

Abstract
The definition of the clinicopathological entity of amyotrophic lateral sclerosis evolved over half a century. Although the definitive term amyotrophic lateral sclerosis that acknowledged both upper and lower motor neuron involvement was attributed to Jean-Martin Charcot in 1874, his initial case was published nearly a decade earlier; and it is accepted that, from at least the 1830s, several others (including Charles Bell, François-Amilcar Aran and Jean Cruveilhier) had already recognized a progressive lower motor neuron-only syndrome within a broader, clinically-defined group of disorders, termed progressive muscular atrophy. Although William Gowers first grouped the three phenotypes of amyotrophic lateral sclerosis, progressive muscular atrophy and progressive bulbar palsy together as part of the same syndrome, the term motor neuron disease, as an over-arching label, was not suggested until nearly a century later by W. Russell Brain. Augustus Jacob Lockhart Clarke (1817–80) is best known for his descriptions of spinal cord anatomy. However, in two detailed case reports from the 1860s, he carried out rigorous post-mortem neuropathological studies of what appear to be classical cases of amyotrophic lateral sclerosis. Furthermore, he recognized the additional involvement of the corticospinal tracts that distinguished this from progressive muscular atrophy. Several aspects of the exquisite clinical histories documented as part of both studies, one by Charles Bland Radcliffe, resonate with contemporary debates concerning the evolution of disease in amyotrophic lateral sclerosis. These ‘past masters’ still have much to teach us.
Keywords: amyotrophic lateral sclerosis, motor neuron disease, Lockhart Clarke, Radcliffe, Charcot
Introduction
Few question Jean-Martin Charcot’s primacy (Charcot, 1874) in the definition of the simultaneous involvement of corticospinal tract and anterior horn cell neurons in the syndrome he termed ‘la sclérose amyotrophique’, or amyotrophic lateral sclerosis (ALS) (Mitchell, 1992). ALS is the most common phenotype of this typically rapidly progressive neurodegenerative disorder. Lower motor neuron-only forms had been described several decades earlier (Bell, 1836; Aran, 1850; Cruveilhier, 1853), often termed progressive muscular atrophy (Visser et al., 2008). At this time the term progressive muscular atrophy, which had formerly been applied to a group of disorders characterized by muscular wasting, was broken down into myopathies and several other recognized causes of generalized and focal muscular wasting. The concept of an upper motor neuron-only phenotype, as a separate entity distinct from the spastic paraplegias and eventually termed primary lateral sclerosis, developed later (Pearce, 2004). On the basis of a common neuropathological signature and clinical overlap in the later stages of each syndrome, most specialist neurologists now accept progressive muscular atrophy as part of the same entity of degeneration within the motor system, albeit marked by clinical heterogeneity (Kim et al., 2009).
In his Manual of Diseases of the Nervous System, William Richard Gowers (1845–1915), citing contemporary literature, concluded that progressive disorders of the motor system involving the lower and upper motor neuron pathways to a variable degree were syndromic variants of a single disorder, although he dealt with what is now termed primary lateral sclerosis under the designation of primary spastic paraplegia (Gowers, 1886). Charcot concurred with this syndromic view, noting that the clinical characteristics of each syndrome could, in general, be correlated with the distribution of the pathological features in the brain. In his textbook, Hermann Oppenheim (1858–1919) likewise considered the different motor degenerations to be related disorders (Oppenheim, 1911). W. Russell Brain, in his popular and influential textbook (Brain, 1933) brought together these concepts and made it clear that, in his view, these three syndromes of upper motor neuron degeneration, lower motor neuron degeneration, and mixed upper motor and lower motor neuron degeneration, should be regarded as manifestations of the same underlying disorder which, in the sixth edition of his book (Brain, 1962), he termed ‘motor neuron disease’. Unfortunately, this change in terminology was followed by considerable international confusion. In the USA, the three syndromes of ALS, progressive muscular atrophy and progressive bulbar palsy tended to be regarded as separate entities, a view largely based on the earlier nomenclature. However, the clinical and pathological literature, even from the 19th century (vide supra), had clearly demonstrated overlap of the clinical and pathological features of these three syndromes. In the UK, the holistic view summarized by Gowers and Brain prevailed, and usually also included the fourth syndrome of primary lateral sclerosis, but this did not preclude recognition of the differing phenotypes since an affected individual’s prognosis seemed to depend principally on the clinical features. In this essay we shall illustrate the state of knowledge extant in the 19th century using contemporary sources, highlighting Lockhart Clarke, and show how the underlying unity of these syndromes was well understood at that time.
Lockhart Clarke’s early clinicopathological studies in amyotrophic lateral sclerosis
Augustus Jacob Lockhart Clarke (1817–80; Fig. 1) is best known for his descriptions of spinal cord anatomy, in particular the ‘posterior vesicular column’ (Clarke’s column), but also for an early account of syringomyelia (Pearce, 1990a, b). He also made important contributions to the early history of muscular dystrophy (Emery and Emery, 2000). It was noted by Pearce that, despite his remarkable achievements, and although he had been elected to membership of the Royal College of Physicians under the ‘Dispensing Clause’ in 1871, he was mysteriously never advanced to fellowship of the college. One of his major achievements was what appears to be an early detailed clinicopathological description of a case of ALS, published with Charles Bland Radcliffe (1822–89) in the British and Foreign Medico-Chirurgical Review (Radcliffe and Lockhart Clarke, 1862)—3 years ahead of Charcot’s description of this disease (Charcot, 1865), and 12 years before Charcot’s first use of the term ALS (Charcot, 1874).
Figure 1.

Jacob Augustus Lockhart Clarke (1817–80) (reproduced courtesy of the National Library of Medicine, Bethesda, MD, USA).
Lockhart Clarke was a physician on the staff of the Hospital for the Paralysed and Epileptic, in Regent’s Park, London. Although engaged in clinical practice, his real skills and interests were in the then new and evolving science of histological technique, anatomical studies of the nervous system and morbid anatomy; sciences that led to the establishment of the modern discipline of neuro-pathology. Lockhart Clarke played an important role in the London Pathological Society, but his clinical activities achieved little reputation in the early development of neurology, at least in terms of original clinical contributions to the subject. He never held an academic position (Emery and Emery, 2000) and his obituary in The Lancet concluded:
He was a man single of purpose, of noble independence and honesty, wholly free from ambition…he will be remembered, not as the popular physician, but on account of his patient and laborious researches so fruitful to medical science.
Radcliffe’s contributions to the development of neurology were regarded as minor by Gordon Morgan Holmes (1876–1965) (Holmes, 1954), but Radcliffe wrote extensively on contemporary neurological topics and his contributions on epilepsy, although unconventional in modern terms, were important in their time (Eadie, 2007). An indication of the esteem Radcliffe had earned may be judged from the observation that at the time of his appointment to The National Hospital at Queen Square, he was preferred to John Hughlings Jackson (1835–1911). Indeed, Holmes states that Hughlings Jackson was twice passed over for election to the staff of the new Hospital (Holmes, 1954). Hughlings Jackson, with the firm support of Jonathan Hutchinson (1828–1913), was established as physician-assistant on the staff of the London Hospital in 1862, and was one of the four founders of Brain in 1878. We consider that Radcliffe’s contributions to neurology have been underestimated.
Case 1: An important case of paralysis and muscular atrophy, with disease of the nervous centres (Radcliffe and Lockhart Clarke, 1862)
Part I: Dr Radcliffe’s observations
Dr Radcliffe began his report with deference to his colleague
The chief interest of the case which I am about to relate is in the light which is thrown upon it by Mr Clarke.
In his initial summary, there are already clear clues that make this case instantly recognizable as ALS with marked bulbar and respiratory involvement:
‘Looking at the clinical facts, it was obvious that there was no material injury in the seat of intelligence, and it was probable that there was some grave injury to the parts which rule the movements of the tongue and pharynx, and the respiratory movements generally. Without this latter injury, indeed, it was difficult to account for the palsied and wasted state of the tongue, for the difficult deglutition, for the occasional trouble of breathing, for the mode of dying’.
Patient F.P. was a male aged 40 years, an age which would now be considered a young-onset case of ALS since the most common age of symptom onset is generally accepted as the late sixth and early seventh decades. Younger-onset ALS is also generally associated with prolonged survival beyond the median of 2–3 years from symptom onset, but Patient F.P.’s illness was very rapidly progressive. Bulbar-onset disease is the feature consistently predictive of more aggressive disease in ALS (Chio et al. 2009), yet Mr FP’s limb-onset disease still ran its course over only 2 years. It is not unlikely that the absence of modern dietary management, and of ventilatory assistance and antibiotics, was contributory to this rapid demise (Traynor et al., 2003). Consistently aggressive forms of ALS with rapid progression in younger patients have been associated with the ‘A4V’ mutation of the superoxide dismutase SOD1 gene (Juneja et al., 1997) and in ALS with FUS positive basophilic inclusions (Baümer et al., 2010). Although younger patients are generally assumed to have a greater genetic component to their ALS, the vast majority of cases are still considered sporadic; and in this case there is no mention of a family history of neurological disease. It was not until nearly 20 years later that Osler recognized a familial ALS pedigree in the Farr family from Vermont, USA (Osler, 1880).
Radcliffe and Clarke’s patient was a former US Army surgeon, who had travelled to England by boat on a surgical placement. An increased incidence of ALS has been noted among military personnel (Weisskopf et al., 2005), and in those with a history of unusual physical activity or physical injury (Erb, 1897). More recently, an increased risk of ALS has been posited in Italian professional footballers (Chio et al., 2005). The role of physical exercise (Harwood et al., 2009), or whether those at risk of ALS simply tend to come from an ‘athletic’ background (Scarmeas et al., 2002) remains an ongoing debate. Radcliffe observed, as many ALS physicians have reported anecdotally, that prior to onset of symptoms his patient had been in excellent health:
‘…had never, so he says, had a day’s illness…’
Recall bias remains a concern in much of the supposed association of ALS with prior life events. Many ALS neurologists have commented on what is assumed to be a natural tendency for patients to identify a clear precipitant to the onset of their ALS symptoms. Patient F.P. linked his illness to an episode of what was described as sun-stroke:
‘One day, after a long ride in the hot sun, he was seized with vertigo and pain in the head, and on dismounting fell down insensible. This insensibility lasted from five to ten minutes. After a week’s interval he quite recovered, with the exception of a slight impairment of vision. A month later he began to find a weakness in his left hand…’
The focal nature of onset and spread of symptoms in ALS has, perhaps surprisingly, only relatively recently been explored systematically (Swash, 1980; Swash et al., 1986; Ravits et al., 2007a, b; Ravits and La Spada, 2009). However, it is no surprise that, 8 months later, the symptoms in this patient had spread from the left arm to the right arm. Although the ipsilateral leg or bulbar region would have been other possible consecutive territories, it is noteworthy in terms of ALS pathogenesis that weakness does not appear to spread consecutively in a ‘diagonal’ direction (Brooks et al., 2000; Turner et al., 2009); i.e. not to the contralateral lower limb in Patient F.P. Radcliffe reported:
‘His countenance is bright and intelligent, his complexion remarkably pale and transparent, his body and limbs greatly emaciated, especially in the arms, which are literally little more than skin and bones’.
The examination revealed profound wasting and weakness of the tongue and both upper limbs, and ‘flickerings’ (fasciculations in modern terminology) were noticed in the deltoid and pectoral muscles as well as in lower limb muscles. Radcliffe noted normal ocular movements, a characteristic feature of ALS in all but those with very prolonged survival, and there was no facial weakness:
‘The eye was intelligent, and the features not inexpressive’.
Tone in the upper limbs was increased:
‘…the relics of muscles being tense and rigid, and altogether disobedient to the will’.
Lower limb tone was not described as increased, but Patient F.P. was unable to stand due to weakness. In 1862 clinical examination was already quite subtle and detailed, but concepts such as ‘spasticity, brisk reflexes’, or the reflex of Joseph Jules François Félix Babinski (1857–1932) (Babinski, 1896) were not yet clinical knowledge. The knee jerk was studied independently in Germany in 1875 by Wilhelm Heinrich Erb (1840–1921) and Karl Friedrich Otto Westphal (1833–90) (Louis, 2002; Pearce, 2003), although it was noted in 1859, 25 years earlier, by Silas Weir Mitchell (1829–1914) (Louis, 2008), and was later studied by him in detail (Mitchell and Lewis, 1886). After 1875, the knee jerk was rapidly adopted as a clinical test, but its sensitivity as a measure of spinal excitability was not fully understood (Bannister, 1878). Abnormal ‘rigidity’ was recognized as a clinical abnormality nonetheless by Radcliffe and Lockhart Clarke, and was an established feature also recognized by Charcot and his contemporaries in France, Germany and England. The full significance of these concepts would be realized only after new ideas concerning the physiology of the nervous system and the effects of abnormal reflex action in disease entered the clinical canon, derived from the work of Charles Scott Sherrington (1857–1952) and his school of neurophysiology, some years later (Sherrington, 1906).
In Radcliffe and Lockhart Clarke’s patient, bulbar involvement was profound with virtual anarthria but, on several occasions, Radcliffe noted that he had apparently fully intact ‘intelligence’. This was confirmed by the patient’s wife. This observation is entirely in keeping with modern clinical experience in patients with ALS, in whom frank dementia is rare despite the recently recognized clinicopathological overlap with frontotemporal dementia (Lomen-Hoerth et al., 2002; Phukan et al., 2007).
‘And Mrs. P, who was standing by the bedside at the time, removed all doubt upon this point by saying that her husband was ‘too intelligent, if anything’, and that he was never tired of hearing read books requiring attention and thought’.
Radcliffe also noted specifically that sphincter functions were intact and that there were no sensory symptoms. The phenotype in this case was an upper limb-predominant ALS. Although apparently symmetrical, the syndrome described by Radcliffe is not commensurate with the ‘flail arm’ variant of ALS originally ascribed to Vulpian (Gamez et al., 1999), as illustrated in Gowers’ text (Gowers, 1886), and shown more recently to be associated with prolonged survival (Hu et al., 1998).
It seems clear that, by the time of admission, the patient was in respiratory failure:
‘All the time of the examination, the breathing was disturbed and hurried, more so at the beginning than afterwards. At first the walls of the chest were almost motionless. Now and then, after every twelfth breath or so, there was a pause, followed by a deep drawn sigh’.
In terms of treatment, Patient F.P. was reported to have already tried:
‘…a course of mercury, pushed to salivation and continued several weeks after his arrival…’
Mercury, strychnine and iodides were frequently used as putative therapies at this time, in a pharmacological tradition that extended for many years, even centuries earlier. These were non-specific treatments. He then went on to receive:
‘continuous current from six simple galvanic cells along the palsied upper extremities…’
He was given a diet that:
‘…consisted of eggs, beef-tea, an extra allowance of bread, and half-a-pint of port wine…’
All of this was, unfortunately, to no avail and he died suddenly overnight on the sixth day after admission. Sudden death in ALS is well recognized and could have resulted from respiratory failure, pulmonary embolism, retained secretions or cardiac arrhythmia.
Part II: Mr. Clarke’s morbid anatomy of the nervous centres
Clarke began his description by stating:
‘All the most prominent symptoms of disease – the extensive paralysis and muscular atrophy described in the history of the patient, are so clearly and satisfactorily explained by the lesions of structure discovered on examination of the nervous centres, that the case now before us must be considered one of the most remarkable and interesting on record’.
Clarke then suggested that:
‘…the ordinary and inefficient method of examining the nervous centres…would perhaps have resulted in ranking the case as one of simple muscular atrophy’.
Lockhart Clarke was well known for his advanced and beautiful histological methods, which provided exceptionally clear sections for microscopy, and utilized early tinctorial methods (vide infra). He described these methods in some detail in an earlier paper (Lockhart Clarke, 1851), and seems to have developed them himself. Fixation of fresh tissue from the cord in spirits of wine caused hardening and he was able to cut thin sections using a very sharp knife. These were placed on a glass slide and treated with one part acetic acid and three parts spirits of wine, covered with thin glass and viewed. A second method placed the tissue into the acetic acid/wine mixture and then into pure spirit, followed by turpentine, put up in Canada balsam and viewed. His superb drawings followed outlines made with a camera lucida.
In this case, Lockhart Clarke first described the lumbar cord, noting that while it was not appreciably thinned externally, transverse section revealed atrophy of the ‘anterior cornu of grey substance’, and that the nerve cells were reduced in number with small cell bodies, some without visible nuclei, and irregular in shape. He noted that his eponymous ‘posterior vesicular columns’ were largely normal in size and structure in this region.
He then observed complete absence of the normal cervical cord enlargement:
‘I was surprised to find that scarcely a vestige could be seen of the large groups of cells which are found in corresponding parts of the healthy cord…the cells were wonderfully altered from their natural appearance…looked like aggregated granules…all more or less atrophied and shrivelled…’ (Fig. 2)
Figure 2.
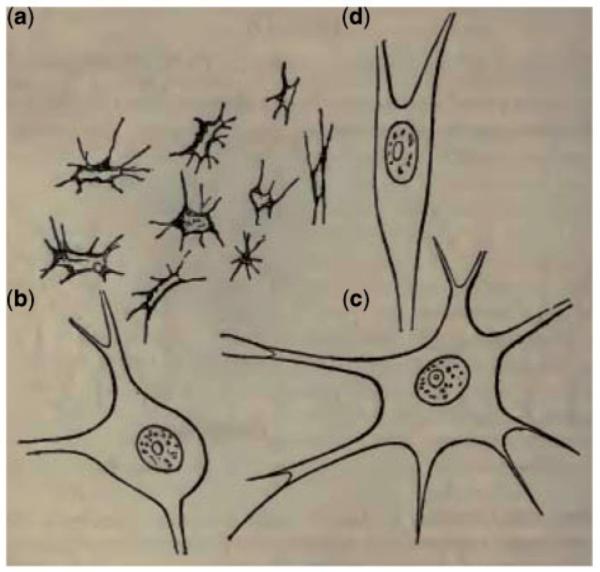
Lockhart Clarke’s original drawings published in Case 1 (Radcliffe and Lockhart Clarke, 1862). He depicts ‘atrophied cells from the cervical enlargement, magnified 420 diameters’ (a) with ‘healthy cells from the same quarter, and magnified to the same extent’ (b-d).
Clarke went on to describe the anterior horn cell and corticospinal tract degeneration characteristic of ALS:
‘All the white columns of the cord in every region, but particularly in the cervical region, had suffered more or less from atrophy or degeneration…the anterior roots of the nerves were decidedly below their average size’.
He then moved to the brainstem:
‘…from about the lower end of the olivary bodies to the commencement of the fourth ventricle, the morbid changes were much greater and more extensive…The hypoglossal or lingual nerve-roots in their course through the medulla, were also in places not more than half their natural size; and in other places could scarcely be discerned’.
He found the brain to be largely unremarkable though he noted some ‘softening’ and adherence of the membranes to the grey matter either side of the middle of the longitudinal fissure, which we interpret as probably corresponding to the region of the primary motor cortex at the midline.
Clarke was clearly fascinated by this patient’s pathology but ended his entry unable to draw firm conclusions from his observations of this single case:
‘…some facts of importance…may be more safely or advantageously considered after a few more cases of a similar nature have been examined with the same care’.
Jean-Martin Charcot (1825–93), and the anatomist Jean Cruveilhier (1791–1874), would later do just that. Localization of the responsible lesion for muscular wasting in progressive muscular atrophy had been made, by Cruveilhier, to the anterior roots in the days prior to systematic histology (Cruveilhier, 1853). This concept was extended by the neurologist Jules Bernard Luys (1828–97) to the anterior horn cell (Luys, 1860). Nonetheless, Radcliffe and Lockhart Clarke’s case report, with the detailed pathological examination provided by Clarke, using the most advanced histological techniques then available, is probably the first recognizable description of ALS. The case report is particularly remarkable for the careful correlation of clinical and pathological observations. Indeed, Charcot acknowledged Lockhart Clarke’s contributions in his Lectures on Diseases of the Nervous System (Charcot, 1881), clearly recognizing their importance.
Case 2: On a case of muscular atrophy with disease of the spinal cord and medulla oblongata (Lockhart Clarke and Hughlings Jackson, 1867)
This second paper was communicated to the Royal Society. Lockhart Clarke was a Fellow of the Royal Society and in recognition of his research on the spinal cord was recipient of its highest honour, the Society’s Royal Medal, in 1864. In this report a 38-year-old female is described, who:
‘…was always delicate’.
Although familial cases of ALS were not yet recognized, it was noted nonetheless that her father had died at age 62 years, but:
‘…neither of apoplexy nor paralysis’.
Patient H had fallen, injuring her right hand, in June 1864, and reported weakness in the right thumb ‘ever after’, although she continued as a needlewoman for another 6 months. This apparent link between trauma and the onset of ALS has been much studied. However, a recent hospital record-based study (perhaps free from the concern of recall bias) did not demonstrate any clear association (Turner et al., 2010). Patient H’s symptoms then appeared to abruptly progress and later to remit:
‘She then began to complain of severe pain in the right arm, neck and shoulder, and found that she could hardly hold a needle or lift the arm to her head. There was little or no pain in the hand and forearm; but she complained that they felt generally much colder than on the left side. At this time the muscles of the hand and forearm were much wasted…in two months her health was greatly improved; she had less pain, and could hold a needle and work, and her hand did not appear so wasted or cold’.
Patient H married and became pregnant, but her husband died suddenly in September 1865, 3 months into the pregnancy, to which she attributed the cause of her rapid decline over the following 6 months, with features that became much more suspicious for ALS:
‘She was much shocked and never felt well after. Her face began to waste and lose expression; she complained of pain in the neck; weakness of the left hand and arm, as well as the right, and her legs dragged after her. Her voice changed; she did not pronounce words as usual; she never complained of any loss of sensation…’
The initial right arm symptoms were reminiscent of neuralgic amyotrophy (Parsonage and Turner, 1948). Interestingly, there are two other reported patients with ALS of similar onset, both young women, one of whom was also pregnant (Weiss et al., 2006; Baümer et al., 2010). ALS in the context of pregnancy (either before or during) has been described and, in a review of the literature, it was concluded that pregnancy did not generally adversely affect the course of the disease (Chio et al., 2003). In Patient H’s case, however, progression nonetheless continued inexorably:
‘Her chin generally rested on the sternum, or the cheek fell on the right shoulder; and she was unable to move her head from either position, except by bending backward or sideways the trunk. She was able to shuffle about the house, but could not dress herself. She had almost completely lost the power of moving her left arm; over the right she had some power, but not much…The whole of the muscles of the back of the right scapula are apparently gone, and there is very little trace of those on the left side. There is little muscular tissue on each side of the bones of the neck. There is a feeble action of each sterno-mastoid under the Faradaic current…Her legs were like sticks…’
The use of electric stimulation as a treatment was introduced in the late 18th century, following Luigi Galvani’s (1737–98) concept of ‘animal electricity’ (Piccolino, 1998). Galvanic and Faradic stimulation of muscle as a clinical test can be said to mark the beginnings of modern electrophysiology and was studied with enthusiasm as a therapy, by Guillaume Benjamin Amand Duchenne (1806–75) in some of Cruveilhier, Francois-Amilcar Aran (1817–61) and Charcot’s cases (Duchenne, 1883). Interestingly, Brain (1962) remarked that he believed electrical treatment could be harmful in motor neuron disease.
There were also graphic descriptions of the bulbar involvement, with bilateral wasting and fasciculation of the tongue:
‘Her deglutition now became difficult…The palate moves little, and more as if blown than as if raised. She does not say ah! but makes a vague noise…The tongue is protruded badly…It is atrophied on each side, and in folds, reminding one of cerebral convolutions. It is also tremulous, and it does not seem to tremble as a whole, but in waves of tremulousness’.
Respiratory compromise was clear:
‘…the action in inspiration is nearly solely abdominal. After the abdominal inspiration the upper part of the thorax seems to move with a jerk’.
In two instances double vision was mentioned. The oculomotor nuclei are relatively spared in ALS, although functional abnormalities can be detected (Jacobs et al., 1981), often later in the course of the disease (Palmowski et al., 1995). Frank diplopia in ALS would be exceptional, but the examination findings later confirmed that:
‘…the eyes move quite well on careful testing’.
Patient H gave birth to a healthy child in March 1866 and died 4 months later.
Unlike the first case, this clinical description gave no clear hint of what we might recognize as an upper motor neuron sign, so critical to the clinical diagnosis of ALS rather than progressive muscular atrophy. If anything, muscular tone appeared flaccid:
‘There is no stiffness nor rigidity anywhere the fingers being quite supple’.
At autopsy, Lockhart Clarke found marked loss of nerve cells in the medulla, particularly in the floor of the fourth ventricle as well as in the anterior horns of the spinal cord, with especially marked atrophy of the anterior cornua in the cervical region:
‘Scarcely a trace could be discovered of anything resembling the large cells that belong to these parts, which were now filled instead with a multitude of small granular masses and minute stellate bodies…’
Crucially, he then noted and illustrated (Fig. 3), extensive damage to the lateral spinal columns:
‘Nor were the lesions of structure, in this case, limited to the grey substance; for the swollen lateral column was very much softened, and in several places was completely destroyed by patches of transparent disintegration’.
Figure 3.
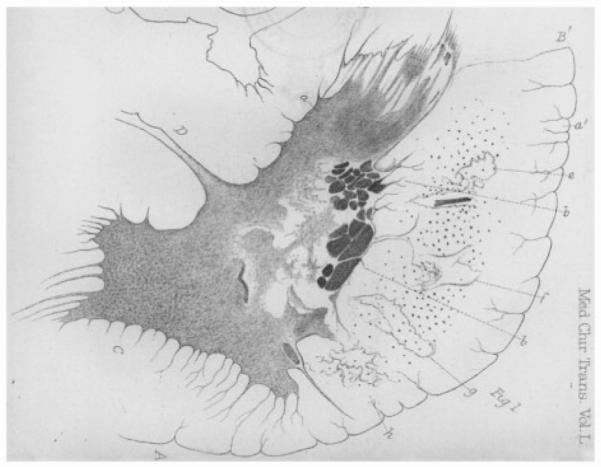
Lockhart Clarke’s original drawings showing the right lateral half of a transverse section of the spinal cord in the second case described (Lockhart Clarke and Hughlings Jackson, 1867). His own legend reads thus, with e, f, g and h clearly demonstrating his recognition of corticospinal tract involvement: ‘A: The anterior white column; B: The lateral white column; B′: The posterior lateral fissure; C: The anterior cornu; D: The transverse commissure; a and a′: The caput cornu posterioris; b and b′: Dark masses enclosed in a network of fibres and blood vessels, along the right border of the grey substance; e-h: Patches of transparent disintegration in the lateral column’.
Lockhart Clarke noted that the right side of the cervical cord showed localized swelling, with a similar swelling noted on the left side of the lumbar cord. These observations are not further delineated. There is no indication from Lockhart Clarke’s reported histological observations that there was any other unexpected pathology, such as infarction or neoplasm. Cord swelling is not a recognized feature of ALS at autopsy or by magnetic resonance imaging, and no cord pathology is recognized in neuralgic amyotrophy. An acute or subacute cord demyelination would not adequately explain the inexorably progressive clinical course (although this would not have been a recognized diagnosis at the time this case was examined). Post-mortem or fixation artefact seems the most likely cause.
Lockhart Clarke also made interesting observations about the pattern of spread of the pathology:
‘Indeed, the whole of that portion of the cord which supplied nerves to the lower extremities was very much less damaged that that which gives origin to the nerves of the upper extremities. The conus medullaris…was perfectly healthy’.
Whilst relative sparing of Onuf’s nucleus in ALS is now well recognized, only recently have rigorous clinicopathological studies confirmed the concept of a ‘graded loss’ of motor neurons from the site of onset (Ravits et al., 2007a), with Lockhart Clarke’s observations entirely in keeping with Patient H’s initial upper limb symptoms.
Lockhart Clarke’s histological techniques
Histopathology was in its infancy at this time and Cruveilhier referred to these efforts within the rubric of anatomic pathology. The first atlas of histopathology by Mandl, a Baillière atlas comparable to Cruveilhier’s, served up a ‘livraison’ that showed CNS cells (Mandl, 1838). It was notable for priority but not for the relatively poor quality and representations of cells drawn by hand. The method used for fixation was an extension of Vicq d’Azyr’s alcohol method but Lockhart Clarke’s drawings of neurons are much more detailed and lucid than the depiction of cells and neural ganglia by Mandl, yet notably in the absence of cellular staining. Lockhart Clarke was a pioneer of histological technique in studying the nervous system, especially the spinal cord. In 1859, he described the methods he had developed (Lockhart Clarke, 1859). He hardened small portions of brain or spinal cord tissue in dilute spirits of wine for 24 h and then in pure spirit of wine, changing the spirit every 5–6 days for a period of about 14 days. Transparency was induced by placing the sections in a 1:3 or 1:5 mixture of strong acetic acid and spirit for 2–10 min, according to their thickness. The sections were then floated in turpentine to clear them, and mounted in Canada balsam under a thin glass slide. He noted that several reapplications of turpentine might be necessary to bring out the fibrous structure of the nervous tissue. Lockhart Clarke described that he had recently replaced spirit of wine with chromic acid before mounting his sections on glass slides in Canada balsam. He did note that carmine was a useful tinctorial agent, although this ‘interfered with the sharpness of the fibres’. Carmine, an intensely red pigment obtained from the carminic acid produced by scale insects, was discovered by the Italian anatomist Marquis Alfonso Giacomo Gaspare Corti (1822–76) in 1851, and, mixed with gelatin, it was popularized as a histological stain in 1858 by the German anatomist Joseph von Gerlach (1820–96). It was to play an important role for Charcot later.
Lockhart Clarke’s technical advances in adequately preparing nervous tissues for study were pioneering. The age of histology had only emerged in the previous decade, through publications such as Allgemeine Anatomie (1841) by the German anatomist Friedrich Gustav Jacob Henle (1809–85), and histology of the nervous system was recognized to be among the most difficult to prepare and interpret (Bracegirdle, 1993). With specific reference to nervous tissue histology, it was 25 years later that the French anatomist Louis-Antoine Ranvier (1835–1922) published his leading textbook of the time Traité technique d’histologie, and the Italian physician Camillo Golgi (1843–1926) developed his silver chromate method for staining neural tissue. The Spanish anatomist Santiago Ramón y Cajal (1852–1934) did not publish his Manuel de histologia normal y técnica micrografica until 1889, and both he and Golgi shared the Nobel Prize in 1906 for their work on the structure of the nervous system.
Pearce, in a recent biographical appreciation of Lockhart Clarke’s contributions, states that many of his preparations were made in his landlady’s house (Pearce, 1990b), so perhaps facilities for preparation of microscopic sections of material from the CNS at Regent’s Park or Queen Square were rather undeveloped. However, the use of private facilities for histological studies was not unusual at this time (Swash, 2008).
Charcot’s interpretation of the clinicopathological features of amyotrophic lateral sclerosis
It is appropriate to note briefly Charcot’s ideas and contributions to this field of study, as others have done (Goetz, 2000). Charcot is generally regarded as the originator of ALS studies, based on his description of the clinical and pathological features of the disease (Charcot and Joffroy, 1869). He referred, in his Lectures XI, XII and XIII, to two patterns of motor system degeneration resulting in muscle atrophy and weakness, which he termed ‘protopathic’ and ‘deuteropathic’ forms (Charcot, 1881). The former consisted of muscle atrophy and weakness with degeneration of the anterior cornua of the spinal cord, of varying extent at different levels. The latter term was used to describe combined degeneration of the anterior cornua of the cord, with associated degeneration of the lateral columns of the cord, i.e. the corticospinal tracts. No specific correlations were established at that time between these pathological features and the pattern of the clinical disorder. Charcot pointed out in these lectures, delivered at La Salpêtrière, that during the period 1858–67 ‘the role of alterations of the nerve cells themselves had not been yet elucidated’ (Charcot, 1881; p. 176, footnote 2).
Charcot’s account of ALS was prepared in the context of his efforts to distinguish various motor system disorders as separate entities. He recognized ALS as a progressive, adult-onset motor disorder. He noted that protopathic spinal amyotrophy often developed in families, and also noted that many patients survived for as many as 18–20 years, but that some ran a more rapid course, with death in 2–5 years. In his Lecture XI on chronic spinal amyotrophies, Charcot (1881) described the clinical features and pathology of progressive muscular atrophy by reviewing the ‘six of seven cases’ reported or with autopsy confirmation up to that time in relation to his own clinical experience. These included the case of Radcliffe and Lockhart Clarke, and two additional patients from the Salpêtrière who died in 1872 and 1874, respectively. He stressed the importance of sparing of the white columns (dorsal columns) and lateral columns (corticospinal columns) in the more recent autopsies and stated that Lockhart Clarke did not clearly describe the state of the white matter in his case. However, the reports we have extracted here show that there was white matter involvement in the cord in Lockhart Clarke’s cases.
Charcot’s clinical description is important in that he described progressive muscular atrophy as a disorder characterized by progressive weakness, often commencing in one arm and spreading to the other arm, involving the legs less often and accompanied by fibrillary twitching of muscles, usually without muscular rigidity. Alterations in nerve cells in the anterior cornua at the affected levels consisted of loss of ‘the great motor cells’ in the anterior grey matter of the cord, pigmentary or sclerous atrophy of remaining nerve cells, and what he interpreted as inflammatory proliferation in the neuroglia. The anterior roots were atrophic, but the lateral and dorsal spinal columns were spared. He commented that vacuolar change in remaining nerve cells might be ‘an artificial product’. By this time, the neuropathological specimens examined at the Salpêtrière are described as prepared from ‘hardened cord’, indicating that a method of tissue fixation was now being employed, which may explain the apparent inconsistency in Lockhart Clarke’s reported ‘softening’ of the lateral cord. Charcot referred to Lockhart Clarke as previously describing the various phases of degeneration and pigmentary atrophy of these spinal nerve cells (Charcot, 1881; p. 156). In accompanying lectures Charcot described the clinical and anatomical features of primary lateral sclerosis and ALS, separating these disorders from various causes of progressive cervical myelopathy, including hypertrophic spinal pachymeningitis and multiple sclerosis. He was at pains to stress that in ALS, muscular atrophy and loss of motor nerve cells was most predominant in the cervical region, but also involved the bulbar nuclei, while relatively sparing the lumbar cord. ‘Rigidity’, on the other hand, was more marked in the lower than in the upper limbs, as modern physicians would expect from involvement of the corticospinal tracts in the cervical region, as was demonstrated in Charcot and Cruveilhier’s autopsy studies (Charcot and Joffroy, 1869).
Both Charcot and Gowers focused on the ‘relation between the degeneration of the two segments, the lateral sclerosis and the affection of the ganglion cells’ (Gowers, 1893). Gowers recognized that there was degeneration of the lateral columns even in the clinical syndrome of progressive muscular atrophy [confirmed more recently (Ince et al., 2003)], and felt that Charcot’s distinction between protopathic and deuteropathic forms of progressive motor degeneration was unwarranted. He concluded that there was ‘every gradation, in degree and distribution, of atonic atrophy, spastic paralysis, and tonic wasting’. It was this concept that later led Brain to bring these clinical syndromes together under the rubric of ‘motor neuron disease’ (Brain, 1962). Gowers also considered the relation of pathological changes in the lateral columns and the anterior horns in the disease, and concluded ‘that the only adequate explanation of the facts is that the degeneration of the upper and lower segment is simultaneous, or if not simultaneous, at least so far independent that neither is the cause or the consequence of the other’.
This issue remains contentious and unresolved to the present day; with the notion that there might be primary involvement of the motor cortex in ALS having once again arisen (Eisen et al., 1992; Vucic and Kiernan, 2006; Eisen, 2009). Gowers also stressed the often forgotten observation that even at death, degeneration and loss of motor cells in affected spinal grey matter was patchy, and some nerve cells remained, apparently unaffected by the degenerative process (Gowers, 1893; Swash et al., 1986; Ravits et al., 2007b). Indeed, as he noted, for ‘rigidity’ to be a feature of a wasted limb in ALS, it is necessary that some anterior horn cells remain functional in the spinal segments innervating the muscles in that limb.
Learning from past masters
We wish to acknowledge here the remarkable advances made to our knowledge of motor neuron diseases in the 19th century. In reading these reports one cannot fail to be impressed by the clarity of the clinical and scientific issues formulated so many years ago. They are as relevant now as they were then; indeed, it is sobering to reflect how many of these issues are still matters of current debate. In some instances, for example discussions relating to the classification of the syndromes of motor system degeneration, it is striking that the issues were fully debated and, indeed, largely resolved 150 years ago. Study of old books and journals, as Sir Andrew Huxley remarked in a Florey Lecture (Huxley, 1982), is something to which everyone involved in science should devote some energy; or the wheel will constantly be reinvented without any real advance in knowledge. It is also important to recognize in the papers described here how technical advances, both in clinical practice and pathological studies, were, and of course remain, of cardinal importance in breaking new ground in old problems.
Acknowledgments
Funding
MRC/MND Association Lady Edith Wolfson Fellowship to M.R.T.
Abbreviations
- ALS
amyotrophic lateral sclerosis
References
- Aran FA. Recherches sur une maladie non encore décrite du système musculaire (atrophie musculaire progressive) Arch Gén Méd. 1850;24:5–35. 172–214. [Google Scholar]
- Babinski J. Sur le réflexe cutané plantaire dans certains affections organique du système nerveux central. Comptes rendus de la Société de Biologie. 1896;48:207–8. [Google Scholar]
- Bannister HM. Diagnostic significance of the tendon reflex. J Nerv Ment Dis. 1878;5:656–63. [Google Scholar]
- Baümer D, Hilton D, Paine S, Turner MR, Lowe J, Talbot K, et al. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology. 2010 doi: 10.1212/WNL.0b013e3181ed9cde. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C. The nervous system of the human body. Longmans; London: 1836. Case of partial wasting of the muscles of the upper extremities; pp. 432–4. [Google Scholar]
- Bracegirdle B. The microscopical tradition. In: Bynum WF, Porter R, editors. Companion encyclopedia of the history of medicine. Vol. 1. Routledge; London: 1993. pp. 102–19. [Google Scholar]
- Brain WR. Diseases of the nervous system. Oxford University Press; Oxford: 1933. Amyotrophic Lateral Sclerosis; pp. 497–508. [Google Scholar]
- Brain WR. Diseases of the nervous system. Oxford University Press; Oxford: 1962. Motor Neurone Disease; pp. 531–43. [Google Scholar]
- Brooks BR, Sanjak M, Belden D, Juhazs-Poscine K, Waclawik A. Natural history of amyotrophic lateral sclerosis—impairment, disability, handicap. In: Brown RH Jr, Meininger V, Swash M, editors. Amyotrophic lateral sclerosis. Martin Dunitz; London: 2000. pp. 31–58. [Google Scholar]
- Charcot J-M. Sclérose des cordons latéraux de la moelle épinière chez une femme hystérique atteinte de contracture permanente des quatre membres. Bull de la Société Méd des Hôpit de Paris. 1865;10:24–35. [Google Scholar]
- Charcot J-M. Amyotrophies spinales deuteropathiques sclérose latérale amyotrophique & Sclérose latérale amyotrophique. Bureaux du Progrès Médical. 1874;2:234–66. [Google Scholar]
- Charcot J-M. Lectures on the diseases of the nervous system. Sydenham Society; London: 1881. [Google Scholar]
- Charcot J-M, Joffroy A. Deus cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antéro-latérale. Arch Physiol. 1869;2:354–67. 744–60. [Google Scholar]
- Chio A, Benzi G, Dossena M, Mutani R, Mora G. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 2005;128:472–6. doi: 10.1093/brain/awh373. [DOI] [PubMed] [Google Scholar]
- Chio A, Calvo A, Di Vito N, Vercellino M, Ghiglione P, Terreni A, et al. Amyotrophic lateral sclerosis associated with pregnancy: report of four new cases and review of the literature. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:45–8. doi: 10.1080/14660820310006724. [DOI] [PubMed] [Google Scholar]
- Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–23. doi: 10.3109/17482960802566824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruveilhier J. La paralysie musculaire, progressive, atrophique. Bull Acad Méd. 1853;18:490–501. 546–83. [Google Scholar]
- Duchenne GBA. Selections from the clinical works of Dr Duchenne (de Boulogne) New Sydenham Society; London: 1883. Progressive muscular atrophy; pp. 42–87. [Google Scholar]
- Eadie MJ. Rigor mortis and the epileptology of Charles Bland Radcliffe (1822–1889) J Clin Neurosci. 2007;14:201–7. doi: 10.1016/j.jocn.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Eisen A. Amyotrophic lateral sclerosis - evolutionary and other perspectives. Muscle Nerve. 2009;40:297–304. doi: 10.1002/mus.21404. [DOI] [PubMed] [Google Scholar]
- Eisen A, Kim S, Pant B. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve. 1992;15:219–24. doi: 10.1002/mus.880150215. [DOI] [PubMed] [Google Scholar]
- Emery AE, Emery ML. Lockhart Clarke (1817–80): his role in the early history of muscular dystrophy. Neuromuscul Disord. 2000;10:530–3. doi: 10.1016/s0960-8966(00)00106-1. [DOI] [PubMed] [Google Scholar]
- Erb WH. Zur Lehre von den Unfallerkrankungen des Rückenmarkes, über Poliomyelitis anterior chronica nach Trauma. Dtsch Nervenheilk. 1897;11:122–42. [Google Scholar]
- Gamez J, Cervera C, Codina A. Flail arm syndrome of Vulpian-Bernhart’s form of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1999;67:258. doi: 10.1136/jnnp.67.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz CG. Amyotrophic lateral sclerosis: early contributions of Jean-Martin Charcot. Muscle Nerve. 2000;23:336–43. doi: 10.1002/(sici)1097-4598(200003)23:3<336::aid-mus4>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Gowers WR. A manual of diseases of the nervous system. Churchill; London: 1886. [Google Scholar]
- Gowers WR. A manual of diseases of the nervous system. Churchill; London: 1893. p. 402. [Google Scholar]
- Harwood CA, McDermott CJ, Shaw PJ. Physical activity as an exogenous risk factor in motor neuron disease (MND): a review of the evidence. Amyotroph Lateral Scler. 2009;10:191–204. doi: 10.1080/17482960802549739. [DOI] [PubMed] [Google Scholar]
- Holmes G. History of the National Hospital 1860–1948. Livingstone; London & Edinburgh: 1954. [Google Scholar]
- Hu MT, Ellis CM, Al Chalabi A, Leigh PN, Shaw CE. Flail arm syndrome: a distinctive variant of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1998;65:950–1. doi: 10.1136/jnnp.65.6.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxley A. The Florey Lecture, 1982. Discovery: accident or design? Proc R Soc Lond B Biol Sci. 1982;216:253–66. doi: 10.1098/rspb.1982.0073. [DOI] [PubMed] [Google Scholar]
- Ince PG, Evans J, Knopp M, Forster G, Hamdalla HH, Wharton SB, et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60:1252–58. doi: 10.1212/01.wnl.0000058901.75728.4e. [DOI] [PubMed] [Google Scholar]
- Jacobs L, Bozian D, Heffner RR, Jr, Barron SA. An eye movement disorder in amyotrophic lateral sclerosis. Neurology. 1981;31:1282–7. doi: 10.1212/wnl.31.10.1282. [DOI] [PubMed] [Google Scholar]
- Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with glu100gly and ala4val mutations in Cu,Zn superoxide dismutase. Neurology. 1997;48:55–7. doi: 10.1212/wnl.48.1.55. [DOI] [PubMed] [Google Scholar]
- Kim WK, Liu X, Sandner J, Pasmantier M, Andrews J, Rowland LP, et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. 2009;73:1686–92. doi: 10.1212/WNL.0b013e3181c1dea3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart Clarke J. Researches into the structure of the spinal chord. Phil Trans R Soc Lond. 1851;141:607–21. [Google Scholar]
- Lockhart Clarke J. Further researches on the grey substance of the spinal cord. Phil Trans R Soc Lond. 1859;149:437–67. [Google Scholar]
- Lockhart Clarke J, Hughlings Jackson J. On a case of muscular atrophy with disease of the spinal cord and medulla oblongata. Med Chir Trans. 1867;50:489–98. doi: 10.1177/095952876705000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–9. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- Louis ED. Erb and Westphal: simultaneous discovery of the deep tendon reflexes. Semin Neurol. 2002;22:385–90. doi: 10.1055/s-2002-36760. [DOI] [PubMed] [Google Scholar]
- Louis ED. Weir Mitchell’s 1859 demonstration of “a peculiar contraction” produced by a percussion hammer. Neurology. 2008;70:969–73. doi: 10.1212/01.wnl.0000305968.03898.fe. [DOI] [PubMed] [Google Scholar]
- Luys JB. Atrophie musculaire progressive; lésions histologiques de la substance grise de la moelle épinière. Gaz Méd Paris. 1860:15. [Google Scholar]
- Mandl L. Anatomie microscopique. Baillière; Paris: 1838. [Google Scholar]
- Mitchell JD. Amyotrophic lateral sclerosis – the Edinburgh disease? J Hist Neurosci. 1992;1:59–63. doi: 10.1080/09647049209525515. [DOI] [PubMed] [Google Scholar]
- Mitchell SW, Lewis MJ. Physiological studies of the knee jerk, and of the reactions of muscles under mechanical and other excitants. Phil Med News. 1886;48:169–73. 198–203. [Google Scholar]
- Oppenheim H. Textbook of nervous diseases; for physicians and students. Otto Schutze & Co.; Edinburgh: 1911. [Google Scholar]
- Osler W. On heredity in progressive muscular atrophy as illustrated in the Farr family of Vermont. Arch Med. 1880;4:316–20. [Google Scholar]
- Palmowski A, Jost WH, Prudlo J, Osterhage J, Kasmann B, Schimrigk K, et al. Eye movement in amyotrophic lateral sclerosis: a longitudinal study. Ger J Ophthalmol. 1995;4:355–62. [PubMed] [Google Scholar]
- Parsonage MJ, Turner JW. Neuralgic amyotrophy; the shoulder-girdle syndrome. Lancet. 1948;1:973–8. doi: 10.1016/s0140-6736(48)90611-4. [DOI] [PubMed] [Google Scholar]
- Pearce JM. Erb, Westphal and the tendon reflexes. Fragments of neuroloigcal history. Imperial College Press; London: 2003. pp. 44–7. [Google Scholar]
- Pearce JM. Primary lateral sclerosis and Pierre Marie. J Neurol Neurosurg Psychiatry. 2004;75:897. doi: 10.1136/jnnp.2003.024737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce JMS. A note on Jacob Augustus Lockhart Clarke. J Neurol Neurosurg Psychiatry. 1990a:53. [Google Scholar]
- Pearce JMS. A note on the work of Clarke of Clarke’s column. J Neurol Neurosurg Psychiatry. 1990b;53:125. [Google Scholar]
- Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6:994–1003. doi: 10.1016/S1474-4422(07)70265-X. [DOI] [PubMed] [Google Scholar]
- Piccolino M. Animal electricity and the birth of electrophysiology: the legacy of Luigi Galvani. Brain Res Bull. 1998;46:381–407. doi: 10.1016/s0361-9230(98)00026-4. [DOI] [PubMed] [Google Scholar]
- Radcliffe CB, Lockhart Clarke J. An important case of paralysis and muscular atrophy with disease of the nervous centres. Brit & Foreign Medico-Chirurgical Rev. 1862;30:215–25. [PMC free article] [PubMed] [Google Scholar]
- Ravits J, Laurie P, Fan Y, Moore DH. Implications of ALS focality: rostral-caudal distribution of lower motor neuron loss postmortem. Neurology. 2007a;68:1576–82. doi: 10.1212/01.wnl.0000261045.57095.56. [DOI] [PubMed] [Google Scholar]
- Ravits J, Paul P, Jorg C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology. 2007b;68:1571–5. doi: 10.1212/01.wnl.0000260965.20021.47. [DOI] [PubMed] [Google Scholar]
- Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009;73:805–11. doi: 10.1212/WNL.0b013e3181b6bbbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Shih T, Stern Y, Ottman R, Rowland LP. Premorbid weight, body mass, and varsity athletics in ALS. Neurology. 2002;59:773–5. doi: 10.1212/wnl.59.5.773. [DOI] [PubMed] [Google Scholar]
- Sherrington C. The integrative action of the nervous system. Charles Scribner’s Sons; New York: 1906. [Google Scholar]
- Swash M. Vulnerability of lower brachial myotomes in motor neurone disease: a clinical and single fibre EMG study. J Neurol Sci. 1980;47:59–68. doi: 10.1016/0022-510x(80)90025-8. [DOI] [PubMed] [Google Scholar]
- Swash M, Leader M, Brown A, Swettenham KW. Focal loss of anterior horn cells in the cervical cord in motor neuron disease. Brain. 1986;109:939–52. doi: 10.1093/brain/109.5.939. [DOI] [PubMed] [Google Scholar]
- Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. Effect of a multi-disciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000. J Neurol Neurosurg Psychiatry. 2003;74:1258–61. doi: 10.1136/jnnp.74.9.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MR, Abisgold J, Yeates DG, Talbot K, Goldacre MJ. Head and other physical trauma requiring hospitalisation is not a significant risk factor in the development of ALS. J Neurol Sci. 2010;288:45–8. doi: 10.1016/j.jns.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Turner MR, Brockington A, Scaber J, Hollinger H, Marsden R, Shaw PJ, et al. Pattern of spread and prognosis in lower limb-onset ALS. Amyotroph Lateral Scler. 2009 doi: 10.3109/17482960903420140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser J, de Jong JM, de Visser M. The history of progressive muscular atrophy: syndrome or disease? Neurology. 2008;70:723–7. doi: 10.1212/01.wnl.0000302187.20239.93. [DOI] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain. 2006;129:2436–46. doi: 10.1093/brain/awl172. [DOI] [PubMed] [Google Scholar]
- Weiss MD, Ravits JM, Schuman N, Carter GT. A4V superoxide dismutase mutation in apparently sporadic ALS resembling neuralgic amyotrophy. Amyotroph Lateral Scler Other Motor Neuron Disord. 2006;7:61–3. doi: 10.1080/14660820500467009. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, O’Reilly EJ, McCullough ML, Calle EE, Thun MJ, Cudkowicz M, et al. Prospective study of military service and mortality from ALS. Neurology. 2005;64:32–7. doi: 10.1212/01.WNL.0000148649.17706.D9. [DOI] [PubMed] [Google Scholar]