

Abstract
There is a tremendous source of entropic energy available from the salinity difference between river water and seawater, but this energy has yet to be efficiently captured and stored. Here we demonstrate that H2 can be produced in a single process by capturing the salinity driven energy along with organic matter degradation using exoelectrogenic bacteria. Only five pairs of seawater and river water cells were sandwiched between an anode, containing exoelectrogenic bacteria, and a cathode, forming a microbial reverse-electrodialysis electrolysis cell. Exoelectrogens added an electrical potential from acetate oxidation and reduced the anode overpotential, while the reverse electrodialysis stack contributed 0.5–0.6 V at a salinity ratio (seawater:river water) of 50. The H2 production rate increased from 0.8 to 1.6 m3-H2/m3-anolyte/day for seawater and river water flow rates ranging from 0.1 to 0.8 mL/ min. H2 recovery, the ratio of electrons used for H2 evolution to electrons released by substrate oxidation, ranged from 72% to 86%. Energy efficiencies, calculated from changes in salinities and the loss of organic matter, were 58% to 64%. By using a relatively small reverse electrodialysis stack (11 membranes), only ∼1% of the produced energy was needed for pumping water. Although Pt was used on the cathode in these tests, additional tests with a nonprecious metal catalyst (MoS2) demonstrated H2 production at a rate of 0.8 m3/m3/d and an energy efficiency of 51%. These results show that pure H2 gas can efficiently be produced from virtually limitless supplies of seawater and river water, and biodegradable organic matter.
Keywords: electrohydrogenesis, microbial electrolysis cell, microbial fuel cell, renewable energy, sustainable energy
Exoelectrogenic bacteria oxidize organic matter and can transfer electrons to electrically conductive materials such as graphite or metal, making it possible to convert waste organic matter into useful energy. In microbial fuel cells (MFCs), exoelectrogens on the anode, coupled with oxygen reduction at the cathode, can generate a potential as large as ∼0.8 V (open circuit; pH 7; 0.2 atm O2), although less voltage (∼0.23 to 0.5 V) is generated in practice (1). Exoelectrogens can also be used to drive electrochemical H2 production in a microbial electrolysis cell (MEC) (2, 3). However, the potential generated by substrate oxidation (-0.30 V vs. Standard Hydrogen Electrode; 1 g/L acetate; pH 7) is not sufficient to drive H2 evolution (-0.41 V vs. Standard Hydrogen Electrode at pH 7) (1). Thus, additional energy (∼0.11 V in theory) is needed to overcome this thermodynamic threshold, and an external voltage of > 0.4 V is typically applied to MECs (4). This additional energy could be provided by a renewable source of energy, such as solar (5), wind, or waste organic matter (6). However, no method has yet been developed to directly achieve H2 production in one process without an external voltage supply.
Reverse electrodialysis (RED) holds great promise as a method for generating electricity from the salinity gradient between seawater and river water (7). RED systems are built as stacks of alternating cation- and anion-exchange membranes situated between two electrodes. When seawater and river water are provided into the RED stack, counter-ions (selected ions) to the membranes are driven from seawater to river water due to the salinity difference, creating an electric potential across the ion-exchange membrane. Each pair of anion- and cation-exchange membranes can in theory create 0.155 V for a salinity ratio of 50 between seawater and river water (based on the open circuit potential). Thus, a RED stack requires a minimum at least eight cell pairs to produce current, based on the thermodynamic threshold for water electrolysis (1.23 V). In practice, however, the required number of cell pairs is much larger due to energy losses by Ohmic resistance and electrode overpotentials. Reported resistivity values in RED systems range from 35 to 65 Ω-cm2 per cell pair for salinity ratios of 30 to 100 (8–10). Assuming roughly a 50% loss in efficiency due to electrode overpotentials, a hypothetical RED system would require ∼20 cell pairs for H2 production at 0.5 mA/cm2. The voltage produced in a RED stack can be increased by adding cell pairs. However, this addition of cell pairs increases capital costs mainly for ion-exchange membranes and reduces energy recovery due to solution pumping. For example, a 50-cell paired RED system produced 3 V, but the energy loss for pumping was ∼25% of the gross power production (10); in a 25-cell paired RED system, the overpotential loss for water electrolysis was 1 to 2 V (11). Alternative redox couples have been suggested to reduce electrode overpotentials in RED systems (10, 12). However, chemicals such as 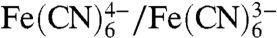 can result in toxic HCN gas production under extreme redox conditions at the electrodes, and chemicals such as Fe2+/Fe3+ can result in undesirable precipitation on electrode surfaces. Thus, it is not possible to generate current or achieve hydrogen gas production using a RED system without a large number of membranes.
can result in toxic HCN gas production under extreme redox conditions at the electrodes, and chemicals such as Fe2+/Fe3+ can result in undesirable precipitation on electrode surfaces. Thus, it is not possible to generate current or achieve hydrogen gas production using a RED system without a large number of membranes.
A unique method of H2 production is demonstrated here based on integrating a very small (five membrane pairs) RED stack into a microbial electrolysis cell, where anodic oxidation of organic matter is driven by exoelectrogenic microorganisms. In this microbial reverse-electrodialysis electrolysis cell (MREC), H2 production is achieved by two driving forces: a thermodynamically favorable oxidation of organic matter by exoelectrogens on the anode; and the energy derived from the salinity gradient between seawater and river water. Individually, neither of these systems can accomplish hydrogen gas generation: the MEC requires an energy input (added voltage); and a small RED stack by itself cannot produce current. As stand-alone systems, a much larger RED stack than that used in this study would be needed to power the MEC. However, in this integrated system, the exoelectrogens on the anode produce current and create a favorable reaction, and thus substantially reduce the electrode overpotential. The added salinity driven energy, provided by using only a small number of RED cell pairs, completely eliminates the need for an external power source for the MEC. The use of a small number of RED cell pairs, coupled with an optimized flow scheme, also minimizes energy losses for pumping solutions. The MREC therefore represents a unique method for generating H2 from relatively unlimited energy sources: organic matter in solution, for example in domestic and industrial wastewaters, and seawater and river water.
Results and Discussion
MREC Operation.
The MREC, constructed with five pairs of seawater and river water cells, produced from 21 to 26 mL of gas over each fed-batch cycle (Fig. 1A). The produced gas was pure H2, with no detectable CH4 or CO2 gases. Increasing the water flow accelerated H2 production, because the salinity gradient was maintained between the seawater and river water cells throughout the RED stack. The increase in H2 production from an increase in water flow from 0.4 to 0.8 mL/ min was smaller than that obtained when increasing the flow from 0.1 to 0.4 mL/ min, however, suggesting that further increases in the flow would not substantially increase H2 production. There was a slightly larger rate of H2 production using the cathode constructed around a stainless steel current collector (SS) than a more expensive carbon cloth (CC) cathode (13).
Fig. 1.

Effects of solution flow (0.1, 0.4, and 0.8 mL/ min) and current collector (SS: stainless steel, CC: carbon cloth) on (A) gas production, (B) current generation.
The maximum electrical current in the MREC was observed at the beginning of the fed-batch cycle, and the current decreased over time as the substrate was depleted (Fig. 1B). Current densities were similar for water flow rates between 0.8 and 0.4 mL/ min, but substantially smaller currents were obtained at 0.1 mL/ min. As a result of slower oxidation of substrate, the fed-batch cycle increased from ∼0.9 to ∼1.4 d with the decreased water flow rate. The current was almost identical in the later part of the cycle (after 0.2 d) with the CC and SS cathodes, but the SS cathode produced slightly greater current in the earlier part of the cycle.
The substrate removal was 85 ± 6% (n = 6) on the basis of COD (chemical oxygen demand) removal over a fed-batch cycle. As the acetate was oxidized, protons were released in the anode chamber, resulting in a pH decrease from 7.0 to 5.2 ± 0.1 even with the use of a pH buffer. This low pH was responsible for the incomplete removal of COD as it is known that exoelectrogenic activity is significantly inhibited at a pH of ∼5 (14). There was a much larger catholyte pH increase over a cycle (from 6.1 ± 0.2 to 12.3 ± 0.1) due to the absence of a buffer. According to the Nernst equation, a unit increase in the catholyte pH increases the equilibrium potential for H2 evolution by 0.06 V. This correlation between pH and equilibrium potential implies, for instance, that further increase in the catholyte pH up to 13 would result in little decrease in MREC performance. Thus, the low anolyte pH is considered to be the main reason for the termination of current production at the end of the fed-batch cycle. In future designs it should be possible to recirculate a smaller volume of a high pH catholyte into a larger-volume anode chamber to better control pH conditions. This catholyte addition would also have the advantage of slightly increasing the solution conductivity and reducing Ohmic losses in the anode chamber.
Voltage Contribution by RED.
The five-cell paired RED stack was estimated to contribute 0.5–0.6 V for H2 production (Fig. 2A) using Eqs. 2–4 and the observed ion flux efficiency (ηflux = 0.81 ± 0.06, n = 6). This ion flux efficiency corresponds to a transport number of the counter-ion (tcounter) of ∼0.91, indicating there was no failure in membrane integrity or permselectivity. During the experiments with water flow rates of 0.4 or 0.8 mL/ min, ∼80% of the H2 was produced at a current between 2 and 4 mA (Fig. 1). Similarly, at a water flow rate of 0.1 mL/ min, the majority of the H2 production was achieved at a current of 1 to 2 mA. For these current ranges, the corresponding voltage contribution by the RED stack was 0.5–0.6 V for all three applied flows (Fig. 2A).
Fig. 2.

The RED stack voltage calculated by Eqs. 2–4 (ηflux = 0.81): (A) net potential from the stack; (B) Ohmic losses; and (C) voltage losses due to a reduction in the initial salinity gradient.
The decrease in the RED voltage with increasing current is due to two factors: a resistive Ohmic loss; and the reduction in the salinity gradient as the water flows through the membrane stack. Ohmic losses increased at higher flow rates due to the influent river water salinity being maintained throughout the RED stack (Fig. 2B). As a result, smaller changes in the salinity gradient along the flow through the stack provide greater energy available for current generation (Fig. 2A). An increase in flow rate from 0.1 to 0.4 mL/ min substantially reduced voltage losses due to less change in the initial salinity gradient as the water flowed through the stack (Fig. 2C), resulting in a significant improvement in the net RED voltage (Fig. 2A). However, with an increase in flow rate from 0.4 to 0.8 mL, the increase in the net RED voltage was relatively small because an improvement in maintaining the overall salinity gradient was offset by an increase in Ohmic losses.
H2 Recovery, Yield, and Rate.
The Coulombic recovery ranged from rCE = 70 to 85% during MREC operation with a SS cathode (Fig. 3A). The increase in rCE at the higher flow rates was due to a shorter fed-batch cycle (Fig. 1). Substrate oxidation in the anode chamber can occur due to the activity of nonexoelectrogenic microorganisms such as methanogens or aerobic microorganisms using oxygen that can leak into the reactor through gaskets and fittings. The relative proportion of substrate oxidation by these nonexoelectrogens will increase with increasing cycle time, resulting in reduced capture of substrate as current and therefore a lower rCE.
Fig. 3.

MREC performance with Pt-SS cathode. (A) Recoveries and energy efficiencies; (B) H2 yields and production rates.
The cathodic H2 recovery (rcat) was somewhat greater than unity (Fig. 3A), suggesting that H2 production was slightly overestimated. This overestimation was likely due to osmotic water transport into the cathode chamber from the adjacent river water cell. This increase in water volume in the cathode chamber reduced the headspace volume, and released additional gas from the headspace through the respirometer. We estimated that this gas volume was ∼0.8 mL, which would have resulted in ∼3–4% overestimation in rcat. If this correction is included in our calculations, then values of rcat would be near unity. This conclusion that osmotic water transport was responsible for this elevated gas production is also consistent with the gas analysis finding that the only gas produced was H2. Thus, the overall H2 recovery (rH2) is consistent with the calculated Coulombic recoveries (Fig. 3A).
The H2 yield (YH2) increased from 1.4 to 1.7 mole-H2/mole-COD with an increase in water flow rate (Fig. 3B). The maximum yield based on stoichiometry is YH2 = 2.0 mole-H2/mole-COD. The maximum H2 production rate varied with the water flow rate, increasing from QH2 = 0.8 to 1.6 m3-H2/m3-Van/day with flow rate (Fig. 3B). The H2 production rate, expressed on the basis of the total empty bed volume (VEB = 80.4 mL), was 0.29 to 0.59 m3-H2/m3-VEB/day.
H2 production achieved with the MREC is consistent with performance expected for an MEC with an externally applied voltage (Eap), indicating the presence of the RED stack did not adversely affect performance. Based on MEC studies performed under similar conditions (Van ∼ 30 mL; acetate as substrate; platinum catalyst; Eap 0.5–0.6 V), hydrogen recoveries were rH2 = 0.04 to 0.9, and gas production rates were QH2 = 0.1 to 2.5 m3-H2/m3-Van/day (15–17). Based on values obtained here (rH2 = 0.7–0.8, and QH2 = 0.8–1.6 m3-H2/m3-Van/day), we conclude that the RED stack voltage (0.5–0.6 V) was efficiently used for H2 production in the MREC.
Energy Recovery and Efficiency.
The energy recovery, or the energy captured based on the total energy input, was as high as rE = 36% at the lowest water flow rate of 0.1 mL/ min (Fig. 3A). A slow water flow rate provides a longer residence time in the stack and thus achieves more complete utilization of the salinity driven energy between the seawater and river water. Based on the total provided energy (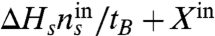 ), the fractional contribution of the salinity driven energy was 53% (47% contributed as substrate) at 0.1 mL/ min, and it increased up to 85% (15% as substrate) at 0.8 mL/ min.
), the fractional contribution of the salinity driven energy was 53% (47% contributed as substrate) at 0.1 mL/ min, and it increased up to 85% (15% as substrate) at 0.8 mL/ min.
The energy efficiency, or the energy captured based on the energy entering and leaving the MREC, was ηE = 65% at 0.8 mL/ min, and ηE = 58% at 0.1 mL/ min (Fig. 3A). Complete utilization of the salinity driven energy in an MREC, based on energy recovery (rE), may be unnecessary because seawater and river water are relatively unlimited energy sources. Thus, we suggest that the process be run at higher flow rates to maximize the energy efficiency (ηE), not energy recovery (rE). In addition, the use of higher flow rates (0.4 and 0.8 mL/ min) improved the H2 yield and production rate. The required energy for pumping seawater and river water through the stack was small (∼4 × 10-5 W), which is only about ∼1% of the energy recovered as H2 (ΔHH2nH2/tB = 3.8 × 10-3 W).
Molybdenum Cathode Catalyst.
The above tests were conducted with a noble metal catalyst in order to evaluate the performance of the system under optimal conditions. However, sustainable H2 production will require the use of nonprecious metal cathode catalysts. MREC tests with a MoS2 catalyst resulted in 20 mL of H2 production (Fig. 4). The Mo-SS cathode required a longer fed-batch cycle ∼1.4 d, compared to ∼0.9 d with SS cathode containing Pt (Pt-SS), resulting in substrate losses to noncurrent generating processes. The corresponding process parameters using the MoS2 catalyst were: hydrogen recovery, rH2 = 0.72; hydrogen yield, YH2 = 1.4 mole-H2/mole-COD; hydrogen production rate of QH2 = 0.8 m3-H2/m3-Van/d; and an energy efficiency of ηE = 51%. Even though H2 production with the MoS2 cathode was less efficient than that achieved with Pt, these results proved that it is possible to use inexpensive and nonprecious metal catalysts in MRECs. Further improvements in catalysts used for H2 evolution will help improve the MREC performance.
Fig. 4.

Comparison of platinum and molybdenum catalysts for H2 production.
Outlook.
Hydrogen has significant potential as an efficient energy carrier and it has many industrial uses, but broader applications are limited due to environmental concerns (if produced from fossil fuels) and costs for production. A relatively unlimited energy source obtained from a salinity gradient, like those between seawater and river water, was used here to produce pure hydrogen gas in a process using exoelectrogenic bacteria. The voltage and current generation from organic matter reduced the required number of seawater and river water cells that would be needed to drive H2 evolution at the cathode, making the combined process much more efficient than an individual RED process. By using a small RED stack, energy consumption for water pumping was negligible compared to the produced energy as H2. Because practical applications of RED have been limited due to the need for stacks with hundreds ion-exchange membranes, this unique MREC process could be used to more efficiently produce H2 from an estimated 1.7 TW of global salinity driven energy (18).
The MREC can be used not only as a method for H2 production, but also as a method of wastewater treatment. Even though this study was conducted under well controlled conditions with acetate as substrate, exoelectrogens can use various sources of organic matter, ranging from domestic and animal wastewaters to industrial wastewaters (19). The anode chamber was operated as a fed-batch reactor here, but wastewater can also be continuously supplied into the anode chamber, as previously demonstrated with MECs (20, 21). The continuous flow system for wastewater would not affect the H2 production, because the anode and cathode chamber are completely separated with a stack of ion-exchange membranes. In addition, this separated cathode chamber from wastewater excludes possible losses of H2 through methanogenesis, which is inevitable in single chamber MECs (17). Thus, this unique type of integrated system has significant potential to treat wastewater and simultaneously produce pure H2 gas without any consumption of electrical grid energy.
Materials and Methods
MREC Construction.
A cubic Lexan block with a cylindrical chamber (∼30 mL; 7 cm2 in cross section) was used for an anode and cathode container, with a glass tube (20 mL) glued to the top of the cathode chamber to collect H2 (22). The SS cathode was prepared with platinum (0.6 mg/cm2; BASF) as the catalyst (except as indicated) in a mixture of carbon black and Nafion on both sides of a 7-cm2 stainless steel mesh (#50) (13). In some tests the SS cathode was prepared with MoS2 (6.3 mg/cm2) as a replacement of the Pt catalyst. The carbon cloth (CC) cathode was made with a Pt catalyst (0.5 mg/cm2) in the same manner using a 7-cm2 piece of carbon cloth (23). The anode (a graphite fiber brush 2.7 cm in diameter and 2.3 cm in length; Mill-Rose Lab Inc.) was inoculated with the effluent from an existing MFC and initially enriched in a single chamber MFC (24). During this start-up stage, the anode microbes were acclimated to a gradual increase in NaCl concentration to avoid salt inhibition effects due to chloride ion transfer into the anode chamber.
A RED stack was sandwiched between the anode and cathode chambers, and it consisted of five pairs of seawater and river water cells made with five cation- and six anion-exchange membranes (Selemion CMV and AMV, AGC Engineering Co.) (Fig. 5A). Each cell had a dimension of 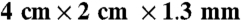 , and the corresponding empty bed volume of the stack was 10.4 mL. Seawater flowed serially through every seawater cell, and river water had a similar flow path but in the opposite direction to seawater (Fig. 5A). Each solution was continuously provided into the MREC at a specified rate from 0.1 to 0.8 mL/ min. The power required for pumping at the highest water flow rate (0.8 mL/ min) was approximated by measuring the head loss through the stack. All experiments were performed at 30 °C in a constant temperature room.
, and the corresponding empty bed volume of the stack was 10.4 mL. Seawater flowed serially through every seawater cell, and river water had a similar flow path but in the opposite direction to seawater (Fig. 5A). Each solution was continuously provided into the MREC at a specified rate from 0.1 to 0.8 mL/ min. The power required for pumping at the highest water flow rate (0.8 mL/ min) was approximated by measuring the head loss through the stack. All experiments were performed at 30 °C in a constant temperature room.
Fig. 5.

(A) Schematic design of MREC for H2 production by integrating exoelectrogens with five-cell paired RED stack. (B) Continuous flow and H2 collection for MREC operation. The pump and effluent reservoirs are not shown.
Solutions.
Synthetic seawater was a 35 g/L NaCl solution, and river water was 0.7 g/L NaCl, creating a salinity ratio of 50. The catholyte (40 mL) was synthetic seawater without any pH buffer, and it was initially purged with N2. The anolyte (30 mL) was prepared with 1.0 g/L sodium acetate in a phosphate buffer (9.16 g/L Na2HPO4; 4.9 g/L NaH2PO4-H2O; 0.62 g/L NH4Cl; 0.26 g/L KCl) with minerals and vitamins (25). The corresponding solution volume of the MREC was 30 + 40 + 10.4 = 80.4 mL. The anolyte and catholyte were replaced every fed-batch cycle, while seawater and river water were continuously supplied (Fig. 5B).
Measurements.
The produced gas volume from the cathode chamber was measured using a respirometer (AER-208, Challenge Environmental Systems) prior to being collected in a gas bag (100 mL capacity; Cali-5-Bond, Calibrated Instruments Inc.). The gas in the bag and in the cathode headspace was analyzed for H2, O2, N2, CH4, and CO2 by gas chromatography (SRI-310C, SRI Instruments) as previously described (17).
The anode and cathode were connected externally to a 10-Ω resistor to measure current based on the voltage drop using Ohm’s law. The voltage drop across the resistor was recorded every 20 min using a multimeter (Keithley Instruments).
Effluent seawater, river water, anolyte, and catholyte solutions were analyzed with conductivity and pH probes (SevenMulti, Mettler-Toledo International Inc.). The anolyte COD was determined according to standard methods (Hach Co.) (26).
Membrane Permselectivity.
The ion flux efficiency (ηflux) is the fractional contribution of the ion transport to the current and thus it represents the permselectivity of the ion-exchange membranes as:
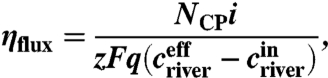 |
[1] |
where NCP is the number of cell pairs, i the current, z the ionic charge, F the Faraday constant, q the solution flow rate, and criver the NaCl molarity in river water. The superscript eff denotes the effluent and in the influent to the MREC.
Calculations on RED.
The voltage added by the RED stack equals the sum of the junction potentials created by the salinity difference minus the sum of Ohmic losses. The junction potential across an ion-exchange membrane (Δϕjct) quantifies the salinity driven energy between seawater and river water as (27)
 |
[2] |
where R is the gas constant and T the absolute temperature. For counter- and co-ions to the ion-exchange membrane, t is the transport number defined as the fractional contribution of the ionic flux to the current density in the membrane, and a is the chemical activity. The chemical activity was calculated by multiplying the molar concentration by the activity coefficient (fi). The activity coefficient was calculated as (28)
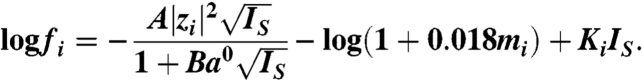 |
[3] |
The Debye-Hückel constants were A = 0.5085 kg1/2/mol1/2 and B = 0.3282 Å kg1/2/mol1/2. The ion size parameter (a0) was 0.78 Å for both sodium and chloride, while KNa = 0.105 and KCl = -0.009 kg2/mol2. IS is the ionic strength in molality, and m the molal concentration. This equation is valid for a NaCl solution up to 1.2 molality (28).
The NaCl concentration was assumed to be homogeneous in each cell, while the concentration change between cells along the flow (Δccell) was determined by:
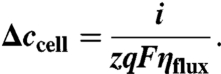 |
[4] |
Ohm’s law was employed for the resistive loss in each of the solution and membrane phases. Effect of the boundary layer near the membrane surface was ignored due to relatively low current densities (29). The membrane resistivity was 3.0 (CMV) and 2.8 Ω cm2 (AMV) (30).
H2 Recovery.
The Coulombic recovery (rCE) is the fraction of the transferred electron to the anode among the total electron released by substrate oxidation (4), and is calculated as
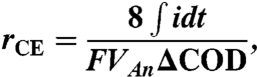 |
[5] |
where Van is the anolyte volume and ΔCOD is the removed COD as O2.
The cathodic H2 recovery (rcat) represents the contribution of the mole-H2 evolution (nH2) to the total cathodic charge transfer as (4)
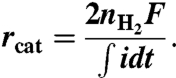 |
[6] |
The overall H2 recovery (rH2) is determined by rH2 = rCErcat, meaning the ratio of the produced H2 to the removed organic matter on the electron basis.
The H2 Yield (YH2) is defined on the mole basis as (4)
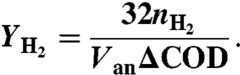 |
[7] |
The maximum volumetric H2 production rate at 1 atm (QH2, m3-H2/m3-Van/d) was calculated with averaged current (iavg) over the first 1 h of a fed-batch cycle as (4)
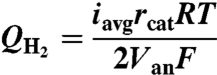 |
[8] |
Energy Recovery and Efficiency.
The energy recovery (rE) is the combustion energy of the produced H2 normalized by the total energy provided to the MREC as:
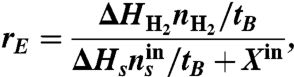 |
[9] |
where ΔH is the heat of combustion (J/mol), tB the time span for each batch cycle, and Xin the theoretical energy (W) estimated by the change in the free energy involving complete mixing of seawater and river water as (31)
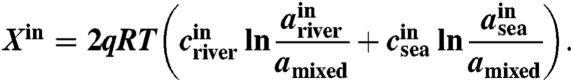 |
[10] |
The energy efficiency (ηE) is defined similarly but by subtracting the energy leaving the MREC (superscript out) from the provided energy as:
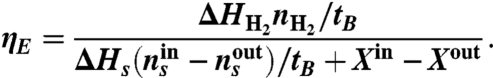 |
[11] |
Acknowledgments.
This research was supported by funding through the King Abdullah University of Science and Technology (KAUST) (Award KUS-I1-003-13).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Logan BE. Microbial fuel cells. Hoboken, NJ: John Wiley & Sons, Inc; 2008. [Google Scholar]
- 2.Liu H, Grot S, Logan BE. Electrochemically assisted microbial production of hydrogen from acetate. Environ Sci Technol. 2005;39:4317–4320. doi: 10.1021/es050244p. [DOI] [PubMed] [Google Scholar]
- 3.Rozendal RA, Hamelers HVM, Euverink GJW, Metz SJ, Buisman CJN. Principle and perspectives of hydrogen production through biocatalyzed electrolysis. Int J Hydrogen Energ. 2006;31:1632–1640. [Google Scholar]
- 4.Logan BE, et al. Microbial electrolysis cells for high yield hydrogen gas production from organic matter. Environ Sci Technol. 2008;42:8630–8640. doi: 10.1021/es801553z. [DOI] [PubMed] [Google Scholar]
- 5.Fang HHP, Liu H, Zhang T. Phototrophic hydrogen production from acetate and butyrate in wastewater. Int J Hydrogen Energ. 2005;30:785–793. [Google Scholar]
- 6.Sun M, et al. An MEC-MFC coupled system for biohydrogen production from acetate. Environ Sci Technol. 2008;42:8095–8100. doi: 10.1021/es801513c. [DOI] [PubMed] [Google Scholar]
- 7.Weinstein JN, Leitz FB. Electric power from differences in salinity: the dialytic battery. Science. 1976;191:557–559. doi: 10.1126/science.191.4227.557. [DOI] [PubMed] [Google Scholar]
- 8.Długołęcki P, Gambier A, Nijmeijer K, Wessling M. Practical potential of reverse electrodialysis as process for sustainable energy generation. Environ Sci Technol. 2009;43:6888–6894. doi: 10.1021/es9009635. [DOI] [PubMed] [Google Scholar]
- 9.Post JW, Hamelers HVM, Buisman CJN. Energy recovery from controlled mixing salt and fresh water with a reverse electrodialysis system. Environ Sci Technol. 2008;42:5785–5790. doi: 10.1021/es8004317. [DOI] [PubMed] [Google Scholar]
- 10.Veerman J, Saakes M, Metz SJ, Harmsen GJ. Reverse electrodialysis: performance of a stack with 50 cells on the mixing of sea and river water. J Membr Sci. 2009;327:136–144. [Google Scholar]
- 11.Veerman J, de Jong RM, Saakes M, Metz SJ, Harmsen GJ. Reverse electrodialysis: comparison of six commercial membrane pairs on the thermodynamic efficiency and power density. J Membr Sci. 2009;343:7–15. [Google Scholar]
- 12.Veerman J, Saakes M, Metz SJ, Harmsen GJ. Reverse electrodialysis: evaluation of suitable electrode systems. J Appl Electrochem. 2010;40:1461–1474. [Google Scholar]
- 13.Zhang F, Cheng S, Pant D, Bogaert GV, Logan BE. Power generation using an activated carbon and metal mesh cathode in a microbial fuel cell. Electrochem. Commun. 2009;11:2177–2179. [Google Scholar]
- 14.He Z, Huang Y, Manohar AK, Mansfeld F. Effect of electrolyte pH on the rate of the anodic and cathodic reactions in an air-cathode microbial fuel cell. Bioelectrochemistry. 2008;74:78–82. doi: 10.1016/j.bioelechem.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Cheng S, Logan BE. Sustainable and efficient biohydrogen production via electrohydrogenesis. Proc Natl Acad Sci USA. 2007;104:18871–18873. doi: 10.1073/pnas.0706379104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selembo PA, Merrill MD, Logan BE. The use of stainless steel and nickel alloys as low-cost cathodes in microbial electrolysis cells. J Power Sources. 2009;190:271–278. [Google Scholar]
- 17.Call D, Logan BE. Hydrogen production in a single chamber microbial electrolysis cell lacking a membrane. Environ Sci Technol. 2008;42:3401–3406. doi: 10.1021/es8001822. [DOI] [PubMed] [Google Scholar]
- 18.Wick GL. Power from salinity gradients. Energy. 1978;3:95–100. [Google Scholar]
- 19.Pant D, Bogaert GV, Diels L, Vanbroekhoven K. A review of the substrates used in microbial fuel cells (MFCs) for sustainable energy production. Bioresoure Technol. 2010;101:1533–1543. doi: 10.1016/j.biortech.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 20.Tartakovsky B, Manuel MF, Wang H, Guiot SR. High rate membrane-less microbial electrolysis cell for continuous hydrogen production. Int J Hydrogen Energ. 2008;34:672–677. [Google Scholar]
- 21.Tartakovsky B, Manuel M-F, Neburchilov V, Wang H, Guiot SR. Biocatalyzed hydrogen production in a continuous flow microbial fuel cell with a gas phase cathode. J Power Sources. 2008;182:291–297. [Google Scholar]
- 22.Mehanna M, Kiely PD, Call DF, Logan BE. Microbial electrodialysis cell for simultaneous water desalination and hydrogen gas production. Environ Sci Technol. 2010;44:9578–9583. doi: 10.1021/es1025646. [DOI] [PubMed] [Google Scholar]
- 23.Cheng S, Liu H, Logan BE. Power densities using different cathode catalysts (Pt and CoTMPP) and polymer binders (Nafion and PTFE) in single chamber microbial fuel cells. Environ Sci Technol. 2006;40:364–369. [PubMed] [Google Scholar]
- 24.Logan BE, Cheng S, Watson V, Estadt G. Graphite fiber brush anodes for increased power production in air-cathode microbial fuel cells. Environ Sci Technol. 2007;41:3341–3346. doi: 10.1021/es062644y. [DOI] [PubMed] [Google Scholar]
- 25.Lovley DR, Phillips EJP. Novel mode of microbial energy metabolism: organic carbon oxidation coupled to dissimilatory reduction of iron or manganese. Appl Environ Microbiol. 1988;54:1472–1480. doi: 10.1128/aem.54.6.1472-1480.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.APHA. Standard Methods for the Examination of Water and Wastewater. Washington DC: American Public Health Association, American Water Works Association, Water Environment Federation; 1998. [Google Scholar]
- 27.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. New York: John Wiley & Sons; 2001. [Google Scholar]
- 28.Zhuo K, Dong W, Wang W, Wang J. Activity coefficients of individual ions in aqueous solutions of sodium halides at 298.15 K. Fluid Phase Equilibr. 2008;274:80–84. [Google Scholar]
- 29.Kim Y, Walker WS, Lawler DF. Electrodialysis with spacers: effects of variation and correlation of boundary layer thickness. Desalination. 2011;274:54–63. [Google Scholar]
- 30.Selemion reference data. AGC Engineering Co, www.selemion.com/SEL3_4.pdf.
- 31.Escobar I, Schäfer A, editors. Sustainable water for the future: Water recycling versus desalination. Amsterdam: Elsevier; 2010. [Google Scholar]