

Abstract
Toll-like receptors are host sentinel receptors that signal the presence of infectious nonself and initiate protective immunity. One of the primary immune defense mechanisms is the recruitment of neutrophils from the bloodstream into the infected tissue. Although neutrophils are important in host defense, they can also be responsible for damaging pathologies associated with excessive inflammation. Here, we report that the di-acylated TLR2 ligand lipoteichoic acid can directly inhibit neutrophil recruitment in vivo. This discovery allowed us to test the concept that conventional proinflammatory TLR2 ligands can be made to act as inhibitors through specific structural modifications. Indeed, lipopeptide TLR2 ligands, when modified at their acyl chains to contain linoleate, lose their capacity to induce inflammation and yield ligands that can directly inhibit the in vivo neutrophil recruitment initiated by a wide range of proinflammatory stimuli. The inhibitory capacity of LTA and these modified ligands requires the expression of TLR2, but is independent of the TLR2 signaling adaptor, MyD88. Instead, this inhibitory effect requires functional activity of the fatty acid and nuclear hormone receptor peroxisome proliferator-activated receptor γ (PPARγ). Therefore, these data support a model in TLR2 biology where structural modifications of these ligands can profoundly influence host–microbial interactions. These inhibitory TLR2 ligands also have broader implications with respect to their potential use in various inflammatory disease settings.
Keywords: inflammation, leukocyte recruitment, bacterial ligands, innate immunity
Toll-like receptors (TLRs) are sentinel receptors of the immune system. Upon ligand binding, these receptors initiate receptor-specific recruitment of a family of TIR-domain-containing adaptor proteins, which further initiate signaling cascades that culminate in the activation of cell-type-specific, and receptor/ligand-specific inflammatory responses (1–3). In particular, TLR2 forms heterodimers with either TLR1 or -6 to initiate inflammatory responses upon stimulation with a wide variety of microbial-derived ligands (4, 5). A unifying feature of many of these TLR2 ligands is their acylation status, where acylation patterns impart specificity to receptor-ligand interactions; di-acylated ligands are recognized by TLR2/6 heterodimers and triacylated ligands are recognized by TLR2/1 heterodimers (6, 7). Principal among these ligands are acylated lipopeptides derived from bacteria, however other acylated bacterial components, such as the Gram-positive bacterial cell wall component lipoteichoic acid (LTA), are also recognized by TLR2 (8).
TLR-induced activation of acute inflammatory responses is ultimately responsible for the eradication of infectious agents, in part through the recruitment of polymorphonuclear neutrophils from the bloodstream into the affected tissue. This recruitment follows a well characterized cascade of successive steps (reviewed in refs. 9 and 10) that allow the neutrophils to deal with the offending microbes through the activities of proteases, bactericidal peptides, and reactive intermediates. In addition to the host-protective role that these cells play in the immune response, excessive or inappropriate neutrophil recruitment results in significant pathophysiology and morbidity (11–13).
Although TLR receptors and their ligands have been consistently characterized as activators of the immune response, we found that the TLR2 ligand LTA can actively inhibit the neutrophil-recruitment-induced in vivo in response to a wide variety of proinflammatory stimuli. We also found that TLR2 lipopeptide ligands can be engineered to exhibit inhibitory properties. This inhibitory mechanism relies on a collaboration between TLR2 and the anti-inflammatory nuclear hormone receptor peroxisome proliferator-activated receptor γ (PPARγ; ref. 14).
Results
TLR2/6 Ligand, LTA, Inhibits Acute Neutrophil Recruitment in Vivo.
In previous studies, we reported that the di-acylated TLR2/6 ligand LTA does not induce neutrophil recruitment in vivo when evaluated in a murine model of acute inflammation (15, 16). This finding is in contrast to other TLR2 lipopeptide ligands, which each induce robust neutrophil recruitment in vivo (16). The inability of LTA to induce neutrophil recruitment was unexpected, especially because many LTA preparations have been shown to contain non-TLR2, proinflammatory stimuli, including lipopolysaccharide (LPS; ref. 17). Despite the presence of these potent proinflammatory components, these preparations, as well as ultrapure preparations of LTA, did not induce neutrophil recruitment in vivo (15, 16). We hypothesized that LTA possesses inhibitory properties that prevent neutrophil recruitment in response to other acute inflammatory stimuli. To test this hypothesis, we used intravital microscopy of the murine cremaster muscle to evaluate the effects of the TLR4 ligand LPS or the cytokine TNFα on neutrophil recruitment in the presence or absence of Staphylococcus aureus LTA. In the presence of LTA, there was a dramatic reduction in the number of neutrophils that emigrated into the cremaster muscle compared with LPS or TNFα alone (Fig. 1A). In fact, the number of emigrated cells in the presence of LTA was similar to baseline, noninflamed conditions (Fig. 1A). This inhibitory response mediated by LTA was not observed in TLR2−/− mice (Fig. 1A), consistent with a functional role for this receptor. Using bone-marrow chimeric mice, we found that TLR2 expression is required on bone-marrow-derived cells in order to observe the inhibitory response (Fig. 1B). To ensure that the reduced emigration observed in wild-type animals was not the result of neutrophil trapping in the liver and lung (18), we evaluated circulating leukocyte counts. In all cases, the number of circulating leukocytes was unchanged from the control saline treatment (Fig. S1A). In addition, the neutrophils were induced to roll slowly along the endothelium in response to LPS and TNFα; this outcome was unaffected by LTA cotreatment (Fig. S1B), suggesting that there were no untoward toxic effects on the neutrophils.
Fig. 1.

S. aureus LTA inhibits the in vivo acute inflammatory responses to a wide array of inflammatory stimuli, in a TLR2-dependent manner. (A) Wild-type and TLR2−/− male mice were given intrascrotal injections of 150 μL of saline or saline containing LTA (5 ng/g), LPS (10 ng/g), TNFα (20 ng/g), either alone or in combination. Neutrophil emigration in the cremaster tissue was monitored using intravital microscopy 4.5 h following intrascrotal injection of the above ligands. (B) Bone marrow chimeric mice were given intrascrotal injections of 150 μL of saline or saline containing LTA (5 ng/g) or TNFα (20 ng/g), either alone or in combination. Neutrophil emigration 4.5 h following intrascrotal injection of the above ligands is shown. (C and D) Male C57BL/6 mice were given a dorsal s.c. inoculum of live luminescent E. coli in the presence or absence of LTA. (C) Luminescence normalized to the detected luminescence at the time of inoculation, observed over 6 h. (D) Image of two examples of these mice (with and without LTA), taken at the time of inoculation and 4 h later. (E) Neutrophil emigration into the cremaster tissue monitored by intravital microscopy following exteriorization of the cremaster and superfusion with buffer containing MIP2 (2.5 μM), MIP2 (2.5 μM) + LTA (200 ng/mL), or LTA (200 ng/mL) alone. (F) Evaluation of the number of neutrophils that had emigrated into the cremaster tissue, 4.5 h following an intrascrotal injection of LPS (10 ng/g) or TNFα (20 ng/g) and 2.5 h following an intrascrotal injection of LTA (100 ng/g).
To further evaluate the inhibitory potential of LTA, a s.c. inoculum of a luciferase reporter E. coli strain was used. When LTA was coadministered with this live bacterial inoculum, the host response to these bacteria was delayed, as seen by a prolonged presence of the reporter strain in animals that had received LTA (Fig. 1 C and D). This observation was not due to a general ability of LTA to promote the growth of E. coli, as the presence of LTA had no effect on bacterial growth in vitro (Fig. S1C). To assess whether LTA could have a direct effect on the ability of neutrophils to respond to a chemotactic stimuli, MIP2 was perfused over the exposed cremaster in the presence or absence of LTA. Neutrophil emigration was consistently inhibited by the presence of LTA (Fig. 1E). To assess whether LTA could abrogate an established acute inflammatory reaction, mice were administered with LPS or TNFα via an intrascrotal injection 2 h before the addition of LTA. Assessment 2.5 h later found a 50% reduction of newly emigrated cells in animals exposed to LTA (Fig. 1F). These data demonstrate that the inhibitory capacity of the LTA is not limited to a particular proinflammatory stimulus or receptor system, and most importantly, that LTA can dampen acute inflammatory responses, even after they have been initiated.
Because LTA is routinely used to study TLR2 biology in vitro, we evaluated the inhibitory capacity in murine bone marrow-derived macrophages. Consistent with the literature (16), we did not see any inhibitory effects and instead found LTA to activate inflammatory responses in these cells (Fig. S1 D and E). Finally, to determine whether this is a general inhibitory property of all TLR2 ligands or a unique characteristic of LTA, LPS was administered with each of the other TLR2 ligands: R-FSL1, S-FSL1, Pam2CSK4, or Pam3CSK4. Each ligand was found to be proinflammatory and did not exhibit any inhibitory properties toward neutrophil emigration (Fig. S1F).
Inhibitory Capacity of LTA Is MyD88-Independent.
It has previously been reported that signaling downstream of TLR2 requires MyD88 (19). Therefore, we used MyD88−/− mice to evaluate the requirement for MyD88 in this TLR2-dependent response and found that MyD88 was dispensable for the inhibitory activity of LTA (Fig. 2A). As a control, we assessed the conventional TLR2-signaling pathway in bone marrow-derived macrophages in vitro and observed a strict requirement for MyD88 for NFκB pathway activation (Fig. 2B). Moreover, to eliminate the possibility that the TLR3/TLR4-specific, MyD88-independent pathway was involved, similar experiments were performed using TRIF-deficient animals (20). Again, LTA inhibited TNFα-induced neutrophil recruitment in these animals, with levels comparable to those observed in wild-type or MyD88−/− animals (Fig. S2).
Fig. 2.
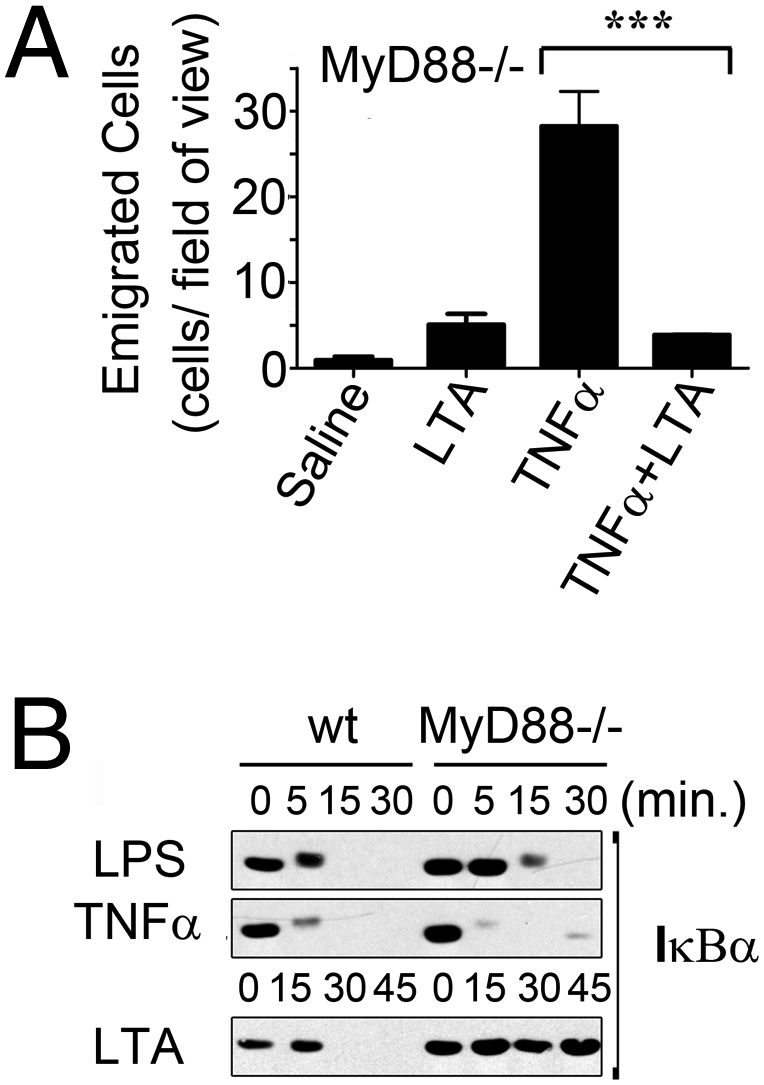
The inhibitory capacity of LTA is independent of conventional TLR2 signaling pathways. (A) MyD88−/− mice were given an intrascrotal injection of 150 μL of saline or saline containing LTA (5 ng/g) and/or TNFα (20 ng/g), either alone or in combination, and evaluated 4.5 h later by intravital microscopy for the number of emigrated cells in the cremaster tissue. (B) Wild-type or MyD88−/− bone marrow-derived macrophages were treated in vitro with LPS (100 ng/mL), TNFα (20 ng/mL), or LTA (1 μg/mL), and analyzed by Western blot for IκBα expression.
Defining the Molecular Characteristics That Render TLR2 Ligands Inhibitory.
LTA is a complex, heterogeneous mixture of up to 50 1,3-linked polyglycerolphosphate subunits linked to a β-gentiobiose core with a lipid anchor (21, 22). To identify the molecular characteristics responsible for the TLR2 inhibitory effects, we focused on the lipid anchor, because fatty acids derived from specific acyl chain modifications can be modified to yield products with anti-inflammatory properties. In particular, oxidized or nitrated derivatives of linoleic acid and nitrated derivatives of oleic acid are known PPARγ ligands (23), and activation of this nuclear hormone receptor has been shown to be anti-inflammatory by limiting neutrophil migration and the production of proinflammatory mediators by macrophages (14, 24, 25). Moreover, the fatty acid linolenic acid can also be metabolized to yield anti-inflammatory mediators of the resolvin and protectin family (26). Because the structure of LTA does not lend itself easily to chemical synthesis, we took the approach of transforming a TLR2 ligand into one with inhibitory properties by incorporating specific fatty acids. We synthesized modified versions of the proinflammatory lipopeptide TLR2 ligand, FSL-1, to contain the lipid modifications di-linoleate, di-oleate, or di-linolenate, in lieu of the conventional di-palmitate (Fig. 3). These modified ligands were then used to determine whether any of these modifications could transform the proinflammatory lipopeptide TLR2 ligand, FSL1, into an anti-inflammatory TLR2 ligand.
Fig. 3.

Acyl chain modifications to FSL1.
Following intrascrotal administration of the ligand containing di-linoleate (FSL1-Lin2), neutrophil recruitment was severely compromised compared with the parent FSL-1 ligand (Fig. 4A). Once again, this was not due to an effect on circulating leukocyte counts (Fig. S3A). By comparison, the other engineered ligands (FSL1-Lnn2 and FSL1-Ole2) maintained their ability to induce emigration to levels comparable to the parent FSL-1 ligand (Fig. 4A). When these ligands were used to stimulate murine macrophages, each ligand similarly activated NFκB and MAP kinase signaling, indicating competence in stimulating the TLR2 signaling axis (Fig. S3B). The small residual capacity of FSL1-Lin2 to initiate neutrophil recruitment remained dependent upon TLR2 (Fig. S3C). To establish whether FSL1-Lin2 is able to inhibit neutrophil recruitment in vivo, the exposed cremaster muscle was superfused with MIP2 in the presence or absence of FSL1-Lin2 for 1 h. In a manner that mimicked the inhibitory capacity of LTA, the presence of FSL1-Lin2 inhibited the emigration of cells into the tissue (Fig. 4B) in a TLR2-dependent manner (Fig. S3D), a response that was not observed using the proinflammatory FSL1-Lnn2 ligand (Fig. S3E). These data indicate that imbedding linoleate within the FSL1-Lin2 ligand imparted anti-inflammatory properties to this TLR2 ligand. FSL1-Lin2 did retain a limited capacity to activate inflammatory responses and recruit neutrophils, and thus did not fully mimic the anti-inflammatory capacity of LTA.
Fig. 4.

Molecular modification of a proinflammatory TLR2 ligand to contain linoleate yields a TLR2 ligand with diminished inflammatory potential and inhibitory characteristics. Mice were evaluated using intravital microscopy for the number of neutrophils emigrated into the cremaster tissue. (A) Mice evaluated 4.5 h following intrascrotal injections of 150 μL of saline or saline containing FSL1-Lin2, FSL1-Ole2, or FSL1-Lnn2 (all at 5 ng/g) in comparison with the parent (R/S)FSL-1 ligands. (A) Mice evaluated during superfusion of the exposed cremaster with buffer containing MIP2 (2.5 μM) or MIP2 (2.5 μM) + FSL1-Lin2 (200 ng/mL). (C) Wild-type mice or MyD88−/− mice evaluated 4.5 h following intrascrotal injections of 150 μL of saline or saline containing FSL1-Lin2 (5 ng/g) and/or TNFα (20 ng/g) as indicated.
Unlike LTA, FSL1-Lin2 retains a low level of neutrophil recruitment when evaluated 4.5 h following intrascrotal injection (Fig. 4A). The 1-h incubation period described above for the inhibition of MIP2-induced neutrophil recruitment is not enough time for the residual proinflammatory capacity of FSL-Lin2 to be a complicating factor. Therefore, we evaluated the inhibitory effect toward TNFα-induced neutrophil recruitment at 4.5 h. Consistent with its residual proinflammatory capacity, FSL1-Lin2 did not exhibit any inhibitory potential in wild-type animals (Fig. 4C). Next, we determined whether removing the residual proinflammatory capacity of this ligand would yield a ligand with purely anti-inflammatory properties. To address this possibility, MyD88−/− mice were used to negate the influence of the proinflammatory signaling capacity of FSL1-Lin2. In the absence of MyD88, FSL1-Lin2 inhibited the TNFα-induced neutrophil emigration (Fig. 4C). Therefore, in the absence of the residual proinflammatory responses induced by this engineered ligand, its inhibitory capacity is absolute. Taken together, these data suggest that, in addition to the acyl chain modifications that yield an inhibitory TLR2 ligand, the structure of LTA must impart additional inhibitory potential. In other words, the structure of a TLR2 ligand must completely impede its proinflammatory capacity to reveal a purely anti-inflammatory TLR2 ligand.
Designing a Synthetic TLR2 Ligand with Purely Anti-Inflammatory Properties: GM1-Targeted, Linoleate-Containing TLR2 Ligand (GML).
We next sought to abrogate the proinflammatory capacity of the TLR2 anti-inflammatory ligand FSL1-Lin2. We previously reported that LTA is not as potently proinflammatory as other TLR2 ligands and that this may be due to differential cellular internalization mechanisms between LTA and the other TLR2 ligands (16). Internalized LTA is targeted to the ER, golgi, and endosomal compartments (27). Artificially retaining LTA on the cell surface allows LTA to activate inflammatory responses much more potently in vitro (27). Therefore, we sought to further refine FSL1-Lin2 to mimic the internalization program of LTA in an attempt to hamper its proinflammatory capacity. To accomplish this, we altered the peptide sequence of the lipopeptide to a sequence that has been shown to bind the plasma membrane ganglioside GM1 (28). This strategy would allow the TLR2 ligand to bind GM1 in addition to TLR2 and to mimic the internalization mechanisms of the GM1 ligand, cholera toxin, which is also known to accumulate in the golgi (29). This so-named, GM1-targeted, linoleate-containing TLR2 ligand (GML; shown in Fig. 5A) was tested for the ability to inhibit neutrophil recruitment. We found that GML mimics the TLR2-dependent, MyD88-independent inhibitory capacity of the LTA preparations (Fig. 5B). Similarly to LTA, GML was found to inhibit the clearance of a s.c. inoculum of live E. coli (Figs. 5C and 4D), but had no beneficial effect on bacterial growth in vitro (Fig. S4A). GML was also found to inhibit the neutrophil recruitment induced by MIP2 (Fig. 6D). When the internalization of FITC-labeled FSL1 and GML ligands was evaluated in bone marrow-derived macrophages, distinct localization patterns were observed. GML accumulated in larger vesicles than the proinflammatory TLR2 ligand FSL1, and GML partially colocalized with the golgi marker Giantin (Fig. S4B). FSL1 was found in much smaller vesicles, which did not colocalize with Giantin. TLR2 is involved in the internalization of both ligands: FSL1 was substantially inhibited with regard to internalization in the absence of TLR2, and GML was no longer associated with the golgi in the absence of TLR2 (Fig. S4B). The residual internalization of GML in the absence of TLR2 may relate to its designed capacity to bind GM1. In sum, we have been able to define the molecular requirements for a TLR2 ligand to be transformed into an inhibitor of neutrophil recruitment and, in the process, we have designed a synthetic anti-inflammatory TLR2 ligand: GML.
Fig. 5.

Engineering of the inhibitory TLR2 lipopeptide ligand - GM1-targeted, linoleate-containing TLR2 ligand (GML). (A) Schematic of GML. (B) Wild-type, TLR2−/−, or MyD88−/− mice treated with an intrascrotal injection of 150 μL of saline alone or saline containing the indicated ligands (37.5 ng/g GML and/or 20 ng/g TNFα) either alone or in combination, and evaluated 4.5 h later for the number of emigrated neutrophils in the cremaster tissue. (C and D) Male C57BL/6 mice were given a dorsal s.c. inoculum of live luminescent E. coli in the presence or absence of GML. (C) Image of two examples of these mice (with and without GML) taken at the time of inoculation and 4 h later. (D) Luminescence normalized to the detected luminescence at the time of inoculation, observed over 6 h.
Fig. 6.

The inhibitory capacity of TLR2 ligands requires functional signaling through PPARγ. Mice were evaluated using intravital microscopy for the number of neutrophils emigrated into the cremaster tissue following intrascrotal injections of 150 μL of saline alone or saline containing GW9662 (0.5 μg) or T0070907 (0.5 μg), as a 1.5 h pretreatment. (A) Followed by an intrascrotal injection of 150 μL of saline alone or saline containing LTA, GML, FSL1-Lin2, or FSL1-Lnn2 (each at 5 ng/g) for 4.5 h. (B) Followed by an intrascrotal injection of 150 μL of saline alone or saline containing TNFα (20 ng/g) or TNFα (20 ng/g) + LTA (5 ng/g) for 4.5 h, (C) Followed by an intrascrotal injection of 150 μL of saline alone or saline containing TNFα (20 ng/g) or TNFα (20 ng/g) + GML (37.5 ng/g) for 4.5 h. (D) Followed by the exteriorization of the cremaster and superfusion of MIP2 (5 μM) in the presence or absence of LTA (5 μg/mL) or GML (5 μg/mL) for 60 min. (E) MPO−/− mice evaluated using intravital microscopy for the number of neutrophils emigrated into the cremaster tissue following intrascrotal injections of 150 μL of saline containing GML (37.5 ng/g), TNFα (20 ng/g), or TNFα + GML.
Inhibitory Activity of LTA and GML Requires Functional PPARγ.
Considering that the linoleate modification renders TLR2 ligands inhibitory, we suspected that in the in vivo proinflammatory microenvironment, linoleate-derived linoleic acid could be modified in the presence of free radicals to form an oxo- or nitro-derivative, and thereby form a ligand for PPARγ (23). Thus, the possibility that functional PPARγ signaling is involved in the inhibitory capacity of these ligands was examined. When mice were pretreated with GW9662, a specific PPARγ antagonist (30), LTA, GML, and FSL1-Lin2 induced significantly more leukocyte recruitment than in the absence of PPARγ inhibition (Fig. 6A). As expected in the case of LTA and GML, the neutrophil emigration remained modest, however, in the presence of GW9662, the emigration induced by FSL1-Lin2 was comparable to that observed in response to the parent FSL-1 compound. Clearly, inhibiting the anti-inflammatory capacity of these ligands by inhibiting PPARγ activation allows the proinflammatory nature of these ligands to dominate in vivo. In line with these data, pretreatment with GW9662 or another PPARγ antagonist, T0070907, abrogated the inhibitory effect of both LTA and GML toward neutrophil recruitment induced by TNFα and MIP2 (Fig. 6 B–D). In a complementary experiment, the PPARγ agonist Rosiglitazone, when coadministered with MIP2, inhibited neutrophil recruitment to the same degree as GML (Fig. S5A), whereas the absence of TLR2 did not affect the capacity for PPARγ to become activated by Rosiglitazone (Fig. S5B). Finally, considering that hydroxy- or oxo-linoleic acid derivatives are the possible PPARγ ligands initiating this inhibitory effect and that the oxidation of linoleic acid in the inflamed in vivo environment is likely to occur in the presence of myeloperoxidase and H2O2 generated by a respiratory burst, we assayed the inhibitory capacity of GML in a myeloperoxidase knockout (MPO−/−) animal. Indeed, Zhang et al. (31) have shown that even in the presence of reactive oxygen species, the oxidation of lipids is reduced in the absence of myeloperoxidase. We clearly demonstrated that the inhibitory capacity of GML is eliminated in the MPO−/− animals (Fig. S5C).
Systemic administration of the proinflammatory TLR2 ligand Pam3CSK4 has recently been shown to increase leukocyte trafficking to the lymph nodes (32). In our model of acute inflammation, neutrophils are the predominate cell type recruited into the tissue (Fig. S5D). Therefore, we evaluated the effect of systemic administration of Pam3CSK4 on neutrophils and found increased numbers of circulating blood neutrophils and increased, albeit low, numbers of neutrophils in the lymph nodes (Fig. S5 E and F). On the other hand, local administration of Pam3CSK4 did not cause any changes in neutrophil numbers in either the blood or the lymph nodes (Fig. S5 E and F), but did result in significant neutrophil recruitment into the cremaster tissue (Fig. S1F). Most importantly, neither systemic nor local administration of GML altered neutrophil numbers in the blood or lymph nodes (Fig. S5 E and F). Therefore, the functional consequences of host recognition of inhibitory TLR2 ligands are distinct from those of proinflammatory TLR2 ligands.
Discussion
Historically, TLRs have been described as sentinel receptors that elicit proinflammatory responses when engaged by their cognate ligands (2, 3). However, we describe a phenomenon where specific acylated TLR2 ligands inhibit the recruitment of neutrophils into an inflamed tissue in vivo. This inhibitory effect requires both TLR2 and the fatty acid and nuclear hormone receptor PPARγ, and not the major TLR2 signaling adaptor, MyD88. Our data suggest that this phenomenon requires the functional involvement of PPARγ and that TLR2 may simply be required for internalization of the ligand. With regard to the structural requirements for a TLR2 ligand to have anti-inflammatory properties, we found that modifications of the acyl chains and the peptide sequence of a lipopeptide TLR2 ligand can yield a TLR2 ligand with purely anti-inflammatory properties. Moreover, we show that the natural ligand, LTA, possesses these inhibitory characteristics. The exact molecular structure and the modification of the acyl chains required to derive a bona fide PPARγ ligand remains to be determined.
We have provided no direct evidence that a MyD88-independent signaling pathway downstream of TLR2 is required for the anti-inflammatory response, and thus TLR2 may serve to mediate a specific means of endocytosis that facilitates the delivery of anti-inflammatory ligands to their intracellular target, PPARγ. We suggest that the targeting of the lipopeptide GML to GM1 alters its internalization in such a way as to mimic the internalization pathway of LTA. This internalization follows an alternative means of endocytosis from that observed with the “classical” proinflammatory TLR2 ligands and effectively limits their proinflammatory potential (16, 27). Distinct spatial requirements for productive signaling is not a unique concept, and is consistent with TLR4 signaling, where Mal/MyD88-driven responses proceed from the plasma membrane, versus Tram/Trif signaling, which originates from complexes forming at the endosome (33). This model would imply that the different classes of TLR2 ligands, pro- versus anti-inflammatory, use spatial and temporal separation of TLR2 receptor complexes to illicit distinct intracellular responses.
Although GML precisely mimics the inhibitory capacity of the LTA, linoleate may not be the inhibitory component within the LTA. Ultimately, the acyl chains of any naturally produced TLR2 ligand would, in part, reflect the fatty acid constituents of the bacterial membranes. Although bacteria typically synthesize only monounsaturated fatty acids, the fatty acid composition of bacteria is markedly affected by the growth conditions (34). Unsaturated fatty acids in the media would be incorporated into the bacterial membranes and could then form part of the bacterial lipoproteins (35, 36); thus, the microbial environment could profoundly influence the production of any anti-inflammatory TLR2 ligands. Nevertheless, it is clear that the inhibitory effect of LTA depends on PPARγ activation, similar to the synthetic inhibitory TLR2 ligand, GML.
These data define a model where a subclass of TLR2 ligands can be considered to have both pro- and anti-inflammatory properties through the functional use of two receptors, namely TLR2 and PPARγ. The preponderance for one biologic effect over the other is related to the specific structural composition of each TLR2 ligand. The data we present herein fundamentally change our conceptual understanding of how bacterial-derived TLR2 ligands can influence neutrophil responses and as such host–microbial interactions. We have provided a mechanistic understanding of how this can occur through the synthesis of a TLR2 ligand with purely anti-inflammatory properties. It is possible that there may be other naturally occurring inhibitory TLR2 ligands besides LTA, and that these ligands may profoundly influence host–pathogen interactions as well as normal interactions with the microbiome. Furthermore, the synthetic ligand described herein, GML, may prove to have therapeutic value in pathological settings where neutrophil recruitment is associated with disease.
Materials and Methods
Mice/Reagents.
C57BL/6 wild-type, TRIF-deficient, and MPO−/− (Jackson Laboratory, Bar Harbor, ME), MyD88−/−, TLR2−/− (kindly provided by Shizuo Akira, Osaka University, Japan), and LysMeGFP (by Thomas Graf, Albert Einstein College of Medicine, Bronx, NY) chimeric mice were made as described (37) and maintained the pathogen-free facility at the University of Calgary's Animal Resource Center. Animal protocols were approved by the University of Calgary Animal Care Committee and met the guidelines of the Canadian Council for Animal Care. Ultra-pure LTA (InvivoGen) was kindly provided by Sonja Von Aulock (Konstanz, Germany). All modified ligands as well as R-FSL1 and S-FSL1 were from EMC Microcollections. Pam2CSK4 and Pam3CSK4 were from Invivogen, TNFα and MIP2 were from R&D Systems, Rosiglitazone was from Cayman Chemical, and T0070907 was from Tocris.
Intravital Microscopy.
Intravital microscopy was performed on male mice as described in ref. 16. Leukocyte emigration was defined as the number of cells in the extravascular space within the field of view adjacent to the observed venule.
Bacterial and Cell Culture.
E. coli (WS2572 Weihenstephan Culture Collection) were cultured in vivo as described (38). Male mice were injected s.c. with 50 μL or 5 × 105 CFU of a 1:1 bacterial/cytodex bead (with or without LTA or GML) mixture between the scapula, centered on the spine. Bacterial growth was monitored using imaging with Xenogen IVIS-200 imaging system (Xenogen Imaging Technologies). Bone-marrow-derived macrophages were differentiated from 7- to 10-wk-old mice as described (16). Raw264.7 cells were grown in RPMI 1640 media (GIBCO) supplemented with 1 mM sodium pyruvate (GIBCO), 100 U/mL penicillin (GIBCO), 100 μg/mL streptomycin (GIBCO), 50 μM 2-mercaptoethanol (Sigma), and 10% (vol/vol) FBS (GIBCO).
SDS/PAGE and Western blotting.
All Western blots were performed as described in detail in ref. 16.
Statistical Analysis.
All results are expressed as mean ± SEM (GraphPad Prism 5). A t test was applied for analysis between two groups, a one-way analysis of variance with Bonferroni multiple comparisons adjustment was applied for multiple comparisons between groups, and a two-way analysis of variance with Bonferroni post test was applied for time-course experiments, (*P ≤ 0.05 **P ≤ 0.01 ***P ≤ 0.001). All results presented n ≥ 3.
Supplementary Material
Acknowledgments
We thank L. Zbytnuik and D. Brown for help in animal care and members of the S.M.R. and P.K. laboratories for valuable discussions. This work was supported by funding from the Alberta Cancer Foundation and the Canadian Institutes of Health Research. S.M.R holds a Canadian Research Chair and is an Alberta Heritage Foundation for Medical Research (AHFMR) Scientist. P.K. holds a Canadian Research Chair and the Snyder Chair in Critical Care Medicine and is an AHFMR Scientist. D.M.M. is an AHFMR Senior Scholar. H.A.P was supported by a postdoctoral fellowship from CAPES (Ministério da Educação, Brasil). E.M.L was supported by studentships from Natural Sciences and Engineering Research Council of Canada and AHFMR.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1100702108/-/DCSupplemental.
References
- 1.Brikos C, O'Neill LA. Signalling of toll-like receptors. Handbk Exp Pharmacol. 2008;183:21–50. doi: 10.1007/978-3-540-72167-3_2. [DOI] [PubMed] [Google Scholar]
- 2.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–1407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manicassamy S, Pulendran B. Modulation of adaptive immunity with Toll-like receptors. Semin Immunol. 2009;21:185–193. doi: 10.1016/j.smim.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozinsky A, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zähringer U, Lindner B, Inamura S, Heine H, Alexander C. TLR2 - promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology. 2008;213:205–224. doi: 10.1016/j.imbio.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Kang JY, et al. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity. 2009;31:873–884. doi: 10.1016/j.immuni.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Jin MS, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Schröder NW, et al. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J Biol Chem. 2003;278:15587–15594. doi: 10.1074/jbc.M212829200. [DOI] [PubMed] [Google Scholar]
- 9.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 10.Petri B, Phillipson M, Kubes P. The physiology of leukocyte recruitment: an in vivo perspective. J Immunol. 2008;180:6439–6446. doi: 10.4049/jimmunol.180.10.6439. [DOI] [PubMed] [Google Scholar]
- 11.Jaeschke H, Hasegawa T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006;26:912–919. doi: 10.1111/j.1478-3231.2006.01327.x. [DOI] [PubMed] [Google Scholar]
- 12.Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol. 2009;40:519–535. doi: 10.1165/rcmb.2008-0348TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown SJ, Mayer L. The immune response in inflammatory bowel disease. Am J Gastroenterol. 2007;102:2058–2069. doi: 10.1111/j.1572-0241.2007.01343.x. [DOI] [PubMed] [Google Scholar]
- 14.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 15.Yipp BG, et al. Profound differences in leukocyte-endothelial cell responses to lipopolysaccharide versus lipoteichoic acid. J Immunol. 2002;168:4650–4658. doi: 10.4049/jimmunol.168.9.4650. [DOI] [PubMed] [Google Scholar]
- 16.Long EM, Millen B, Kubes P, Robbins SM. Lipoteichoic acid induces unique inflammatory responses when compared to other toll-like receptor 2 ligands. PLoS ONE. 2009;4:e5601. doi: 10.1371/journal.pone.0005601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morath S, Geyer A, Spreitzer I, Hermann C, Hartung T. Structural decomposition and heterogeneity of commercial lipoteichoic Acid preparations. Infect Immun. 2002;70:938–944. doi: 10.1128/iai.70.2.938-944.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kerfoot SM, Kubes P. Local coordination verses systemic disregulation: Complexities in leukocyte recruitment revealed by local and systemic activation of TLR4 in vivo. J Leukoc Biol. 2005;77:862–867. doi: 10.1189/jlb.1004607. [DOI] [PubMed] [Google Scholar]
- 19.Akira S. Toll-like receptors: lessons from knockout mice. Biochem Soc Trans. 2000;28:551–556. doi: 10.1042/bst0280551. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 21.Morath S, Geyer A, Hartung T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J Exp Med. 2001;193:393–397. doi: 10.1084/jem.193.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deininger S, et al. Use of synthetic derivatives to determine the minimal active structure of cytokine-inducing lipoteichoic acid. Clin Vaccine Immunol. 2007;14:1629–1633. doi: 10.1128/CVI.00007-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villacorta L, Schopfer FJ, Zhang J, Freeman BA, Chen YE. PPARgamma and its ligands: therapeutic implications in cardiovascular disease. Clin Sci (Lond) 2009;116:205–218. doi: 10.1042/CS20080195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy RC, et al. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-gamma. Blood. 2008;112:4250–4258. doi: 10.1182/blood-2007-12-128967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 26.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nilsen NJ, et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol. 2008;84:280–291. doi: 10.1189/jlb.0907656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsubara T, Ishikawa D, Taki T, Okahata Y, Sato T. Selection of ganglioside GM1-binding peptides by using a phage library. FEBS Lett. 1999;456:253–256. doi: 10.1016/s0014-5793(99)00962-x. [DOI] [PubMed] [Google Scholar]
- 29.Tarragó-Trani MT, Storrie B. Alternate routes for drug delivery to the cell interior: pathways to the Golgi apparatus and endoplasmic reticulum. Adv Drug Deliv Rev. 2007;59:782–797. doi: 10.1016/j.addr.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leesnitzer LM, et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41:6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- 31.Zhang R, Shen Z, Nauseef WM, Hazen SL. Defects in leukocyte-mediated initiation of lipid peroxidation in plasma as studied in myeloperoxidase-deficient subjects: systematic identification of multiple endogenous diffusible substrates for myeloperoxidase in plasma. Blood. 2002;99:1802–1810. [PubMed] [Google Scholar]
- 32.McKimmie CS, et al. A TLR2 ligand suppresses inflammation by modulation of chemokine receptors and redirection of leukocyte migration. Blood. 2009;113:4224–4231. doi: 10.1182/blood-2008-08-174698. [DOI] [PubMed] [Google Scholar]
- 33.Kagan JC, et al. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'leary WM. The fatty acids of bacteria. Bacteriol Rev. 1962;26:421–447. doi: 10.1128/br.26.4.421-447.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe K, Ishikawa C, Inoue H, Cenhua D, Yazawa K, Kondo K. Incorporation of exogenous docosahexaenoic acid into various bacterial phospholipids. J Am Oil Chem Soc. 1994;71:325–330. [Google Scholar]
- 36.Tibor A, Decelle B, Letesson JJ. Outer membrane proteins Omp10, Omp16, and Omp19 of Brucella spp. are lipoproteins. Infect Immun. 1999;67:4960–4962. doi: 10.1128/iai.67.9.4960-4962.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carvalho-Tavares J, et al. A role for platelets and endothelial selectins in tumor necrosis factor-alpha-induced leukocyte recruitment in the brain microvasculature. Circ Res. 2000;87:1141–1148. doi: 10.1161/01.res.87.12.1141. [DOI] [PubMed] [Google Scholar]
- 38.Georgel P, et al. A toll-like receptor 2-responsive lipid effector pathway protects mammals against skin infections with gram-positive bacteria. Infect Immun. 2005;73:4512–4521. doi: 10.1128/IAI.73.8.4512-4521.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.