iron absorption than a C282Y mutation. Although fairly uncommon, people can also exhibit signs and symptoms of HH if they possess only one mutated gene (termed heterozygous), but generally they are only carriers in this case [2]. Most people absorb roughly one milligram of iron per day from their diets, which is then equally excreted through sweat, sloughing skin cells, urine, and the gastrointestinal tract. In contrast, people with HH can absorb anywhere from two to four milligrams per day, but the excretion rate remains the same [3]. Premenopausal women tend to have lower iron levels and fewer complications from HH than men since they lose blood (and iron) during menstruation [4]. Over time the body begins to deposit excess iron in organs such as the liver, heart, brain, pancreas, and lungs [2]. Common symptoms of HH include extreme fatigue, impotence and infertility, skin hyperpigmentation (also referred to as bronzing), depression, abdominal pain, arthritis/joint pain, diabetes, liver disease, and heart disease [5]. Diagnosis typically does not occur until between the ages 40 to 60, when iron levels have already reached toxic, damaging levels [2]. Since 1996, several studies have been performed in order to evaluate the H63D and C282Y frequencies in different populations and to provide information about the worldwide HFE mutations distribution. We notified that no or few information was found about the H63D and C282Y frequencies in Libyan and in North Africa generally. The aims of our study are to determine the two missense mutations frequencies in Libyan population living in Benghazi trying to use this phenotyping disease (correlation between phenotyping and genotyping) in positive samples in Human identification for forensic application and to compare these results to other studies.
Population
Libya, a Northern African country, was first inhabited by Berbers, followed by Phoenicians, Greeks, Romans, Arabs and Ottomans. Libya became independent in 1951 after a brief period as an Italian colony; it had been invaded by Italy in 1911. In February 2011 an uprising against the government occurred in the city. Benghazi is the second largest city in Libya and the main city (or capital) of the Cyrenaica region (or ex-Province), located in the North of Africa. Benghazi is located half way between Tripoli in the West (a distance of approximately 1000 Km between these cities) and Cairo in the East (also approximately 1000 Km) (Figure 1). Cyrenaica is surrounded by desert on three sides; hence in ancient times the most accessible civilization was to the North, across the Mediterranean, in Crete and Greece, only 400 km away. The population of Benghazi was 500, 120 in 1995 (census) and increased to 670, 797 in the 2006 census. As with other cities in Libya, there is a reasonable amount of ethnic diversity in Benghazi. The people of eastern Libya, Benghazi included, have in the past always been of predominantly Arab descent. In recent times, however, there has been an influx of African immigrants into Benghazi. There are also many Egyptian immigrants in Benghazi. A small Greek community also exists in Benghazi; the Greek island of Crete is a short distance from Benghazi and many families in Benghazi today bear Cretian surnames.
Figure 1.
Map of Libya showing the city of Benghazi (http://en.wikipedia.org/wiki/Benghazi).
In modern times, Benghazi has seen a lot of Libyans from different parts of the country move into the city, especially since the Kingdom era (1951-1969). Many Libyans came to Benghazi from Misrata (About 60% of the population have roots from Misrata, West of Benghazi).
Materials and methods
Informed consent was obtained from100 unrelated Libyan individuals (Benghazi region). Fifty of those samples are male and fifty are female.
DNA extraction
DNA was extracted from blood stains collected on FTA® cards (Whatman, Kent, UK) using FTA® Purification Reagent (Whatman) following the manufacturer's protocol and from buccal swabs using QIAamp®DNA Blood Mini Kit (QIAGEN, Hilden, Germany). DNA was quantified using a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, USA).
PCR amplification
About 1.2 mm FTA® disc and 1 ng of DNA purified from buccal swabs was used .Separate PCR are conducted for the two mutations using the pairs of primers previously described by Feder, et al; 1996 [6]. The PCR mixtures contained 16 pmol of primers, 200 μmol of each desoxy nucleotide triphosphate, 100 ng of genomic DNA, 2.5 μl 10×buffers and 0.2 U of Taq polymerase in a final volume of 25 μl. These amplifications were done for 5-min denaturizing at 95 °C followed by 30 cycles of 30-s denaturation at 94° C, 30-s annealing at 58°C and 1-min extension at 72°C. Finally an extension cycle of 5 min at 72°C. Then RFLP analysis [7, 8]. Were made in order to determine the HFE genotype of these samples.
We have tested some amplification of these mutations with PCR conditions used to amplify Autosomal and Y Chromosomal kits and good results were obtained.
PCR was carried out in a Thermocycler 9700 from Applied Biosystems. PCR conditions were as follows: starting temperature was 95°C for 11 min, (denaturing temperature 95°C for 1min, annealing 59°C for 1min, extension 60° C for 1min, 27 cycles), then 60°C for 80 minutes and hold at 4°C, according to the manufacturer's protocol (AmpFlSTR® Yfiler™ and Identifiler Amplification KitsUser's Manual).
C282Y and H63D mutations were screened using enzymatic digestion of which PCR products encompassing the mutation sites.' The C282Y mutation creates a found in new RsaI restriction site. The 390 bp PCR reaction product (forward primer 5'-TGGCAAGGGTAAACAG ATCC-3' and reverse primer 5' CTCAGGCACTCCT-CTCAACC-3') digested with RsaI (E coli strain, gene from Rhodopseudomonas shaeroides) shows cites of two fragments of 249 and 141 bp in normal DNA, while mutant DNA generates two new fragments (112 and 29 bp). The H63D mutation destroys an MboI (E coli strain, gene from Moraxella bovis) site 294 bp PCR product (forward primer 5'-ACATGGTTAAGGCCTGTTGC-3' and reverse primer 5'-CTTGCTGTGGTTGTGATT TTCC-3'), while normal DNA generates three fragments of 138, 99, and 57 bp.
13 μl of the PCR product of 208 bp (H63D) and 390 bp (C282Y) were digested using, respectively, 4 U of MboI (Biolabs, R0147S) and 2U RsaI (Biolabs, R0167S) enzyme. Added to 4U NEBuffer 4 In a final step, the digested products were incubated at 37°C. Over night and run on a 2 % agarose gel for 1 h and photographed under UV light.
Results
Two hundred Libyan chromosomes from Benghazi were analysed. H63D and C282Y mutation were observed at frequencies of 17% and 0 %, respectively.
Of 100 samples from Benghazi (50 females and 50 males) analysed for both H63D and C282Y mutation.
For H63D mutation 17% were positive for site 294 bp and other samples Amplify normal H63d sites (138, 99, and 57 bp) (Figure 2).
Figure 2.
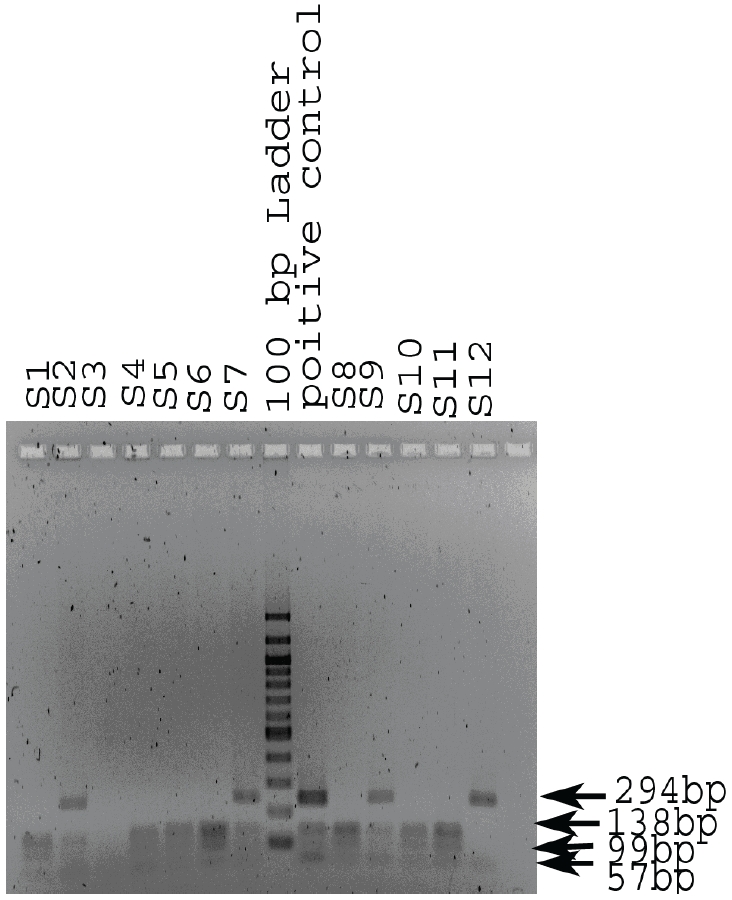
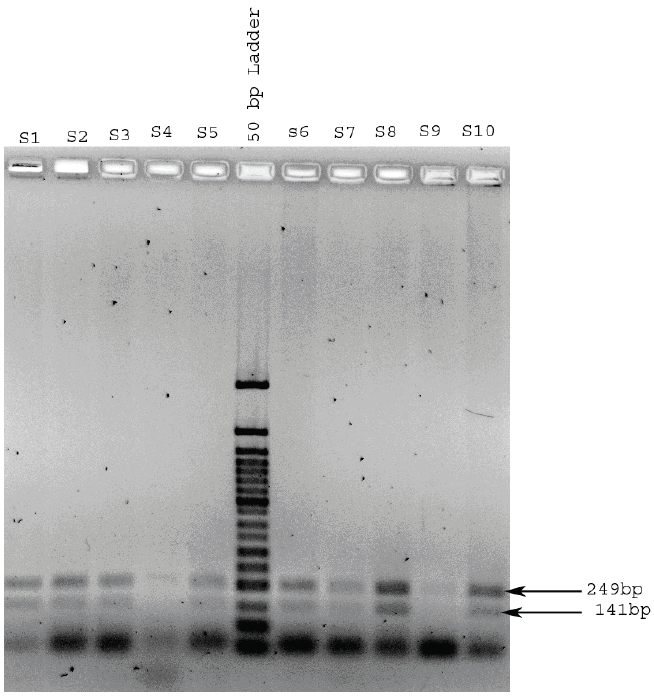
Determination of genotype in the HFE gene codon 282 region. The normal genotype gives rise to 2 RsaI fragments of the codon 282 region PCR product (203bp and 152bp) these were seen for all samples tested on this and other gels. The C282Y mutation, if present, introduces a second RsaI site resulting in three bands (203bp, 123bp and 29bp).
And 0% for C282Y mutation (in all 100 samples only normal two sites observed 141bp and 249bp) positive mutation sites not detected (112 and 29bp) (Figure 3).
Figure 3.
Determination of genotype in the HFE gene codon 63 region. The normal genotype gives rise to 3 MboI fragments of the codon 63 region PCR product (138bp, 99bp and 57bp) (H1, H3, H4, H5, H7, H9 and H10). The H63D mutation abolishes one MboI site resulting in the 237bp band. Heterozygotes give all 4 bands (H2, H6, H8 and the positive control). H11 is homozygous for H63D (237bp and 57bp only).
Discussion and conclusion
The discovery of the HFE gene and the C282Y, H63D mutations implicated in the onset of HH have been pursued researchers to study the prevalence of these two missense mutation around the world (Table 1). However, few information was found about the H63D and C282Y mutation distribution in North Africa. [9-12] and in the Arabian population [13]. This indicated the low prevalence or the absence of the C282Y mutation in these populations. Our study included 100 samples collected from Benghazi (Eastern Libya). The H63D allele frequency in Libyan population seems to be higher compared with Algerian Mzab population (9%) [11] and that found in Saudi Arabians population (8.5%) [13]. that frequency is getting closer to that reported from European population like British Irish (18.9%) [13] but remains lesser than the highest European frequency found in Spain (Table 1). Our results agree with the fact that the mutation responsible for the His 63 Asp substitution is not restricted to the European population.
Table 1.
Estimated prevalence of Haemochromatosis mutation in the general population
| N | H63D allele frequency | C282Y frequency | References | |
|---|---|---|---|---|
| Europe | ||||
| North | ||||
| North Ireland | 404 | 14.10% | 9.9% | Murphy, 1998 |
| Denmark | 200 | 12.75% | 6.75% | Steffen, 1998 |
| Norway | 94 | 11.20% | 6.4% | Clarke, 1997 |
| Sweden | 117 | 12.39% | 3.84% | Cardoso, 1998 |
| South | ||||
| France | 503 | 12.23% | 1.07% | Piperno, 1998 |
| Germany | 153 | 14.07% | 2.96% | Gottschalk, 1998 |
| Greece | 196 | 13.50% | 1.3% | Clarke, 1997 |
| Spain | 420 | 20.40% | 3.09% | Sanchez, 1998 |
| Turkey | 70 | 13.57% | 0% | Clarke, 1997 |
| Africa/Middle East | ||||
| Middle East | ||||
| Saudi Arabia | 118 | 8.5% | 0% | Clarke, 1997 |
| North | ||||
| North Africa | 171 | 13.2% | 9.0% | Aguilar, 1997 |
| Benghazi-Libya | 100 | 17% | 0% | Current study |
| South | ||||
| Gambia | 39 | 1.3% | 0% | Clarke, 1997 |
| Senegal | 130 | 0% | 0% | Clarke, 1997 |
| Kenya | 78 | 1.3% | 0% | Clarke, 1997 |
| Nigeria | 80 | 1.9% | 0% | Clarke, 1997 |
| Zambia | 76 | 0.7% | 0% | Clarke, 1997 |
| Asia | ||||
| China | 72 | 2.8% | 0% | Clarke, 1997 |
| Indonesia | 90 | 2.8% | 0% | Clarke, 1997 |
| Taiwan | 80 | 1.9% | 0% | Clarke, 1997 |
| Australasia | ||||
| Australia (aboriginals) | 93 | 0% | 0% | Clarke, 1997 |
| Australia (vanuatuans) | 90 | 0.6% | 0% | Clarke, 1997 |
| America | ||||
| Mexico | 54 | 6.5% | 0% | Clarke ,1997 |
| Jamaica | 90 | 2.2% | 1.1% | Clarke, 1997 |
| United States | 5171 | 13.5% | 5.4% | Karen, 2001 |
Roth et al. thought that the presence of the H63D around the world and the restriction of the C282Y to European populations may suggest that appears of the first preceded the second.
We have found that the H63D allele frequency is 17% and the C282Y allele frequency is 0%. More detailed studies are needed to identify the correlation between genotype and the phenotype of HH in our population and to determine whether the H63D mutation itself can results in clinical haemochromatosis, or whether other associated risk factors are required in Human identification.
Acknowledgments
The Libyan government and the School of Natural and Applied Sciences - University of Lincoln -UK funded this research as a part of a PhD scholarship in Forensic genetics.
References
- 1.Phatak P, Bonkovsky H, Kowdley K. Hereditary hemochromatosis: Time for targeted screening. Ann Intern Med. 2008;149:270–272. doi: 10.7326/0003-4819-149-4-200808190-00009. [DOI] [PubMed] [Google Scholar]
- 2.Plaut D, McLellan W. Hereditary hemochromatosis. J Contin Educ Topics & Issues. 2009;11:18–21. [Google Scholar]
- 3.Schrier S, Bacon B. Clinical manifestations of hereditary hemochromatosis. 2008 Retrieved on June 26, 2011, from UpToDate database licensed to Yakima Valley Memorial Hospital. [Google Scholar]
- 4.Bring P, Partovi N, Ford J, Yoshida E. Iron overload disorders: treatment options for patients refractory to or intolerant of phlebotomy. Pharmacother. 2008;28:331–342. doi: 10.1592/phco.28.3.331. [DOI] [PubMed] [Google Scholar]
- 5.Weinberg ED. Nashville, TN: Cumberland House Publishing, Inc; 2004. Exposing the hidden dangers of iron. [Google Scholar]
- 6.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, Wolff RK. A novel MHC classI like gene is mutated in patients with hereditary hemochromatosis. Nat Genet. 1996;14:249–251. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 7.Aguilar P, Martinez PH. Jeanfeau C, Masmejean A, Guillard C, Biron H, Robesandratana J, Schved. Simple and rapid detection of the newly described mutations in the HLA-H gene. Blood. 1997;89:1835–1836. [PubMed] [Google Scholar]
- 8.Lynas C. A cheaper and more rapid polymerase chain reaction restriction fragment polymorphism method for the detection of the HLA-H gene mutations occurring in hereditary hemochromatosis. Blood. 1997;90:4235. [PubMed] [Google Scholar]
- 9.Aguilar-Martinez P, Picot MC, Becker F, Boulot P, Montoya F, Mares P, Bachelard B, Henry Y, Delarbre JL, Sarda P, Schved JF. Prevalence of HFE mutations in people from North Africa living in southern France. Br J Haematol. 2001;114:914–916. doi: 10.1046/j.1365-2141.2001.03005.x. [DOI] [PubMed] [Google Scholar]
- 10.Merryweather Clarke AT, Pointon JJ, Joanolle AM, Rochette J, Robson KJ. Geography of HFE C282Y and H63D mutations. Genet Test. 2000;4:183–198. doi: 10.1089/10906570050114902. [DOI] [PubMed] [Google Scholar]
- 11.Roth M, Giraldo P, Hariti G, Polni ES, Sanchez-Mazas A, Stefane GF, Digoujou GM, Coppin H. Absence of the hemochromatosis gene Cys 282 Tyr mutation in three ethnic groups from Algeria (Mzab), Ethiopia, and Senegal, Immunogenetics. 1997;46:222–225. doi: 10.1007/s002510050265. [DOI] [PubMed] [Google Scholar]
- 12.Zorai A, Hartveld CL, Rachdi R, Dellagi K, Abbes S, Delbini P, Giordano PC. Frequency and spectrum of hemochromatosis mutations in Tunisia. Hematol J. 2003;4:433–435. doi: 10.1038/sj.thj.6200337. [DOI] [PubMed] [Google Scholar]
- 13.Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative hemochromatosis mutations. J Med Genet. 1997;34:275–278. doi: 10.1136/jmg.34.4.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy S, Curran MD, McDougall N, Callender ME, O'Brien CJ, Middleton D. High incidence of the Cys 282 Tyr mutation in the HFE gene in Irish population implications for haemochromatosis. Tissue Antigens. 1998;52:484–488. doi: 10.1111/j.1399-0039.1998.tb03076.x. [DOI] [PubMed] [Google Scholar]
- 15.Steffen R, Varning K, Jersild C. Determination of gene frequencies for two common haemochromatosis mutations in the Danish population by a novel polymerase chain reaction with sequence-specific primers. Tissue Antigens. 1998;52:230–235. doi: 10.1111/j.1399-0039.1998.tb03037.x. [DOI] [PubMed] [Google Scholar]
- 16.Cardoso EM, Stål P, Hagen K, Cabeda JM, Esin S, de Sousa M, Hultcrantz R. HFE mutations in patients with hereditary haemochromatosis in Sweden. J Int Med. 1998;243:203–208. doi: 10.1046/j.1365-2796.1998.00270.x. [DOI] [PubMed] [Google Scholar]
- 17.Piperno A, Sampietro M, Pietrangelo A, Arosio C, Lupica L, Montosi G, Vergani A, Fraquelli M, Girelli D, Pasquero P, Roetto A, Gasparini P, Fargion S, Conte D, Camaschella C. Heterogeneity of hemochromatosis in Italy. Gastroenterology. 1998;114:996–1002. doi: 10.1016/s0016-5085(98)70319-1. [DOI] [PubMed] [Google Scholar]
- 18.Gottschalk R, Seidl C, Löffler T, Seifried E, Hoelzer D, Kaltwasser JP. HFE codon 63/282 (H63D/C282Y) dimorphism in German patients with genetic hemochromatosis. Tissue Antigens. 1998;51:270–275. doi: 10.1111/j.1399-0039.1998.tb03101.x. [DOI] [PubMed] [Google Scholar]
- 19.Sánchez M, Bruguera M, Bosch J, Rodés J, Ballesta F, Oliva R. Prevalence of the Cys 282 Tyr and His 63Asp HFE gene mutations in Spanish patients with hereditary hemochromatosis and in controls. J. Hepatol. 1998;29:725–728. doi: 10.1016/s0168-8278(98)80252-3. [DOI] [PubMed] [Google Scholar]
- 20.Steinberg KK, Cogswell ME, Chang JC, Caudill SP, McQuillan GM, Bowman BA, Grummer-Strawn LM, Sampson EJ, Khoury MJ, Gallagher ML. Prevalence of C282Y and H63D mutations in the hemochromatosis (HFE) gene in the United States. J Am Med Assoc. 2001;285:2216–2222. doi: 10.1001/jama.285.17.2216. [DOI] [PubMed] [Google Scholar]