

Abstract
A disulfide-linked nitroxide side chain (R1) is the most widely used spin label for determining protein topology, mapping structural changes, and characterizing nanosecond backbone motions by site-directed spin labeling. Although the internal motion of R1 and the number of preferred rotamers are limited, translating interspin distance measurements and spatial orientation information into structural constraints is challenging. Here, we introduce a highly constrained nitroxide side chain designated RX as an alternative to R1 for these applications. RX is formed by a facile cross-linking reaction of a bifunctional methanethiosulfonate reagent with pairs of cysteine residues at i and i + 3 or i and i + 4 in an α-helix, at i and i + 2 in a β-strand, or with cysteine residues in adjacent strands in a β-sheet. Analysis of EPR spectra, a crystal structure of RX in T4 lysozyme, and pulsed electron-electron double resonance (ELDOR) spectroscopy on an immobilized protein containing RX all reveal a highly constrained internal motion of the side chain. Consistent with the constrained geometry, interspin distance distributions between pairs of RX side chains are narrower than those from analogous R1 pairs. As an important consequence of the constrained internal motion of RX, spectral diffusion detected with ELDOR reveals microsecond internal motions of the protein. Collectively, the data suggest that the RX side chain will be useful for distance mapping by EPR spectroscopy, determining spatial orientation of helical segments in oriented specimens, and measuring structural fluctuations on the microsecond time scale.
Keywords: pulsed EPR, protein dynamics
Site-directed spin labeling (SDSL) is a general method for characterizing protein topography, local and global structure, and dynamics by electron paramagnetic resonance (EPR) spectroscopy (1–5). SDSL can be applied to both soluble and membrane proteins of arbitrary molecular weight under physiological conditions. In traditional SDSL, a unique cysteine residue is introduced into a recombinant protein via site-directed mutagenesis, and subsequently reacted with a sulfhydryl-specific nitroxide reagent to generate a paramagnetic side chain. In addition, an SDSL strategy based on a genetically encoded unnatural amino acid was recently reported, a method that allows labeling in the presence of native thiols (6).
The most widely used spin label for SDSL studies is a disulfide-linked side chain designated R1, in part because its inherent flexibility allows for introduction at virtually any site within a protein—even buried ones—typically with no more energetic cost than a natural amino acid substitution at that site (7, 8). Indeed, R1 has proven useful for determining protein topology (2, 9), characterizing nanosecond backbone motions (4,10,11), mapping structural changes (3, 12–14), and predicting protein structure de novo (15). Although the amplitude of nanosecond internal motion and the number of preferred rotamers of R1 are limited (16), these features nevertheless make it problematic to translate interspin distances measured by EPR into structural constraints, to interpret orientation information (17), or to directly measure slow (i.e., micro- to millisecond) protein dynamics. For these applications, a spin label with more restricted internal motions and fewer rotamers than R1 (i.e., more rigid) would be a useful alternative.
One strategy to restrict internal motion and limit the number of allowed rotamers of R1 is to introduce a bulky substituent on the nitroxide ring (18). Indeed, the utility of such derivatives has been explored (19, 20), and structures in proteins have been determined [Protein Data Bank (PDB) ID codes 1ZUR, 1ZWN, and 2A4T]. An alternative approach is to generate a second covalent bond between the nitroxide ring and the protein (i.e., cross-link), and a series of bifunctional nitroxide reagents capable of cross-linking have been reported (21) and used for EPR studies (22–24). In this study, we used the bifunctional nitroxide reagent HO-1944 to selectively introduce a cross-linked side chain designated RX (Fig. 1A) into both α-helical (T4 Lysozyme, T4L) and β-sheet (rat intestinal fatty acid-binding protein, iFABP) proteins. EPR spectra of all RX mutants indicate that the internal motion within the side chain is highly restricted in both topologies. A crystal structure of T4L containing the RX side chain in an α-helix reveals a single, well-ordered, and energetically relaxed rotamer, indicating the RX may be advantageous for distance mapping. Indeed, interresidue distance measurements between pairs of RX side chains demonstrate that the nitroxide is more localized than R1.
Fig. 1.

The RX side chain, sites of introduction in T4 Lysozyme (T4L) and intestinal fatty acid-binding protein (iFABP), and the corresponding EPR spectra. (A) Reaction of a protein containing two cysteines with the homobifunctional, reagent (HO-1944) results in the cross-linked side chain designated “RX.” (B and C) Ribbon models of wild-type T4L [B, PDB ID code 1L63 (26)] and iFABP [C, PDB ID code 2IFB (27)] highlighting representative solvent-exposed sites used in this study with spheres at their α-carbons. (D and E) Normalized EPR spectra of representative RX mutants of T4L (D) and iFABP (E) attached to CNBr-activated Sepharose in buffer. Overlaid on the Sepharose-bound spectrum for T4L 5-9RX is nonlinear least-squares fit to a MOMD model (red-dashed trace). A vertical scaling factor (Left) was applied to the spectrum of 5-9RX for display purposes. The magnetic field scan width is 100 G. The effective hyperfine splitting (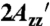 ) and the parallel (A||) and perpendicular (A⊥) components of the axially symmetric hyperfine tensor for the 5-9RX spectrum are shown.
) and the parallel (A||) and perpendicular (A⊥) components of the axially symmetric hyperfine tensor for the 5-9RX spectrum are shown.
Because of its highly restricted internal motion, RX monitors motion of the backbone to which it is attached without complications arising from the internal modes of the side chain. This feature enables an elegant pulsed ELDOR experiment (25) to measure microsecond fluctuations in protein structure based on spectral diffusion rates. Collectively, the results indicate that RX will be a useful alternative to R1 for de novo structure prediction and for detecting structural changes via distance mapping, and for characterizing protein dynamics in a time domain that is challenging for other methods.
Results
In contrast to traditional SDSL, in which a single cysteine is used to introduce a spin label, two cysteines are used to incorporate RX into a protein. Based on molecular modeling, the RX side chain can adopt an energetically relaxed conformation when the second cysteine is located at the i + 3 or i + 4 position of an α-helix, at the i + 2 position of a β-strand, or at the nearest neighboring site of an adjacent β-strand. Accordingly, RX was introduced, one at a time, into eight helices of T4L and both β-sheets of iFABP (three intra- and two interstrand). The locations of representative cysteine pairs at solvent-exposed sites in T4L and iFABP used for introduction of RX are shown in Fig. 1 B and C, respectively. The complete set of sites investigated is shown in Fig. S1. Each double cysteine mutant was labeled with a tenfold molar excess of HO-1944. For comparison, a single cysteine mutant of T4L (T4L 131C) was also labeled with HO-1944. RX mutants are designated as, for example, 5-9RX, indicating that the spin label is covalently linked to cysteines at positions 5 and 9. All RX mutants were found to be predominantly monomeric by nonreducing SDS-PAGE and contain less than 2% free thiols (Table S1). These results indicate that two intramolecular disulfide bonds are formed within a single monomeric protein for all of the spin-labeled mutants tested.
EPR spectra of all RX spin-labeled mutants studied here in 30% wt/wt sucrose reflect strong immobilization of the nitroxide (Fig. S1), as expected from previous studies (23). In 30% sucrose, the correlation time for proteins the size of T4L and iFABP is on the order of τ ≈ 30 ns (28). For a strongly immobilized RX, even this slow rotational diffusion influences the lineshape. To effectively restrict protein tumbling and directly observe the internal motions of RX, each labeled mutant was covalently attached to CNBr-activated Sepharose, a method previously shown to restrict protein tumbling on the X-band EPR time scale (29).
EPR spectra of representative proteins attached to Sepharose with RX in α-helices and β-sheets are given in Fig. 1 D and E, respectively; the spectra of additional T4L RX mutants on Sepharose along with spectra recorded in 30% wt/wt sucrose are given in Fig. S1. In each case, the spectrum is indicative of a highly ordered nitroxide motion, as evidenced by well-resolved parallel (A||) and perpendicular (A⊥) components of the (axially symmetric) hyperfine tensor and a large effective hyperfine splitting, 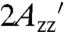 , which serves as a measure of nitroxide mobility, larger values corresponding to lower mobility (30). Individual values of
, which serves as a measure of nitroxide mobility, larger values corresponding to lower mobility (30). Individual values of 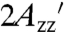 are given in Table S2.
are given in Table S2.
There is no evidence of spin-spin interactions in the EPR spectra that would indicate the presence of two bifunctional spin labels attached to a single protein except in the case of T4L 131-135RX, in which extreme spectral broadening is observed; the broadening is essentially eliminated by labeling with a 1∶1 rather than a 10∶1 ratio of HO-1944 to protein (Fig. S1). When only one of the two reactive functional groups of HO-1944 has reacted with the protein, as in the case of reaction with T4L 131C, the nitroxide has increased mobility (as evidenced by the smaller 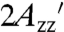 ) compared to the analogous cross-linked case [i.e., T4L 127-131RX (Fig. S1)].
) compared to the analogous cross-linked case [i.e., T4L 127-131RX (Fig. S1)].
For the RX mutants attached to Sepharose in frozen solution (i.e., at -50 °C), where both rotary diffusion of the protein and any internal modes of the side chain are frozen out, the average effective hyperfine splitting, 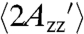 , is 74.9 ± 0.4 G, corresponding to a completely rigid nitroxide. For the Sepharose-attached proteins at room temperature,
, is 74.9 ± 0.4 G, corresponding to a completely rigid nitroxide. For the Sepharose-attached proteins at room temperature, 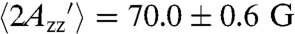 . The smaller value compared to the frozen solution apparently reflects the internal motion of the RX side chain, although contributions from protein fluctuations cannot be ruled out. For the same proteins in 30% sucrose solution,
. The smaller value compared to the frozen solution apparently reflects the internal motion of the RX side chain, although contributions from protein fluctuations cannot be ruled out. For the same proteins in 30% sucrose solution, 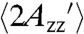 is 66.6 ± 1.1 G; the smaller
is 66.6 ± 1.1 G; the smaller 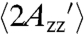 in solution is due to protein rotational diffusion.
in solution is due to protein rotational diffusion.
As an independent means to isolate the contribution from RX internal motion, 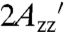 for T4L 5-9RX was plotted as a function of solution viscosity. Extrapolation to infinite viscosity according to the analysis of Timofeev gives the
for T4L 5-9RX was plotted as a function of solution viscosity. Extrapolation to infinite viscosity according to the analysis of Timofeev gives the 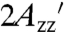 value corresponding to the pure internal motion of RX in the protein (31). The extrapolated value of
value corresponding to the pure internal motion of RX in the protein (31). The extrapolated value of 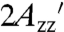 is 70.8 G (Fig. S2), close to that for T4L 5-9RX attached to Sepharose (70.7 G; Table S2), supporting the contentions that (i) the protein is completely immobilized on the support on the EPR timescale, and (ii) the reduction in
is 70.8 G (Fig. S2), close to that for T4L 5-9RX attached to Sepharose (70.7 G; Table S2), supporting the contentions that (i) the protein is completely immobilized on the support on the EPR timescale, and (ii) the reduction in 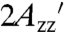 at ambient temperature relative to -50 °C is due to internal motions within RX.
at ambient temperature relative to -50 °C is due to internal motions within RX.
The amplitude and rate of RX internal motion can be assessed by spectral simulations using the macroscopic order with microscopic disorder (MOMD) model (32). The dashed red trace in Fig. 1D shows a best-fit to the spectrum of T4L 5-9RX attached to Sepharose for a simple anisotropic motion of the nitroxide in a cone; the order parameter and correlation time for the fit are S = 0.9 and τR = 2 ns. Thus, the internal motion is reasonably well approximated by a fast, highly restricted motion in a cone with angular amplitude of approximately 15°. Indeed, the spectrum can also be well-fit with a time-independent Hamiltonian model wherein the internal motion is sufficiently fast to effectively average the magnetic anisotropies over the spatial extent of the motion (SI Materials and Methods and Fig. S3).
Crystal Structure of T4L 115-119RX.
To determine the structural origin of the highly ordered motion and the rotamers of RX attached to an α-helix, the X-ray crystal structure of T4L 115-119RX at 100 K was solved to 1.8 Å and refined to an R factor of 18.7% (Table S3). The 2Fo–Fc electron density map reveals a single conformation of the side chain, with nearly all non-hydrogen atoms resolved (Fig. 2). The nitroxide magnetic z axis makes an angle with the helical axis of approximately 80°. Like those of other nitroxide side chains, the dihedral angles of the RX side chain are generally consistent with an energetically relaxed conformation (16, 33); the only strained dihedral appears to be 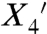 at 122° (see Fig. 1A for definition of the dihedrals and Fig. 2 legend for values). Interestingly, the orientations of both disulfide bonds of the RX side chain are the same as that of the most commonly observed {X1,X2} conformer of the R1 side chain: {-60°,-60°} (16). There is little difference in the backbone dihedral angles of helix G between the T4L 115-119RX and wild-type structures (Fig. S4), showing that the RX spin label does not distort the helical geometry; the greatest difference observed was for residue 115 (Δψ = 15°).
at 122° (see Fig. 1A for definition of the dihedrals and Fig. 2 legend for values). Interestingly, the orientations of both disulfide bonds of the RX side chain are the same as that of the most commonly observed {X1,X2} conformer of the R1 side chain: {-60°,-60°} (16). There is little difference in the backbone dihedral angles of helix G between the T4L 115-119RX and wild-type structures (Fig. S4), showing that the RX spin label does not distort the helical geometry; the greatest difference observed was for residue 115 (Δψ = 15°).
Fig. 2.

Crystal structure of T4L 115-119RX. Electron density for T4L 115-119RX calculated as an unweighted 2Fo - Fc composite omit map (blue mesh) and contoured at 1.0 σ. For clarity, only a stick model and electron density for RX side chain and selected nearby residues are shown with the protein backbone displayed as a ribbon model. Dihedral angles of the RX side chain from 115: {X1 = -94°,X2 = -61°,X3 = -81°,X4 = -160°,X5 = 105°} from 119 : {X1 = -70°,X2 = -56°,X3 = 106°,X4 = 122°,X5 = -90°} using the convention in Fig. 1A.
Comparison of RX to R1 for Distance Mapping by Pulsed EPR Spectroscopy.
The spectroscopic methods double electron–electron resonance (DEER) spectroscopy (34–36) [also known as pulsed electron double resonance (PELDOR)] and double quantum coherence (DQC) (37) have revolutionized interspin distance measurement by extending the distance range of detectable dipole–dipole interactions to approximately 70 Å, making them suitable strategies for structure mapping in large proteins and complexes. An important feature of DEER and DQC is that they provide not only most probable distances, but also distance distributions. The primary data obtained in DEER spectroscopy are background-corrected dipolar evolution functions, from which the distance distributions are derived (35, 36). For spin-labeled proteins using R1, the distance probability distribution is the result of nitroxide delocalization, partly arising from multiple rotamers of R1. On the other hand, the crystal structure of 115–119RX reveals a single rotamer. If this proves general, then the RX label will be an important alternative to R1 for distance mapping by EPR.
To explore the utility of the RX side chain for distance mapping, mutants of T4L and iFABP containing pairs of RX labels were studied with DEER. For comparison, analogous mutants containing a pair of R1 spin labels at approximately the same locations were studied. Fig. 3 shows the dipolar evolution function (Left) and the interspin distance distribution (Right) for these mutants. The most probable interspin distance for the T4L mutant 109-113RX/127-131RX at 80 K is 29.4 Å, which is in excellent agreement with the 31 Å predicted from modeling the crystallographically determined rotamer onto the corresponding sites in the wild-type structure. Remarkably, both RX mutants exhibit narrower distance distributions than the analogous R1 mutants, showing that the nitroxide is more localized within RX than R1.
Fig. 3.

DEER data of T4L mutants bearing either two RX side chains (black traces) or two R1 side chains (red traces). Background-subtracted dipolar evolutions of the indicated mutants (Left) and their corresponding area-normalized distance probability distributions from Tikhonov regularization (Right) for (A) T4L 109R1/131R1 and T4L 109-113RX/127-131RX and (B) iFABP 48R1/116R1 and iFABP 48-50RX/114-116RX. The width of the interspin distance distribution (between 16 and 84% probability) is 6.5 Å for T4L 109R1/131R1, 2.8 Å for T4L 109-113RX/127-131RX, 4.7 Å for iFABP 48R1/116R1, and 2.0 Å for iFABP 48-50RX/114-116RX.
Direct Measurement of Microsecond Dynamics with RX and ELDOR.
Functionally important protein conformational fluctuations occur on the micro- to millisecond time scale (38–40), but the number of techniques capable of detecting and characterizing microsecond protein motions is limited (41, 42). Robinson and coworkers (25) described an elegant EPR double resonance method to measure microsecond rotational diffusion of nitroxides in media of high viscosity. Here, we show that RX is sufficiently immobilized on an α-helix to employ this method for monitoring microsecond fluctuations in protein structure and measuring protein rotational diffusion.
The principle is discussed qualitatively with reference to Fig. 4A, which shows the absorption lineshape for a nitroxide in the slow motional limit (i.e., with correlation time, τR, > ∼ 100 ns); the reader is referred to Haas et al. (25) for a detailed quantitative analysis. In the slow motional limit, the lineshape is insensitive to nitroxide motion and the magnetic field axis also corresponds to the orientation of the nitroxide 2p orbital with respect to the laboratory frame. In the experiments described here, a short saturating pulse of microwave radiation (the “pump” pulse) is applied near 0° in the low-field 14N hyperfine manifold (MI = 1), which corresponds to nitroxides with their 2p orbitals aligned with the external field. Complete saturation results in zero signal amplitude at the pump position. At the end of the pulse, saturation effectively “diffuses” throughout the manifold via rotational motion of the nitroxide with correlation time τR, and diffuses to other hyperfine manifolds via nuclear relaxation with characteristic time T1n. In addition to these spectral diffusion processes, saturation is transferred to the surroundings via spin lattice relaxation with time constant T1e. The arrival and dissipation of saturation can be monitored as the time dependence of the signal amplitude with a weak continuous-wave (CW) microwave source of frequency corresponding to resonance at any desired position in the spectrum (the “observe” source). The above description assumes a pump pulse that is short compared to T1e, T1n, and τR. If the pump and observe frequencies are the same, the experiment is called saturation recovery (SR); if these frequencies differ, the experiment is electron–electron double resonance (ELDOR). Robinson and coworkers have shown that all three time constants can be determined by global fitting of both SR and ELDOR data (25).
Fig. 4.

ELDOR studies of T4L mutants. (A) EPR absorption lineshape for an immobilized nitroxide showing the fields (and nitroxide 2p-orbital orientations, θ) at which the saturating pulse was applied (pump) and at which the arrival and recovery of saturation were observed (observe 1 or observe 2); (B) Intra- (red) and intermanifold (blue) ELDOR curves and global fits to the spectra using both SR and ELDOR data (gray; SI Materials and Methods) for 5–9RX (Upper) and 44-48RX/T26E (Lower) attached to Sepharose in buffer. (C) Intra- (red) and intermanifold (blue) ELDOR curves and global fits to the spectra (gray) of the T4L 5-9RX mutant in buffer (Upper) and in 80% glycerol (Lower) at 298 K. The time constants yielded by the fits in B and C are as follows (T1e, T1n, τR, respectively, in μs): 5–9RX on Sepharose; 7.79 ± 0.02, 0.91 ± 0.02, not applicable (N/A); 44-48RX/T26E on Sepharose; 7.90 ± 0.04, 0.56 ± 0.09, 6.2 ± 0.3; 5–9RX in buffer; 7.12 ± 0.01, N/A, N/A; 5–9RX in 80% glycerol; 11.1 ± 0.1, 0.22 ± 0.07, 1.2 ± 0.6. Each curve spans 40 μs.
If the pump and observe frequencies in an ELDOR experiment correspond to resonance at different angles (positions) within the same hyperfine manifold, say at 0 and 90°, respectively (pump and observe 1 in Fig. 4A), the only mechanism by which saturation can arrive at the observe frequency is by rotational diffusion of the nitroxide, so this is a powerful means of directly observing spatial reorientation. Spectral diffusion and spin lattice relaxation occur simultaneously, so the longest value of τR that can be measured is limited to approximately 50 μs by T1e, which is typically 8–10 μs for an immobilized nitroxide (23).
Fig. 4B Upper shows ELDOR data for Sepharose-attached T4L 5-9RX that illustrate the main points to be made. The red trace shows the signal detected by an observer at 90° (observe 1) following a pump pulse at 0° within the same hyperfine manifold; the lack of change in signal amplitude means that saturation has not spread from the pump to the observe position. Similar results were obtained for the Sepharose-attached 68-72RX and 109-113RX mutants (Fig. S5B). These results permit the conclusions that: (i) the protein is immobilized with respect to the solid support, and (ii) that RX is immobilized with respect to the protein, both with reference to an approximately 50-μs time scale and rotations on the order of 90°. If the pump and observe are both at 0°, but are in different nuclear manifolds (observe 2), arrival of saturation by nuclear relaxation (the rapid initial decrease in signal) and subsequent recovery by T1e are clearly seen (Fig. 4B Upper, blue trace).
Analogous experiments were carried out for T4L 5-9RX in buffer where the protein undergoes rotary diffusion. In this case, both τR and T1n are short, estimated to be approximately 12 ns (28) and 30 ns (25), respectively. With the 100-ns pump pulse used in all experiments, spectral diffusion due to nuclear relaxation and rotational motion are nearly complete within the pulse time and are suppressed in the recovery (43). Thus, T1e relaxation is observed as a single exponential recovery, independent of the observer position (Fig. 4C, Upper). When the rotational diffusion is slowed in approximately 80% glycerol, arrival of saturation and subsequent relaxation to the lattice is clearly resolved at both observe positions (Fig. 4C Lower). Values for the relevant time constants from global fits to SR and ELDOR data according to Haas et al. (25) are given in the legend to Fig. 4. For the 80% glycerol case, τR = 1.2 μs, close to the value of 0.7 μs estimated from the Stokes–Einstein equation.
Unlike the case for T4L 5-9RX, ELDOR data for Sepharose-attached T4L 44-48RX reveal intramanifold spectral diffusion from the pump at 0° to the observe position at 90° (Fig. 4B Lower, red trace). Global fits of SR and ELDOR data give τR ∼ 6 μs for the motion responsible for spectral diffusion. The mutant employed for this experiment was T4L 44-48RX/T26E. Upon reaction with substrate, the T26E mutant becomes covalently bound to a substrate adduct (44) that predominantly favors the closed state (45), thereby eliminating the hinge bending mode of T4L as a contributor to the observed motion; an identical result is obtained for 44-48RX in the WT* background (Fig. S5C). Assuming that the structure and dynamics of RX are similar to those in T4L 5-9RX, as suggested by the CW spectra, the origin of this motion is not internal motion of RX itself. A possible origin of the motion is discussed below.
Discussion
In this study, we investigated the incorporation, structure, internal motion, and utility of a cross-linked nitroxide side chain (RX, Fig. 1A) for distance mapping and for measuring microsecond protein motions by pulsed EPR spectroscopy. In contrast to previously developed, heterobifunctional spin labels that are photo-cross-linked to the protein—and therefore only useful for measuring protein rotational motions (46, 47)—the homobifunctional reagent used in this study can be used to site-specifically introduce a cross-linked spin label within a secondary structural element.
Three lines of experimental evidence indicate that the RX side chain is readily formed in all of the double cysteine mutants studied here, whether in an α-helix or β-structure. First, an assay for free thiol indicates that < 2% is present after spin labeling. This observation cannot be explained by two labels attached to a single protein, because there is no evidence in the EPR spectra of broadening due to spin–spin interactions that would occur in the doubly labeled protein; the single exception is discussed below. Second, the EPR spectra of the product of HO-1944 reaction with single and double cysteine mutants are distinct, the latter being more immobilized, as expected. Finally, the X-ray crystal structure of an RX mutant (Fig. 2) is entirely consistent with the chemical structure of RX shown in Fig. 1A.
Formation of the RX side chain occurs stepwise; presumably, the rate of the second disulfide bond formation is significantly faster than the first at the concentration of reagent employed because, as mentioned, there is no evidence of doubly labeled proteins in the spectra of the mutants studied except for the single case of T4L 131-135RX, where spin–spin interactions are clearly evident. It is likely that strain or a change in local structure in this mutant must accompany the formation of the second disulfide bond, slowing the reaction. In crystal structures of T4L (e.g., PDB ID code 1L63), residue 135 is not formally located in helix H, but rather in the loop between helices H and I. Even though the crystallographically determined RX conformer can be modeled onto sites 131 and 135, it is likely that formation of the cross-link in 131-135RX leads to strain in forming a proper helix extending to 135. This result suggests the interesting possibility that the cross-linking rate to form RX, readily assayed via changes in lineshape, might be used to identify dynamically or statically “frayed” helical or β-structures in proteins.
All data presented here suggest spatially restricted internal motion of RX. For example, the spectrum of T4L 5-9RX attached to Sepharose is well-fit by a simple anisotropic motion with S = 0.9, τR ≈ 2 ns; the amplitude of angular motion corresponding to this order is θ ≈ 15°, consistent with the restricted amplitude of motion (< 90°) shown by ELDOR (Fig. 4). In addition, the value of 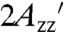 in the absence of protein rotation, obtained by extrapolation of the viscosity dependence, is essentially identical to that for the Sepharose-bound protein, making it unlikely that the restricted motion of RX is due to interactions with the solid support. The very similar EPR spectra for all RX mutants studied indicate that these conclusions are likely to be general.
in the absence of protein rotation, obtained by extrapolation of the viscosity dependence, is essentially identical to that for the Sepharose-bound protein, making it unlikely that the restricted motion of RX is due to interactions with the solid support. The very similar EPR spectra for all RX mutants studied indicate that these conclusions are likely to be general.
The limited internal motion makes RX an ideal label for investigating protein dynamic modes, for determining interresidue distances, and for determining the orientation of structural elements to which it is attached. For these purposes, a number of laboratories are exploring the use of an amino acid with a 6-membered nitroxide ring called TOAC (2,2,6,6-tetramethyl-piperidine-1-oxy-4-amino-4-carboxylic acid), which can be incorporated into peptides during synthesis and is directly fused to the peptide backbone (48–50). Unlike RX, however, TOAC cannot be easily incorporated into selected sites of large proteins. Both of these highly ordered labels have relatively low sensitivity to small differences in nanosecond backbone dynamics in stable structures compared to R1 (SI Materials and Methods and Fig. S6). Thus, R1 or its 4-methyl analog may be the side chains of choice for monitoring relative backbone dynamics on the nanosecond time scale in folded proteins (10, 18).
On the other hand, RX promises to be an elegant sensor for slow (microsecond) protein fluctuations by monitoring spectral diffusion via ELDOR. For this application, protein rotational diffusion of correlation times shorter than the characteristic time of the structural fluctuation must be eliminated. In the present experiments on small soluble proteins covalent attachment to a Sepharose support was used, and this may be a generally applicable strategy; for larger soluble and membrane-bound proteins this may not be necessary. The first application of the SR–ELDOR method is the observation of slow (approximately 6 μs) motions involving helix B of T4L as detected by 44-48RX. Because RX residues introduced into other helices detect no such motion (e.g., the 5-9RX mutant on helix A), overall motion of the protein is eliminated as the responsible mode. As mentioned above, the covalent enzyme-substrate adduct form of this RX mutant—in which hinge bending motions that open and close the enzyme are restricted—gives a similar SR–ELDOR result as 44-48RX without substrate; thus, the 6-μs mode likely represents motion of helix B relative to the rest of the protein. Indeed, the crystallographic B factors in this helix are larger than average (51), consistent with the relatively large solvent-exposed area of this helix.
An alternative strategy for detecting fluctuations on the microsecond time scale makes use of the R1 side chain and an SR-exchange method for which rotational diffusion of the protein is not an issue (23). However, this method requires that the fluctuations move the spin label side chain between two distinct environments with different T1es. Together, the SR exchange and SR–ELDOR spectral diffusion methods offer powerful new strategies for investigation of conformational fluctuations on a functionally important time scale.
The crystal structure of RX in a helix reveals a single rotamer with a well-localized nitroxide (Fig. 2). If this proves to be general, RX will be an important alternative to R1 for de novo structure prediction by EPR. Interresidue distance measurements between pairs of RX or R1 residues in helices confirm that the nitroxide is more localized in the former (Fig. 3A), and yield a most probable distance that agrees with a model in which the crystallographically determined rotamer of RX is attached to the corresponding sites in the wild-type structure. A similar comparison of R1 and RX in the β-sheets of iFABP also shows that the nitroxide is more localized in the latter (Fig. 3B). When RX is attached to unstable β-strands or α-helices that have large fluctuations in backbone dihedral angles, or are in equilibrium between conformations, it is expected that the RX side chain itself will modify the local structure and dynamics and hence influence the distance distributions; whether or not this is an issue may be revealed in the reaction kinetics of HO-1944 (see above). In any event, distance distributions between RX pairs wider than those found here likely result from structural heterogeneity in the protein.
Taking advantage of the high order of RX at a helical site, it was recently found that the orientation of the domain within which a labeled helix resides could be determined in a macroscopically oriented system by conventional EPR spectroscopy (52). It should be also possible to characterize the relative orientation of two RX-labeled helices with respect to one another by high-frequency pulsed EPR methods (53, 54). To design such experiments and interpret the data, the angle subtended by the magnetic tensor z axis of the nitroxide and the helical axis is required, and this is shown by the crystal structure to be approximately 80°.
Materials and Methods
Construction, Purification, and Spin Labeling of Cysteine Mutants.
Mutants were generated by QuikChange site-directed mutagenesis of the pET11a-T4L (6) or the pET11d-riFABP (rat iFABP) genetic construct (a generous gift from Alan K. Kleinfeld, Torrey Pines Institute for Molecular Studies, San Diego, CA) with primers designed according to ref. 54 and reactions carried out according to manufacturer’s suggested protocol; mutations were verified by DNA sequencing. All T4L cysteine mutants contain the pseudo-wild-type mutations C54T and C97A (56). Cysteine mutants of T4L and riFABP were expressed, purified, and then exchanged into a buffer suitable for spin labeling as previously described (6, 29). For spin labeling, a tenfold molar excess of HO-1944 (21) was immediately added directly to the desalted mutant, and the reaction was allowed to proceed at 4 °C overnight (HO-1944 is now available from Toronto Research Chemicals). Excess HO-1944 was removed by desalting using a HiPrep 26/10 (GE Healthcare). RX mutants were covalently attached to CNBr-activated Sepharose (Sigma) as previously described for R1 mutants (29). See additional information in SI Materials and Methods.
EPR Spectroscopy.
CW and DEER experiments were performed as previously described (6). SR experiments were performed as described earlier (23). For ELDOR experiments, the 1,500-mW, 100-ns pump pulse was set to the rising edge of the low-field resonance (3 G downfield of the θ = 0° orientation). The 100-μW CW detection was set to a frequency corresponding to resonance at a field corresponding to the θ = 90° orientation in the same hyperfine manifold, or to the θ = 0° position in the MI = 0 manifold (Fig. 4A). Global analysis of SR and ELDOR data was carried out as described by Robinson and coworkers (25). Additional details are found in SI Materials and Methods. Spectral simulations were carried out with the MOMD model (32) using software available at www.acert.cornell.edu.
X-Ray Crystallography.
Diffraction quality crystals of T4L 115-119RX were grown by the hanging drop method using phosphate as the precipitant (16). For the cryogenic diffraction study, a single crystal cryoprotected in mineral oil was exposed to X-rays generated by an FRD generator, and the reflections collected on an R-AXIS IV++ detector (Rigaku). Data processing were performed as previously described (16). A molecular replacement solution was found using 1C6T (57) as a starting model, and refined using COOT (58) and PHENIX (59).
Supplementary Material
Acknowledgments.
We thank Christian Altenbach, Carlos López, and Ned Van Eps for careful reading of the manuscript, Christian Altenbach for enlightening discussions on the pulsed ELDOR technique, and Carlos López for assistance with the T4L 128C/131C mutant. This work was supported by National Institutes of Health Grants EY05216, T32 EY07026, and 5P41EB001980; the Jules Stein Professorship endowment; and the Hungarian National Research Fund OTKA K81123.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and structure factors for T4L 115-119RX have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3L2X).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1111420108/-/DCSupplemental.
References
- 1.Todd A, Crozel V, Levinthal F, Levinthal C, Hubbell WL. Site-directed mutagenesis of colicin E1 provides specific attachment sites for spin labels whose spectra are sensitive to local conformation. Proteins. 1989;6:294–305. doi: 10.1002/prot.340060312. [DOI] [PubMed] [Google Scholar]
- 2.Hubbell WL, Gross A, Langen R, Lietzow MA. Recent advances in site-directed spin labeling of proteins. Curr Opin Struct Biol. 1998;8:649–656. doi: 10.1016/s0959-440x(98)80158-9. [DOI] [PubMed] [Google Scholar]
- 3.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat Struct Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 4.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 5.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Fleissner MR, et al. Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc Natl Acad Sci USA. 2009;106:21637–21642. doi: 10.1073/pnas.0912009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mchaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 8.Lietzow MA, Hubbell WL. Motion of spin label side chains in cellular retinol-binding protein: Correlation with structure and nearest-neighbor interactions in an antiparallel beta-sheet. Biochemistry. 2004;43:3137–3151. doi: 10.1021/bi0360962. [DOI] [PubMed] [Google Scholar]
- 9.Hubbell W, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: A perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- 10.Columbus L, Hubbell WL. Mapping backbone dynamics in solution with site-directed spin labeling: GCN4-58 bZip free and bound to DNA. Biochemistry. 2004;43:7273–7287. doi: 10.1021/bi0497906. [DOI] [PubMed] [Google Scholar]
- 11.Van Eps N, Oldham WM, Hamm HE, Hubbell WL. Structural and dynamical changes in an α-subunit of a heterotrimeric G protein along the activation pathway. Proc Natl Acad Sci USA. 2006;103:16194–16199. doi: 10.1073/pnas.0607972103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong J, Yang G, Mchaourab HS. Structural basis of energy transduction in the transport cycle of MsbA. Science. 2005;308:1023–1028. doi: 10.1126/science.1106592. [DOI] [PubMed] [Google Scholar]
- 13.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci USA. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Eps N, et al. Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc Natl Acad Sci USA. 2011;108:9420–9424. doi: 10.1073/pnas.1105810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander N, Bortolus M, Al-Mestarihi A, Mchaourab H, Meiler J. De novo high-resolution protein structure determination from sparse spin-labeling EPR data. Structure. 2008;16:181–195. doi: 10.1016/j.str.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleissner MR, Cascio D, Hubbell WL. Structural origin of weakly ordered nitroxide motion in spin-labeled proteins. Protein Sci. 2009b;18:893–908. doi: 10.1002/pro.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Risse T, Hubbell WL, Isas JM, Haigler HT. Structure and dynamics of annexin 12 bound to a planar lipid bilayer. Phys Rev Lett. 2003;91:188101. doi: 10.1103/PhysRevLett.91.188101. [DOI] [PubMed] [Google Scholar]
- 18.Columbus L, Kálai T, Jeko J, Hideg K, Hubbell WL. Molecular motion of spin labeled side chains in alpha-helices: Analysis by variation of side chain structure. Biochemistry. 2001;40:3828–3846. doi: 10.1021/bi002645h. [DOI] [PubMed] [Google Scholar]
- 19.Fleissner MR. Los Angeles: Univ of California; 2007. X-ray structures of nitroxide side chains in proteins: A basis for interpreting distance measurements and dynamic studies by electron paramagnetic resonance. PhD thesis. [Google Scholar]
- 20.Fawzi N, et al. A rigid disulfide-linked nitroxide side chain simplifies the quantitative analysis of PRE data. J Biomol NMR. 2011 doi: 10.1007/s10858-011-9545-x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kálai T, Balog M, Jeko J, Hideg K. Synthesis and reactions of a symmetric paramagnetic pyrrolidine diene. Synthesis Stutt. 1999:973–980. [Google Scholar]
- 22.Thompson AR, Naber N, Wilson C, Cooke R, Thomas DD. Structural dynamics of the actomyosin complex probed by a bifunctional spin label that cross-links SH1 and SH2. Biophys J. 2008;95:5238–5246. doi: 10.1529/biophysj.108.138982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bridges MD, Hideg K, Hubbell WL. Resolving conformational and rotameric exchange in spin-labeled proteins using saturation recovery EPR. Appl Magn Reson. 2010;37:363–390. doi: 10.1007/s00723-009-0079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rayes RF, Kálai T, Hideg K, Geeves MA, Fajer PG. Dynamics of tropomyosin in muscle fibers as monitored by saturation transfer EPR of bi-functional probe. PLoS One. 2011;6:e21277. doi: 10.1371/journal.pone.0021277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haas DA, Mailer C, Sugano T, Robinson BA. New developments in pulsed electron paramagnetic resonance: Direct measurement of rotational correlation times from decay curves. Bull Magn Reson. 1992;14:35–41. [Google Scholar]
- 26.Nicholson H, Anderson DE, Dao Pin S, Matthews BW. Analysis of the interaction between charged side chains and the alpha-helix dipole using designed thermostable mutants of phage T4 lysozyme. Biochemistry. 1991;30:9816–9828. doi: 10.1021/bi00105a002. [DOI] [PubMed] [Google Scholar]
- 27.Sacchettini JC, Gordon JI, Banaszak LJ. Crystal structure of rat intestinal fatty-acid-binding protein. Refinement and analysis of the Escherichia coli-derived protein with bound palmitate. J Mol Biol. 1989;208:327–339. doi: 10.1016/0022-2836(89)90392-6. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, et al. Multifrequency electron spin resonance study of the dynamics of spin labeled T4 lysozyme. J Phys Chem B. 2010;114:5503–5521. doi: 10.1021/jp910606h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.López CJ, Fleissner MR, Guo Z, Kusnetzow AK, Hubbell WL. Osmolyte perturbation reveals conformational equilibria in spin-labeled proteins. Protein Sci. 2009;18:1637–1652. doi: 10.1002/pro.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freed JH. Theory of slow tumbling ESR spectra for nitroxides. In: Berliner LJ, editor. Spin Labeling: Theory and Applications. New York: Academic; 1976. pp. 53–132. [Google Scholar]
- 31.Timofeev VP, Tsetlin VI. Analysis of mobility of protein side-chains by spin label technique. Biophys Struct Mech. 1983;10:93–108. [Google Scholar]
- 32.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg–Marquardt algorithm. J Magn Reson A. 1996;120:155–189. [Google Scholar]
- 33.Warshaviak DT, Serbulea L, Houk KN, Hubbell WL. Conformational analysis of a nitroxide side chain in an alpha-helix with density functional theory. J Phys Chem B. 2011;115:397–405. doi: 10.1021/jp108871m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larsen RG, Singel DJ. Double electron-electron resonance spin-echo modulation—Spectroscopic measurement of electron-spin pair separations in orientationally disordered solids. J Chem Phys. 1993;98:5134–5146. [Google Scholar]
- 35.Jeschke G, Pannier M, Spiess HW. Double electron-electron resonance. In: Berliner LJ, Eaton SS, Eaton GR, editors. Biological Magnetic Resonance 19: Distance Measurements in Biological Systems by EPR. New York: Kluwer Academic/Plenum; 2000. pp. 493–512. [Google Scholar]
- 36.Schiemann O, Prisner TF. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q Rev Biophys. 2007;40:1–53. doi: 10.1017/S003358350700460X. [DOI] [PubMed] [Google Scholar]
- 37.Borbat PP, and Freed JH. Double-quantum ESR and distance measurements. In: Berliner LJ, Eaton SS, Eaton GR, editors. Biological Magnetic Resonance 19: Distance Measurements in Biological Systems by EPR. New York: Kluwer Academic/Plenum; 2000. pp. 383–459. [Google Scholar]
- 38.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 39.Palmer AG, 3rd, Massi F. Characterization of the dynamics of biomacromolecules using rotating-frame spin relaxation NMR spectroscopy. Chem Rev. 2006;106:1700–1719. doi: 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- 40.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 41.Lange OF, et al. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–5. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 42.Hansen DF, Feng H, Zhou Z, Bai Y, Kay LE. Selective characterization of microsecond motions in proteins by NMR relaxation. J Am Chem Soc. 2009;131:16257–16265. doi: 10.1021/ja906842s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Percival PW, Hyde JS. Pulsed EPR spectrometer 2. Rev Sci Instrum. 1975;46:1522–1529. [Google Scholar]
- 44.Kuroki R, Weaver LH, Matthews BW. Structural basis of the conversion of T4 lysozyme into a transglycosidase by reengineering the active site. Proc Natl Acad Sci USA. 1999;96:8949–8954. doi: 10.1073/pnas.96.16.8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mchaourab HS, Oh KJ, Fang CJ, Hubbell WL. Conformation of T4 lysozyme in solution. Hinge-bending motion and the substrate-induced conformational transition studied by site-directed spin labeling. Biochemistry. 1997;36:307–316. doi: 10.1021/bi962114m. [DOI] [PubMed] [Google Scholar]
- 46.Wilcox MD, Parce JW, Thomas MJ, Lyles DS. A new bifunctional spin-label suitable for saturation-transfer EPR studies of protein rotational motion. Biochemistry. 1990;29:5734–5743. doi: 10.1021/bi00476a014. [DOI] [PubMed] [Google Scholar]
- 47.Lösel RM, Philipp R, Kálai T, Hideg K, Trommer WE. Synthesis and application of novel bifunctional spin labels. Bioconjugate Chem. 1999;10:578–582. doi: 10.1021/bc980138v. [DOI] [PubMed] [Google Scholar]
- 48.Nesmelov YE, Karim CB, Song L, Fajer PG, Thomas DD. Rotational dynamics of phospholamban determined by multifrequency electron paramagnetic resonance. Biophys J. 2007;93:2805–2812. doi: 10.1529/biophysj.107.108910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McNulty JC, et al. Electron spin resonance of TOAC labeled peptides: Folding transitions and high frequency spectroscopy. Biopolymers. 2000;55:479–485. doi: 10.1002/1097-0282(2000)55:6<479::AID-BIP1023>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 50.Van Eps N, et al. Electron paramagnetic resonance studies of functionally active, nitroxide spin-labeled peptide analogues of the C-terminus of a G-protein alpha subunit. Biochemistry. 2010;49:6877–6886. doi: 10.1021/bi100846c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo Z, Cascio D, Hideg K, Hubbell WL. Structural determinants of nitroxide motion in spin-labeled proteins: Solvent-exposed sites in helix B of T4 lysozyme. Protein Sci. 2008;17:228–239. doi: 10.1110/ps.073174008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson AR. Minneapolis: Univ of Minnesota; 2009. The structural dynamics of force generation in muscle, probed by electron paramagnetic resonance of bifunctionally labeled myosin. Ph.D. Thesis. [Google Scholar]
- 53.Jeschke G, Polyhach Y. Distance measurements on spin-labeled biomacromolecules by pulsed electron paramagnetic resonance. Phys Chem Chem Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- 54.Schiemann O, Cekan P, Margraf D, Prisner TF, Sigurdsson ST. Relative orientation of rigid nitroxides by PELDOR: Beyond distance measurements in nucleic acids. Angew Chem. 2009;48:3292–3295. doi: 10.1002/anie.200805152. [DOI] [PubMed] [Google Scholar]
- 55.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsumura M, Matthews BW. Control of enzyme activity by an engineered disulfide bond. Science. 1989;243:792–794. doi: 10.1126/science.2916125. [DOI] [PubMed] [Google Scholar]
- 57.Quillin ML, Breyer WA, Griswold IJ, Matthews BW. Size versus polarizability in protein-ligand interactions: Binding of noble gases within engineered cavities in phage T4 lysozyme. J Mol Biol. 2000;302:955–977. doi: 10.1006/jmbi.2000.4063. [DOI] [PubMed] [Google Scholar]
- 58.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 59.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.