

Abstract
Hippocampal and cortical neurons have been used extensively to study central nervous system (CNS) neuronal polarization, axon/dendrite outgrowth, and synapse formation and function. An advantage of culturing these neurons is that they readily polarize, forming distinctive axons and dendrites, on a two dimensional substrate at very low densities. This property has made them extremely useful for determining many aspects of neuronal development. Furthermore, by providing glial conditioning for these neurons they will continue to develop, forming functional synaptic connections and surviving for several months in culture. In this protocol we outline a technique to dissect, culture and transfect embryonic mouse hippocampal and cortical neurons. Transfection is accomplished by electroporating DNA into the neurons before plating via nucleofection. This protocol has the advantage of expressing fluorescently-tagged fusion proteins early in development (~4-8hrs after plating) to study the dynamics and function of proteins during polarization, axon outgrowth and branching. We have also discovered that this single transfection before plating maintains fluorescently-tagged fusion protein expression at levels appropriate for imaging throughout the lifetime of the neuron (> 2 months in culture). Thus, this methodology is useful for studying protein localization and function throughout CNS development with little or no disruption of neuronal function.
Keywords: Neuroscience, Issue 47, hippocampus, cortex, transfection, nucleofection, electroporation, primary culture
Protocol
1. Preparation of Coverslips and Chambers
Preparation of clean coverslips and chambers is essential for healthy cultures. Shortcuts should not be taken at any of these steps.
Wash the coverslips (12mm or 22mm round, German glass - Carolina Assistant Brand) overnight in concentrated nitric acid (HNO3) in a dedicated glass jar or beaker.
Remove the coverslips from nitric acid and wash extensively (5-7x) in deionized water.
Separate the coverslips and dry in a laminar flow hood or biosafety cabinet. When dry, sterilize them with UV light for 30 minutes. Place sterilized coverslips into a sterile petri dishes for storage. If plating neurons on 12mm coverslips directly place coverslips in a sterile 35mm dish and proceed to Section 2.
Imaging chambers are constructed by drilling a 15mm hole in the bottom of 35mm petri dishes (remove all burrs) and attaching the cleaned coverslips with a 3:1 mixture of paraffin and petroleum jelly.
Melt the paraffin/petroleum jelly mixture in a conical tube inside a boiling water bath. Use a small paint brush and coat the underside of the dish around the 15mm hole. Make sure to keep stirring the paraffin/petroleum jelly mixture as it will separate. This usually results in chambers glued together with a higher concentration of petroleum jelly, which will become viscous when dishes are placed in the incubator, resulting in coverslip detachment. The remaining paraffin/petroleum jelly can be stored at room temperature.
Place the dish upside down on a flat tray and place the coverslip over the hole. Heat in 80°C oven until paraffin mixture is melted (~10 minutes). Remove dishes onto a flat surface and let the paraffin mixture set.
Turn the dishes over and sterilize both the insides of the covers and the bottoms of chambers with UV light.
Coat coverslips or glass regions of chambers with 1.0mg/mL Poly-d-lysine (30kDa) in borate buffer (0.1M sodium borate, pH 8.5) for one hour. Rinse 3-5 times with copious amounts of tissue culture grade deionized water. Make sure to remove all traces of borate buffer. Dry and use immediately or store chambers/coverslips for later use. We usually use cleaned coverslips within one month of preparation.
2. Preparation of Neuronal Dissection and Culture Medium
Prepare the dissection medium (DM) by adding appropriate amounts of 10x HBSS and 100X HEPES to tissue culture grade water. Store at 4°C. Keep on ice during the dissection.
The day before the dissection prepare the plating medium (PM) and serum-free medium (SFM). PM consists of Neurobasal Medium, B27 supplement, 2 mM glutamine, 0.3% glucose, 37.5 mM NaCl and 5% fetal bovine serum (FBS). SFM consists of Neurobasal Medium, B27 supplement, 2 mM glutamine, 0.3% glucose and 37.5 mM NaCl.
Make only enough for the dissection and store in a tissue culture incubator overnight with the cap slightly ajar so that the temperature and CO2 content of the medium equilibrates. We add extra glucose and increase the osmolality to approximately 310mOsm with NaCl. We find the cultures do better at a more physiological osmolality (Neurobasal osmolality is typically 205-245mOsm).
3. Cortical Glial Feeder Layer Preparation for Long-term Cultures
If long term cultures are to be prepared, perform this part of the protocol two to three weeks before continuing with the cortical or hippocampal dissections.
Prepare glial medium (GM) with MEM, 0.3% glucose, penicillin/streptomycin and 10% horse serum.
Euthanize P1-P3 mouse pups by cooling on ice for 5 minutes. Remove each pup from the ice and spray with 70% ethanol. Quickly decapitate with scissors. Remove the entire brain to a dish containing cold DM (step 2.1).
Remove the two cerebral hemispheres and meninges. Excise the neocortex and remove to a new dish containing no media. Prepare cortices from 4 brains total.
Mince the cortices with a clean, sterile razor blade as fine as possible and remove chopped tissue with a plastic pipette to a 50 mL conical tube containing 12 mL of cold DM. Add trypsin and DNase to final concentrations of 0.25% (1.5 mL) and 0.1% (1.5 mL), respectively. Incubate in a 37°C water bath for 10 minutes with intermittent swirling.
Remove the tube containing the cortical tissue and clean thoroughly with 70% ethanol before bringing into the tissue culture hood. Pipet cortical tissue up and down with a 10 mL pipette approximately 10-15 times, or until most chunks disappear.
Return the tube to 37°C in a water bath for another 10 minutes with intermittent swirling.
Thoroughly clean the tube with 70% ethanol and bring it back to the tissue culture hood. Pipet the cortical tissue up and down with a 5 mL pipette approximately 10-15 times, or until the chunks disappear.
Add 15 mL of warm GM and centrifuge at 200xg (1000rpm) for 10 minutes.
Discard supernatant, resuspend the pelleted cells in 20 mL of fresh GM and count with a hemocytometer. Plate 5-7.5x106 cells in 15 mL of GM per 75cm2 flask.
After one day and every 2-3 subsequent days in culture, dislodge loose cells by knocking the flask against your hand. Remove the medium along with any dislodged cells and replace with 15 mL of fresh GM.
Glia can be harvested after 1-2 weeks of growth in the flasks, when they are about 70-100% confluent. To prepare individual coverslips coated with glia, place 6 nitric acid cleaned and sterilized 25mm round coverslips in a 10cm dish, and place 3 dots of the 3:1 mixture of paraffin/petroleum jelly onto each coverslip in a triangular pattern with a small paint brush. Treat open dishes with UV light for 30 minutes. Coat the coverslips with 0.1 mg/ mL Poly-d-lysine (30kDa) in borate buffer for one hour, then wash extensively (3-5x) with sterile tissue culture grade deionized water and let dry.
Remove the glial-containing flask from incubator, discard the medium and rinse with 5 mL of pre-warmed trypsin/EDTA solution. Remove the trypsin/EDTA solution from the flask and pipette 3 mL of fresh pre-warmed trypsin/EDTA into the flask. Incubate the flask for 1 minute at 37°C before adding 5 mL of GM to stop the trypsinization.
Remove the glia from flask by repeated pipetting 10-15 times, and then transfer the media to a 15 mL conical tube. Centrifuge at 200xg (1000rpm) for 8 minutes. Remove the supernatant and add 10 mL of GM, count cells, and plate 5x105 cells in 12.5 mL of GM per 10cm dish containing the coverslips.
Exchange the medium with fresh pre-warmed GM every 2-3 days. The day before the neuron dissection, remove the GM and replace with SFM (section 2.2). Use this glial-conditioned SFM in step 4.12 when flooding cortical or hippocampal cultures.
4. Cortical And/or Hippocampal Dissection and Electroporation
Remove appropriate amounts of nucleofection solutions (Lonza), combine, and warm to room temperature before starting the dissection. Since Nucleofection solution has a limited lifetime when combined, we only combine the needed amount for each preparation (100 μL per transfection).
Euthanize a pregnant mouse at E15.5 with CO2 (day of plug is E0.5) and remove the uterus to a 10cm petri dish. Remove fetuses and decapitate into cold DM (section 2.1).
Remove the entire brain into a separate dish of cold DM and with a bent tungsten needle, remove both neocortices. Remove the meninges with microforceps and place cortices into new dish of cold DM. With a small iris or Wecker scissors, remove the cortex or hippocampus and place in a 1.5 mL Eppendorf tube filled with 1.0 mL of cold DM. Keep this tube on ice.
After dissecting all cortices or hippocampi, add 110 μL of 2.5% trypsin to the Eppendorf tube containing the tissue and place tube in a 37°C incubator for 20 minutes.
Remove the supernatant and wash cortices or hippocampi with 1.0 mL PM (section 2.2) by gently inverting the Eppendorf tube. Repeat the wash twice, leaving 1 mL of PM in the tube.
Triturate the chunks 15 times with a P1000 pipette, and remove supernatant/cells to a new 15 mL conical tube containing 4 mL of PM, leaving any chunks that remain in Eppendorf tube.
Spin the 15 mL tube at 20xg (350rpm) for 7 minutes with the brake off. Discard the supernatant and add 100 μL of premixed, room temperature nucleofection solution (Lonza) for each transfection. Triturate 5 times with a gentle up and down motion of the P1000 pipette.
Remove 100 μL of the nucleofection solution/cell mixture to each new Eppendorf tube and add the appropriate amount of DNA. For long term cultures we generally use 1-2μg of DNA per transfection. However, this only labels a small <10% proportion of neurons in the culture. We generally use 5-10μg of DNA per transfection if want higher transfection efficiency for short term culture. We have used up to a total of 40μg of DNA when transfecting with two different plasmids. Plasmids are stored in TE buffer at 1μg/ μL.
Add cell suspension/DNA to the cuvette (Lonza) and electroporate the cells in the Nucleofector (Lonza), using program O-005 (Mouse CNS neurons).
Working quickly, add 500 μL of pre-warmed and equilibrated PM to the cuvette and remove solution/cells to a new 1.5 mL Eppendorf tube. Add enough PM to bring the volume of each transfection to 1.0 mL. Count the cells with a hemocytometer and plate at 3-5x103 cells/cm2 for young cultures, or 5-10x103 cells/cm2 for long-term cultures.
For short-term cultures, flood the 35mm culture dishes with 2.0 mL of warmed, CO2-equilibrated SFM after one hour of plating. If using coverslips, we remove one-half of the PM and replace it with SFM, then repeat two more times. Either flooding the imaging chambers or washing the coverslips results in a very low serum content (<0.5%). Short-term cultures do not need to be cultured with a glial feeder layer and do not need to be re-fed.
For long-term cultures, we remove a glia covered coverslip containing three dots of paraffin/petroleum jelly and invert it over the 15mm hole in the 35mm dish, one hour after initial plating. Two milliliters of the conditioned SFM from the glial dish is then added to the imaging chamber. To feed the long-term cultures, we remove one-third of the SFM every 2-3 days and replace it with fresh, pre-warmed and CO2-equilibrated SFM.
5. Representative Results:
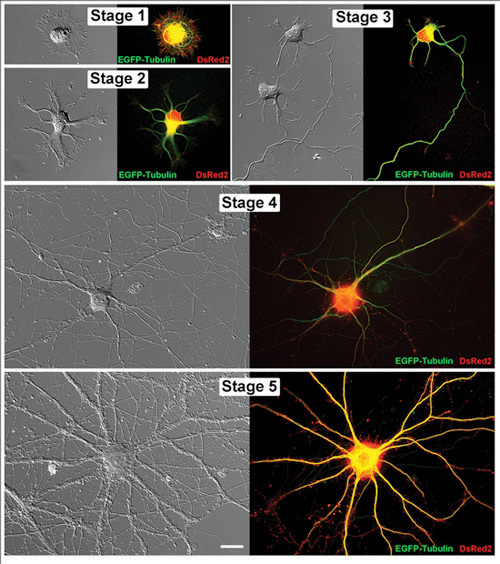 Figure 1. Living hippocampal neurons in successive stages of development. Paired images of representative living hippocampal neurons are shown as both a differential interference contrast image and a corresponding fluorescent micrograph. Each of these cells has been transfected with EGFP-Tubulin and DsRed2 in pCAX vectors. The neurons were imaged at the following days in vitro (DIV): Stage 1(1DIV), Stage 2 (1DIV), Stage 3 (2DIV), Stage 4 (11DIV) and Stage 5 (32DIV). Scale bar is 20μm.
Figure 1. Living hippocampal neurons in successive stages of development. Paired images of representative living hippocampal neurons are shown as both a differential interference contrast image and a corresponding fluorescent micrograph. Each of these cells has been transfected with EGFP-Tubulin and DsRed2 in pCAX vectors. The neurons were imaged at the following days in vitro (DIV): Stage 1(1DIV), Stage 2 (1DIV), Stage 3 (2DIV), Stage 4 (11DIV) and Stage 5 (32DIV). Scale bar is 20μm.
Discussion
This protocol for culturing embryonic hippocampal and cortical mouse neurons was developed as a modification of the Banker protocol, which uses rat neurons1,2. We have used this protocol for culturing mouse and hamster neurons as well3,4,5,6,7. This protocol works equally well for both hippocampal and neocortical neurons and is similar to a protocol published by Meberg and Miller8. Generally, we use hippocampal neurons for long-term culture because they are well characterized and a more established model system. Furthermore, they are likely to contain a more homogeneous population of neurons than neocortex. However, neocortical neurons cultured using this protocol also survive and differentiate similarly (unpublished data). We routinely use hippocampal and neocortical neurons for short term culture. Dissection of neocortex also results in substantially more neurons (1.5x106 neurons per pair of cortices) than hippocampal dissection (2.5x105 neurons per pair of hippocampi), which makes it a better choice of material for Western blotting, for example.
As with any primary culture, it is essential to minimize the time that it takes from the death of the animal to the plating of the cells. It will generally take 10-20 dissections to become consistently fast at dissection and plating. Also, when working with the Lonza Nucleofector, it is critical to work quickly during the electroporation procedure, as the viability of the neurons decreases rapidly if they are left in the nucleofection buffer.
Much of our imaging is conducted with total internal reflectance fluorescence microscopy (TIRFM). This type of microscopy is only capable of imaging several hundred nanometers beyond the coverslip. Therefore, the areas of the neurons that we frequently image, the axonal growth cone and dendritic spines, need to be adhered directly to the coverslip. Thus, we use low density cultures that require glial feeding for long-term culture. We have used higher density cultures (>2x104 cells/cm2), without glial feeder layers, for long-term cultures and found that they survive very well with little feeding. However, the dendritic spines of these neurons are oftentimes too far away from the substrate to image in TIRFM, although they can be readily detected with wide-field microscopy or confocal microscopy.
In most of our studies we transfect neurons before plating, and have imaged fluorescently-labeled proteins for up to three months in culture. This long-term expression of fluorescently-labeled proteins gives us confidence that by using low concentrations of DNA (1-2μg) we are not producing overexpression artifacts in the neurons. However, this procedure can also be used to study overexpression of proteins if high amounts of DNA are used (10-20μg). The plasmids that we use to transfect neurons usually contain EGFP or mCherry fusion proteins, although we also label the neuronal cytoplasm with DsRed2 or EGFP alone. This electroporation technique works well with a number of vectors. We prefer plasmids that contain a β-actin promoter with a CMV enhancer and β-globin poly-A tail (pCAGGs or pCAX plasmids)9, due to the relatively high levels of expression, and the fact that they are well tolerated by the neurons in both short- and long-term culture. Generally, proteins begin to express within about 4 hours of plating and reach levels sufficient for imaging within 10-24hrs10. We have successfully used CMV-promoter-driven plasmids in short term cultures, but have found that they can cause high levels of overexpression that kill neurons in long-term culture. Nevertheless, we have found that the glial conditioning of low density cultures helps with the survival of neurons transfected with CMV-promoter driven plasmids, compared to higher density (non-glial fed) cultures.
Disclosures
No conflicts of interest declared.
Acknowledgments
All procedures were approved by the University of Wisconsin Committee on Animal Care and were in accordance with NIH guidelines. We thank Dr. Katherine Kalil for the generous use of her Nucleofector device. We also thank members of the Dent lab for comments on the protocol. This work was supported by grants NIH R01-NS064014, Dana Foundation and Whitehall Foundation to E.W.D.
Christopher Viesselmann, Jason Ballweg and Derek Lumbard contributed equally to this paper.
References
- Goslin K, Asmussen H, Banker G. Chapter 13. In: Goslin K, Banker G, editors. Culturing Nerve Cells. The MIT Press; 1998. pp. 339–370. [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Dent EW, Callaway JL, Szebenyi G, Baas PW, Kalil K. Reorganization and movement of microtubules in axonal growth cones and developing interstitial branches. J Neurosci. 1999;19:8894–8908. doi: 10.1523/JNEUROSCI.19-20-08894.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Kalil K. Dynamic imaging of neuronal cytoskeleton. Methods Enzymol. 2003;361:390–407. doi: 10.1016/s0076-6879(03)61020-7. [DOI] [PubMed] [Google Scholar]
- Dent EW. Filopodia are required for cortical neurite initiation. Nat Cell Biol. 2007;9:1347–1359. doi: 10.1038/ncb1654. [DOI] [PubMed] [Google Scholar]
- Hu X, Viesselmann C, Nam S, Merriam E, Dent EW. Activity-dependent dynamic microtubule invasion of dendritic spines. J Neurosci. 2008;28:13094–13105. doi: 10.1523/JNEUROSCI.3074-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrand C. Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron. 2004;42:37–49. doi: 10.1016/s0896-6273(04)00108-4. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Miller MW. Chapter 7. In: Hollenbeck PJ, Bamburg JR, editors. Neurons: Methods and Applications for the Cell Biologist. Academic Press; 2003. pp. 112–129. Volume 71. [Google Scholar]
- Osumi N, Inoue T. Gene transfer into cultured mammalian embryos by electroporation. Methods. 2001;24:35–42. doi: 10.1006/meth.2001.1154. [DOI] [PubMed] [Google Scholar]
- Zeitelhofer M. High-efficiency transfection of mammalian neurons via nucleofection. Nat Protoc. 2007;2:1692–1704. doi: 10.1038/nprot.2007.226. [DOI] [PubMed] [Google Scholar]