

Abstract
In mammalians, stem cells acts as a source of undifferentiated cells to maintain cell genesis and renewal in different tissues and organs during the life span of the animal. They can potentially replace cells that are lost in the aging process or in the process of injury and disease. The existence of neural stem cells (NSCs) was first described by Reynolds and Weiss (1992) in the adult mammalian central nervous system (CNS) using a novel serum‐free culture system, the neurosphere assay (NSA). Using this assay, it is also feasible to isolate and expand NSCs from different regions of the embryonic CNS. These in vitro expanded NSCs are multipotent and can give rise to the three major cell types of the CNS. While the NSA seems relatively simple to perform, attention to the procedures demonstrated here is required in order to achieve reliable and consistent results. This video practically demonstrates NSA to generate and expand NSCs from embryonic day 14-mouse brain tissue and provides technical details so one can achieve reproducible neurosphere cultures. The procedure includes harvesting E14 mouse embryos, brain microdissection to harvest the ganglionic eminences, dissociation of the harvested tissue in NSC medium to gain a single cell suspension, and finally plating cells in NSA culture. After 5-7 days in culture, the resulting primary neurospheres are passaged to further expand the number of the NSCs for future experiments.
Protocol
1. Basic Set Up Before Proceeding to Dissection:
Appropriate volume of complete NSC medium is prepared by mixing NeuroCult NSC Basal Medium and NeuroCult NSC Proliferation Supplements at a 9:1 ratio, respectively.
The medium is warmed up in a 37°C water bath.
Cold HEPES-buffered minimum essential medium (HEM) with high concentration of antibiotics (10%) is prepared for dissection and washing purpose. Alternatively, NSC basal medium with antibiotics supplementation may also be used for this purpose.
25-30 mL of cold HEM containing antibiotics is dispensed into sterile 50 mL tubes for collection of the embryos.
Several 10cm plastic Petri dishes are needed to hold the embryos and brains during dissection and also to hold dissected tissue.
The surgical tools, needed to remove the embryos (large scissors, small pointed scissors, large forceps, small curved forceps) or for embryonic brain dissection (small forceps, curved fine forceps, 45° angled fine forceps, and small scissors) are sterilized using glass bead sterilizer at 250°C or other available autoclave methods.
Dissection microscope is wiped with 70% alcohol and set up inside a laminar flow or PC2 hood.
2. Harvesting E14 Mouse Brain and Micro-dissection:
A time mated pregnant mouse is anesthetized on day 14th of gestation according to one's institutional approved animal protocol.
Cervical dislocation is performed to make sure the animal does not suffer pain and distress.
The anesthetized mouse is laid on its back on an absorbent tissue paper, and the abdomen is rinsed with 70% ethanol to sterilize the area.
The skin over the abdomen is grasped using a large forceps, and then the skin and the underlying fascia is cut with large scissors to expose the abdominal cavity and the uterine horns.
The uteri are removed with small forceps and scissors and are transferred into a 50‐ml conical tube-containing cold HEM.
The uterine tissues are transferred to the hood and then rinsed once or twice with enough volume of fresh sterile cold HEM to remove possible contaminants like blood and hair.
The uterine tissues are then transferred to a 10cm Petri dish containing cold HEM.
The uterine horns are opened using small curved forceps and scissors and the embryos are transferred to a new 10cm dish, which contains cold HEM.
The heads of the embryos are separated at the cervical spinal cord level and transferred to another Petri dish containing cold HEM.
The Petri dish is transferred under a dissecting microscope to remove the brain from the skull.
The heads are held with a fine curved forceps from the caudal side so as the dorsal side of the heads are facing upwards. Using micro-scissors, first a horizontal cut is made above the eyes and then continued in the midline from the forehead towards the back of the head. Make sure to cut through the skin and the skull and not to damage the underlying brain.
The brain is removed from the skull by pushing the edges of the cut section in a backward motion using the curved forceps. This procedure is continued until all brains are removed from the skulls.
The brain is held steady using the curved forceps so that the dorsal side is facing upwards and then using microscissors a cut is performed through the cortex of each hemisphere extending from the olfactory bulbs to the back of the hemisphere to expose the ganglionic eminences.
Using the curved forceps in one hand and the 45° angled forceps in the other, the cut flaps of cortices are spread and the ganglionic eminences are dissected out.
The dissected tissue is placed in a sterile 10cm Petri dish, and this procedure is repeated until all the brains have been micro-dissected.
Tissue pieces are collected using 1 mL of NSC medium in a 1 mL pipette tip and transferred to a 15 mL centrifuge tube.
Tissue is then dissociated thoroughly, but gently by pressing the pipette tip to the bottom of the tube and pipetting the suspension up and down. This way the clumps are broken up into single cells. Pipetting is performed 3-4 times so that a milky like suspension is achieved. Then, the suspension is allowed to settle for 1-2 minutes so as the non-dissociated clumps precipitates.
Almost the entire cell suspension is transferred to the other tube and then another 1 mL of NSC medium is added to the remaining clumps and dissociated to single cell as described.
The content of the tubes are pooled and centrifuged for 5 minutes at room temperature, at 700rpm (110g).
The supernatant is vacuumed off down to the actual pellet, and the cells are resuspended in 1 mL of complete NSC medium.
The medium is gently pipetted up and down to have a homogeneous single cell suspension.
10 μL of the cell suspension is mixed with 90 μL of trypan blue to perform a cell count.
Finally, the cells are plated at the density of 2x105 cells/ mL in complete NSC medium supplemented with 20ng/ mL epidermal growth factor (EGF). 5 mL medium is used for T25, 20 mL for T75 and 40 mL for T175 flasks.
Neurospheres are formed in 5-7 days when incubated at 37° C in a humidified incubator with 5% CO2. In 6-7 days, the spheres should measure between 150-200 microns and will be ready to subculture (passage).
3. Passaging and Expansion of Embryonic NSCs:
When the neurospheres are ready for subculture (150-200 μm in diameter), the medium with suspended spheres is removed from the flasks, placed in an appropriate size sterile tissue culture tube, and centrifuged at 700 rpm (110 g) for 5 min at room temperature.
The supernatant is discarded and the spheres are resuspended in 1 mL of %0.05 trypsin-EDTA.
The cell suspension is then incubated in a 37°C water bath for 2-3 min, then an equal volume of soybean trypsin inhibitor is used to stop the trypsin activity.
The cell suspension is gently pipetted up and down to ensure that the trypsin has been completely inactivated.
The cell suspension is centrifuged at 700 rpm (110g) for 5 min. Then, the supernatant is removed and the cells are resuspended in 1 mL of complete NSC medium.
10 μL of the cell suspension is mixed with 90 μL of trypan blue to perform a cell count.
The cells are plated at a concentration of 5x104 cells/ mL in complete NSC medium supplemented with 20ng/ mL EGF in an appropriate size tissue culture flask. 5 mL medium is used for T25, 20 mL for T75 and 40 mL for T175 flasks.
Secondary neurospheres are formed in 5-7 days when incubated at 37° C in a humidified incubator with 5% CO2.
4. Representative Results:
In primary embryonic NSC culture, the majority of the cells will become hypertrophic and attach to the tissue culture dishes upon plating. While the majority of cells will either die or differentiate, after 2-3 days, proliferative cells make small clusters of cells that will detach from the substrate (Figure 1). Formation of large spheroidal aggregates in the first 48-hour of culture should not be mistaken for primary spheres. Aggregate formation mainly depends on the amounts of debris and non-dissociated tissue clumps in the culture. True neurospheres are phase bright and become more spherical as size increases (Figure 2). Small microspikes appears on the outer surface of viable and healthy spheres (Figure 3). After 5-7 days, the spheres must be round but not compacted; and should measure between 150 and 200 μm in diameter. If neurospheres are allowed to grow too large (after 9-10 days in culture), they might form aggregates or become dark in color because of cell death at the center of the spheres (see video). Large neurospheres might eventually begin to differentiate in situ (attaching to the substrate and migrating toward the periphery). It is also difficult to dissociate large neurospheres and subculture them.
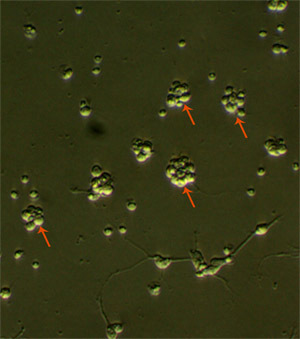 Figure 1. Primary E14 NSC culture 3 days after plating. Arrows show the proliferating clusters of NSCs. Original magnification; 20x
Figure 1. Primary E14 NSC culture 3 days after plating. Arrows show the proliferating clusters of NSCs. Original magnification; 20x
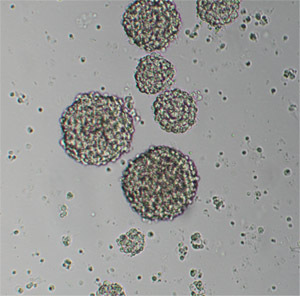 Figure 2. Primary E14 NSC culture 7 days after plating. Original magnification; 10x
Figure 2. Primary E14 NSC culture 7 days after plating. Original magnification; 10x
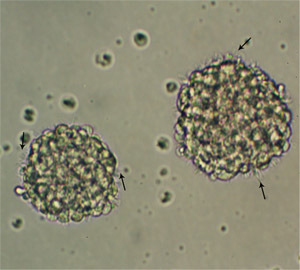 Figure 3. Passage one E14 neurospheres 5 days after plating. Note the micro-spikes (arrows) at the periphery of the spheres. Original Magnification; 20x
Figure 3. Passage one E14 neurospheres 5 days after plating. Note the micro-spikes (arrows) at the periphery of the spheres. Original Magnification; 20x
Discussion
The neurosphere assay is the method of choice for the isolation and expansion of neural stem cells 1-5 because of its simplicity and reproducibility. This assay is an invaluable tool for large-scale generation of undifferentiated CNS precursor cells, which could be used for both in vitro and in vivo studies. It should be emphasized that neurospheres could be generated from both the bona fide neural stem cells and more restricted progenitors. Therefore, calculating the neurosphere forming frequency simply overestimates the number of bona fide NSCs in any given neural cell population 6. To estimate the frequency of bona fide NSCs, it is strongly recommended to use the Neural Colony Forming Cell Assay (N-CFCA), which has been developed for this purpose7.
To have a consistent high quality neurosphere culture of E14 NSCs, we recommend:
To prepare needed items before starting to harvest the embryos.
To keep the embryos in cold HEM or basal NSC medium throughout dissection.
To perform dissection as quickly as possible (within 1 h), as tissue becomes soft and sticky over time and may be difficult to dissect.
Not to let the sphere grow too much. Usually they should be sub-cultured every 5-7 days depending on the size of spheres.
To trypsinize the spheres at the size of 150-200 μm for 2-3 minute. Leaving trypsin for more than 3 minutes causes damage to the cells and the sphere forming efficiency will decrease and the cells will tend to attach the culture dishes and differentiate.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by funding from the Overstreet Foundation.
References
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Morshead CM. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Craig CG. In vivo growth factor expansion of endogenous subependymal neural precursor cell populations in the adult mouse brain. J Neurosci. 1996;16:2649–2658. doi: 10.1523/JNEUROSCI.16-08-02649.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golmohammadi MG. Comparative analysis of the frequency and distribution of stem and progenitor cells in the adult mouse brain. Stem Cells. 2008;26:979–987. doi: 10.1634/stemcells.2007-0919. [DOI] [PubMed] [Google Scholar]
- Azari H, Rahman M, Sharififar S, BA Reynolds. Isolation and Expansion of the Adult Mouse Neural Stem Cells Using the Neurosphere Assay. J Vis Exp. 2010 doi: 10.3791/2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Rietze RL. Neural stem cells and neurospheres--re-evaluating the relationship. Nat Methods. 2005;2:333–336. doi: 10.1038/nmeth758. [DOI] [PubMed] [Google Scholar]
- Louis SA. Enumeration of neural stem and progenitor cells in the neural colony-forming cell assay. Stem Cells. 2008;26:988–996. doi: 10.1634/stemcells.2007-0867. [DOI] [PubMed] [Google Scholar]