

Abstract
Using the three-dimensional structure of biological macromolecules to infer how they function is one of the most important fields of modern biology. The availability of atomic resolution structures provides a deep and unique understanding of protein function, and helps to unravel the inner workings of the living cell. To date, 86% of the Protein Data Bank (rcsb-PDB) entries are macromolecular structures that were determined using X-ray crystallography.
To obtain crystals suitable for crystallographic studies, the macromolecule (e.g. protein, nucleic acid, protein-protein complex or protein-nucleic acid complex) must be purified to homogeneity, or as close as possible to homogeneity. The homogeneity of the preparation is a key factor in obtaining crystals that diffract to high resolution (Bergfors, 1999; McPherson, 1999).
Crystallization requires bringing the macromolecule to supersaturation. The sample should therefore be concentrated to the highest possible concentration without causing aggregation or precipitation of the macromolecule (usually 2-50 mg/ mL). Introducing the sample to precipitating agent can promote the nucleation of protein crystals in the solution, which can result in large three-dimensional crystals growing from the solution. There are two main techniques to obtain crystals: vapor diffusion and batch crystallization. In vapor diffusion, a drop containing a mixture of precipitant and protein solutions is sealed in a chamber with pure precipitant. Water vapor then diffuses out of the drop until the osmolarity of the drop and the precipitant are equal (Figure 1A). The dehydration of the drop causes a slow concentration of both protein and precipitant until equilibrium is achieved, ideally in the crystal nucleation zone of the phase diagram. The batch method relies on bringing the protein directly into the nucleation zone by mixing protein with the appropriate amount of precipitant (Figure 1B). This method is usually performed under a paraffin/mineral oil mixture to prevent the diffusion of water out of the drop.
Here we will demonstrate two kinds of experimental setup for vapor diffusion, hanging drop and sitting drop, in addition to batch crystallization under oil.
Protocol
Materials:
Protein sample - Lysozyme (50 mg/ mL)
Hanging drop 24-well tray
Sitting drop 24-well tray
Microbatch crystallization 96 well tray
Crystallization solutions (either commercial available or home made)
Silicon grease
5 mL syringe without Luer-lock
Siliconized cover slides
Optical sealing tape
Paraffin
0.1-2 μL micropipette with low-retention tips
Tweezers
Professional wipes
1. Hanging/sitting Drop Procedure:
For an initial screen one can use commercially available screens that exploit the sparse matrix incomplete factorial method of trial conditions. The sparse matrix incomplete factorial method is biased by and selected from known crystallization conditions for macromolecules (Carter and Carter, 1979; Cudney et al., 1994; Jancarik and Kim, 1991; Jancarik et al., 1991). If the crystallization condition is already known, a narrow screen can be assembled, varying the precipitant concentration and pH around the original hit. The most commonly successful precipitant is polyethylene glycol (PEG), followed by ammonium sulfate. Together, these two precipitants account for approximately 60% of all recorded macromolecular precipitants used for crystallization (Gilliland et al., 1994; Kimber et al., 2003; Page et al., 2003).
- Prepare your crystallization solutions. Usually these conditions are combined from a precipitating agent, salt of some kind and a buffer to set the pH. In some cases additives may be added specifically for the protein of interest. All stock solution should be filtered using 0.22 μm filter. Some of the stock solution might be very viscous (50% PEGs, glycerol, PEG-MME, etc ).
- Prepare 1.5 mL of 0.6, 0.8, 1.0, 1.2, 1.4, 1.6 M NaCl each in 0.1 M NaOAc pH 4.0, 4.3, 4.6 and 4.9 (total of 24 solutions)
Thaw your protein sample and keep on ice. Protein stocks should be stored at -80°C unless the protein of interest is sensitive to freeze/thaw cycles (if so, store at 4°C). Typical protein concentrations are in the range of 5-50 mg/ mL, with higher concentrations usually giving better results.
Spin the sample 15min/18,000xg/4°C - some proteins tend to partially aggregate and precipitate after a thaw step. These aggregates can promote amorphous precipitation of the rest of the protein molecules therefore thorough spin down will ensure you have only soluble molecules in your sample. Keep protein on ice until setting up the tray.
Determine the protein concentration from its absorbance at 280 nm using a UV spectrophotometer. The protein concentration can be calculated from the absorbance reading and the protein's extinction coefficient. The extinction coefficient can be calculated with the ProtParam tool on the Expasy website: http://ca.expasy.org/tools/protparam.html
Fill wells of the 24-well hanging/sitting drop tray with 500 μL of precipitant solution according to the tray setup scheme (Figure 2B). Create a silicone grease ring around the edge of the well. The ring should have a small gap to prevent air pressure buildup as the well is sealed. In sitting drops, there is no need for grease since the well is sealed with tape (Figure 2A).
Take a cover slide and clean it with condensed air spray, or a professional wipe - avoiding dust will ease the interpretation of the crystallization drop and eliminate contaminations. For sitting drops, cover slide are not used. Instead, the well contains a shelf on which the protein and precipitant are mixed.
- Put equal volumes of the protein sample and reservoir solution on the cover slide. Different volumes of protein and precipitant may be tried as part of optimization process. Using more protein than precipitant results in a net concentration of the protein in the drop after equilibration, which can be desirable for dilute protein samples, and to slow crystal nucleation or growth. Any other nonvolatile substance in the drop solution will also be concentrated by the same factor. When pipetting the protein and reservoir solutions onto the cover slide, be extremely careful to avoid bubble formation. This often occurs when air is blown out of the pipette.
- Hanging drop: Load 2 μL of 50 mg/ mL lysozyme in 0.02 M NaOAc pH 4.9 to the center of the cover slide and add to it 2 μL of the reservoir solution.
- Sitting drop: Load 2 μL of 50 mg/ mL lysozyme solution to the center of the shelf and add to it 2 μL of reservoir solution.
Flip the cover slide gently and lay it down on the grease ring on top of the well. Press down gently so that air can escape from the well through the notch in the grease to prevent air pressure buildup in the well and keep the well sealed. Air pressure buildup in the well can raise the cover slide, compromising the well seal. In the sitting drop method, optically clear transparent tape is used to cover the wells. Hence, in this method sealing will take place every row/column.
Move on to the next well until tray is complete.
Check the tray upon setup - eliminating dust particles and other contaminations can reduce the false positive identification of protein crystals.
Use a scoring sheet to document your experiment.
Place the tray at the desired incubation temperature, generally between 4°C and room temperature. 20°C is most commonly successful. Take care to handle the tray gently and avoid any shaking. Be careful while opening and closing the incubator door. Vibrations or temperature changes during transfer or incubation can prevent crystal growth or negatively affect crystal quality. A shock absorbing material (e.g. packing foam) can be used as padding under the crystal trays.
Check the tray for crystals the following day, and then every few days, always handling the trays with care. Document any findings and drop morphologies in a tray specific scoring sheet. Crystals typically take 2-5 days to appear, although crystals occasionally appear much sooner (almost immediately) or later (up to several months!). Once crystallization conditions have been identified and the growth rate of the crystals is known, it is best to leave the trays undisturbed until crystals have appeared and grown to at least half of their final size. Leaving the trays undisturbed can reduce the number of crystal nucleation events, hence producing smaller numbers of larger crystals.
2. Microbatch Procedure:
Protein sample and precipitant preparation are as described above.
Air spray a new microbatch tray to avoid dust and other large particles.
In the microbatch tray, fill paraffin oil up to 3 mm high (just enough to cover the wells). Remove access of oil.
Load 1 μL of protein solution (50 mg/ mL lysozyme) into the oil pre-filled well - make sure to pipette the solution directly to the bottom of the well (Figure 2A).
Load 1 μL of precipitant solution into the well according to the tray setup scheme (Figure 2B) - make sure the drop sinks to the bottom of the well and fuses with the protein droplet.
Move to the next well until tray is complete.
Follow steps 11-14 of the hanging/sitting drop procedure.
3. Representative Results:
Crystallization is usually referred to as the bottleneck of X-ray crystallography. A sparse matrix incomplete factorial screen of precipitating conditions typically produces many different types of protein aggregation and precipitation, among them large single crystals. If the protein or precipitant concentrations are too high one can see brown matter with no distinct shape and size (amorphous precipitation). When the solution is undersaturated, the drop will often be completely clear and devoid of any kind of precipitation. Figure 3 shows several examples for precipitation phenomena and crystals (Dessau et al., 2006). More precipitation phenomena with more detailed interpretation can be found at http://xray.bmc.uu.se/terese/tutorials.html.
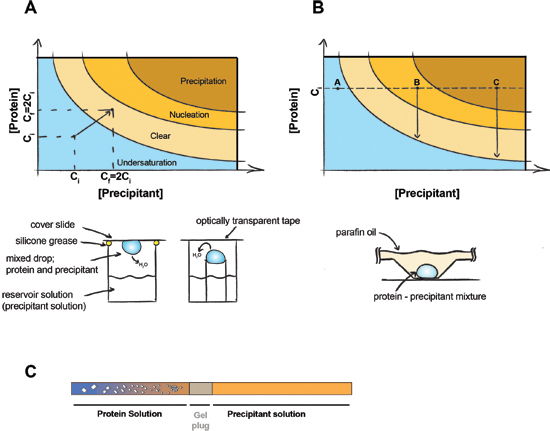 Figure 1. The principle of protein crystallization.
The principle of protein crystallization. In a vapor diffusion experiment (A) equal volumes of precipitant and protein are present in the drop. Water will diffuse out and both the precipitant and protein concentration will be doubled until equilibrium is achieved between the drop and the reservoir solution. In batch crystallization (B) the precipitant and protein concentration do not change during the experiment. Point A - Protein stays undersaturated no crystals can be formed, Point B - Protein nucleation occur, crystals began to form and the concentration of protein in solution drops to saturation. Point C - Protein precipitates, but crystals may still grow. (C) Dialysis crystallization using a gel-plugged capillary. Crystals appear as salt diffuses out of the protein sample (and/or precipitant diffuses into the protein sample) across the gel plug.
Figure 1. The principle of protein crystallization.
The principle of protein crystallization. In a vapor diffusion experiment (A) equal volumes of precipitant and protein are present in the drop. Water will diffuse out and both the precipitant and protein concentration will be doubled until equilibrium is achieved between the drop and the reservoir solution. In batch crystallization (B) the precipitant and protein concentration do not change during the experiment. Point A - Protein stays undersaturated no crystals can be formed, Point B - Protein nucleation occur, crystals began to form and the concentration of protein in solution drops to saturation. Point C - Protein precipitates, but crystals may still grow. (C) Dialysis crystallization using a gel-plugged capillary. Crystals appear as salt diffuses out of the protein sample (and/or precipitant diffuses into the protein sample) across the gel plug.
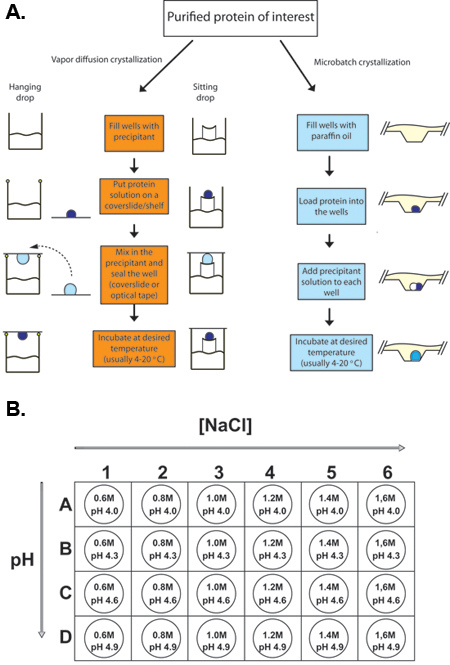 Figure 2. Outline of typical protein crystallization experiments.
(A) Procedures for hanging drop vapor diffusion, sitting drop vapor diffusion, and microbatch protein crystallization. In each case, a small volume of concentrated protein sample is mixed with an equal or smaller volume of precipitant solution and allowed to equilibrate. (B) For the crystallization of chicken lysozyme, a 24-well tray is set up with various concentrations of sodium chloride (the precipitant) and with 0.1 M sodium acetate as the buffer at various pHs (4.0 - 4.9).
Figure 2. Outline of typical protein crystallization experiments.
(A) Procedures for hanging drop vapor diffusion, sitting drop vapor diffusion, and microbatch protein crystallization. In each case, a small volume of concentrated protein sample is mixed with an equal or smaller volume of precipitant solution and allowed to equilibrate. (B) For the crystallization of chicken lysozyme, a 24-well tray is set up with various concentrations of sodium chloride (the precipitant) and with 0.1 M sodium acetate as the buffer at various pHs (4.0 - 4.9).
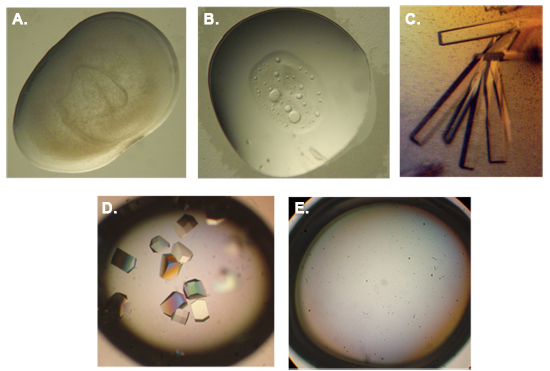 Figure 3. Typical outcomes of a protein crystallization experiment.
(A) Amorphous precipitation. When protein or precipitant (or both) are in high concentration. (B) Phase separation. Protein or detergent may separate to a different phase when mixed with certain precipitants at high concentration. (C) Rod shaped protein crystals of AtCSN7 obtained in polyethylene glycol 8000 and magnesium acetate (Dessau et al., 2006). (D) Lysozyme crystals obtained in 1.0 M NaCl and sodium acetate pH 4.9. (E) Under saturated drops usually remain clear. Photography by Moshe Dessau.
Figure 3. Typical outcomes of a protein crystallization experiment.
(A) Amorphous precipitation. When protein or precipitant (or both) are in high concentration. (B) Phase separation. Protein or detergent may separate to a different phase when mixed with certain precipitants at high concentration. (C) Rod shaped protein crystals of AtCSN7 obtained in polyethylene glycol 8000 and magnesium acetate (Dessau et al., 2006). (D) Lysozyme crystals obtained in 1.0 M NaCl and sodium acetate pH 4.9. (E) Under saturated drops usually remain clear. Photography by Moshe Dessau.
Discussion
In this article we describe and demonstrate general current protocols for protein crystallization. Since it a multi-step procedure there are few considerations one needs to be aware of. When working with very small volumes (0.5-2 μL), drying of the drop due to evaporation is a major concern. Hence, it is recommended to work in a well-controlled environment (with low air flow, high humidity and tight temperature control) and to adopt a technique that minimizes exposure of the drop to the open air. For this reason, it is essential to operate in a well-organized work environment. All reagents should be prepared prior to setting up the tray and lab equipment should be within easy reach (pipetors, tips, tubes, wipes, etc ).
As a general rule, take your protein out of storage as close as possible to the time you will be setting up the tray. That way, there are fewer environmental influences on your sample. It is very important to hold the protein tube by the neck of the tube and not on the bottom where the protein is, so the sample will not get heated from your body temperature. With the above precautions, the experimental margins of error can be minimized, thus increasing the reproducibility of the results.
There are many possible modifications to the protocols described here. One can alter protein concentration, protein/precipitant ratio, temperature (4°C - 37°C), pH and various additives can be added. Another relatively commonly used method for protein crystallization is dialysis. In this method, the protein solution is dialyzed against a precipitant solution through a semi-permeable membrane. Dialysis trials are more cumbersome to set up, but one advantage of dialysis is that precipitations conditions of low ionic strength can easily be tested. This is desirable if the protein is poorly soluble at low salt concentrations, since the protein may then crystallize as the salt is dialyzed away from the protein buffer. A solid phase (gel) can be used to control the rate of dialysis. For example, an agarose gel inside a capillary can be used as the diffusion interface between the protein and precipitant solutions (Figure 1C).
Increasing the concentration of a precipitating agent to a point just below supersaturation and then adjusting the pH or temperature to reduce the solubility of the protein can be used to crystallize samples with low protein concentration. Modification of pH can be accomplished very well with the vapor diffusion technique, when volatile acids and bases such as acetic acid, ammonium hydroxide or ammonium acetate are used (McPherson, 2004).
In the micro-batch technique a whole array of oil mixtures can be tested to cover the wells. Different oils have different evaporation permeabilities, and hence produce different rates of concentration changes of the protein/precipitant mixture under the oil.
Crystallization is a requisite step for obtaining structural information of macromolecules at near-atomic resolution. The methods described here are therefore fundamental to studying enzyme mechanisms and protein-protein or protein-nucleic acids interactions, and are ultimately one of the most important tools in structure-based drug design and development.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by a Burroughs Wellcome Investigator Award to YM and by a Brown-Coxe Postdoctoral Fellowship from Yale University to MD.
References
- Bergfors TM. Protein crystallization : techniques, strategies, and tips : a laboratory manual. La Jolla, Calif: International University Line; 1999. [Google Scholar]
- Carter CW, Carter CW. Protein crystallization using incomplete factorial experiments. J Biol Chem. 1979;254:12219–12223. [PubMed] [Google Scholar]
- Cudney R, Patel S, Weisgraber K, Newhouse Y, McPherson A. Screening and optimization strategies for macromolecular crystal growth. Acta Crystallogr D Biol Crystallogr. 1994;50:414–423. doi: 10.1107/S0907444994002660. [DOI] [PubMed] [Google Scholar]
- Dessau M, Chamovitz DA, Hirsch JA. Expression, purification and crystallization of a PCI domain from the COP9 signalosome subunit 7 (CSN7) Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:1138–1140. doi: 10.1107/S1744309106041959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland GL, Tung M, Blakeslee DM, Ladner JE. Biological Macromolecule Crystallization Database, Version 3.0: new features, data and the NASA archive for protein crystal growth data. Acta Crystallogr D Biol Crystallogr. 1994;50:408–413. doi: 10.1107/S0907444994002003. [DOI] [PubMed] [Google Scholar]
- Jancarik J, Kim SH. Sparse matrix sampling: a screening method for crystallization of proteins. J Appl Cryst. 1991;23:409–411. [Google Scholar]
- Jancarik J, Scott WG, Milligan DL, Koshland DE, Kim SH. Crystallization and preliminary X-ray diffraction study of the ligand-binding domain of the bacterial chemotaxis-mediating aspartate receptor of Salmonella typhimurium. J Mol Biol. 1991;221:31–34. doi: 10.1016/0022-2836(91)80198-4. [DOI] [PubMed] [Google Scholar]
- Kimber MS, Vallee F, Houston S, Necakov A, Skarina T, Evdokimova E, Beasley S, Christendat D, Savchenko A, Arrowsmith CH. Data mining crystallization databases: knowledge-based approaches to optimize protein crystal screens. Proteins. 2003;51:562–568. doi: 10.1002/prot.10340. [DOI] [PubMed] [Google Scholar]
- Lorber B, Sauter C, Theobald-Dietrich A, Moreno A, Schellenberger P, Robert MC, Capelle B, Sanglier S, Potier N, Giege R. Crystal growth of proteins, nucleic acids, and viruses in gels. Prog Biophys Mol Biol. 2009;101:13–25. doi: 10.1016/j.pbiomolbio.2009.12.002. [DOI] [PubMed] [Google Scholar]
- McPherson A. Crystallization of biological macromolecules. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. [Google Scholar]
- McPherson A. Introduction to protein crystallization. Methods. 2004;34:254–265. doi: 10.1016/j.ymeth.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Page R, Grzechnik SK, Canaves JM, Spraggon G, Kreusch A, Kuhn P, Stevens RC, Lesley SA. Shotgun crystallization strategy for structural genomics: an optimized two-tiered crystallization screen against the Thermotoga maritima proteome. Acta Crystallogr D Biol Crystallogr. 2003;59:1028–1037. doi: 10.1107/s0907444903007790. [DOI] [PubMed] [Google Scholar]