

Abstract
Mutations in amyloid precursor protein (APP) are associated with familial Alzheimer’s disease. Recent development suggests that homo- and heterodimerization of APP and APP-like proteins (APLPs), which are enhanced by heparan sulfate binding, may play a role in signal transduction and cell adhesion. Despite efforts to model heparin binding based on known apo crystal structures, the mechanism of heparin-induced APP/APLP dimerization has not been established experimentally. Here we report the crystal structure of a complex between heparin and the E2 domain of APLP1, which harbors the conserved high affinity heparin binding site of the full-length molecule. Within the asymmetric E2:heparin complex, the polysaccharide is snugly bound inside a narrow groove between the two helical subdomains of one protein protomer. The nonreducing end of the sugar is positioned near the protein’s 2-fold axis, making contacts with basic residues from the second protomer. The inability of the E2 dimer to accommodate two heparin molecules near its symmetry axis explains the observed 2∶1 binding stoichiometry, which is confirmed by isothermal titration calorimetric experiment carried out in solution. We also show that, at high concentrations, heparin can destabilize E2 dimer, probably by forcing into the unoccupied binding site observed in the 2∶1 complex. The binding model suggested by the crystal structure may facilitate the design of heparin mimetics that are capable of modulating APP dimerization in cells.
The amyloid β-peptide (Aβ) implicated in Alzheimer’s disease is derived from a large type I membrane protein precursor called amyloid precursor protein (APP) through sequential proteolysis by β- and γ-secretases (1). Mutations in APP and γ-secretase can cause familial Alzheimer’s disease (2). The biological function of APP, however, is not completely clear. Gene manipulation in intact animals showed that the APP family of proteins is essential for survival (3–6) and suggested that they may play a role in signal transduction and cell adhesion (6–9). The soluble ectodomain of APP (sAPP), which can be proteolytically released from the membrane, shows robust trophic activities in vitro: In cultured fibroblasts, where the activity was first described, sAPP promotes cell growth in an autocrine fashion (10); in neuronal cells sAPP not only stimulates neurite extension (11, 12), but also has potent neuroprotective activities (13); sAPP also stimulates the proliferation of epithelial cells (14). Recently it was suggested that APP may trigger axon pruning and neurodegeneration through death receptor 6 (15).
A number of independent studies have shown that APP and its two mammalian homologs, the APP-like proteins (APLPs), can form homo- and heterodimers inside the cell (16–20). Initial evidence suggested that dimerization affects Aβ production (16, 20, 21) and may play a role in the neuroprotective function of sAPP (19). Solution studies of isolated APP ectodomain showed that the protein is monomeric, but dimerizes in the presence of heparin (22). This observation raises the possibility that heparan sulfate proteoglycans, abundantly present in the extracellular matrix, may play a crucial role in regulating APP and APLP dimerization. In consistence with this hypothesis, it has been shown that heparinase treatment greatly reduces the number of APP dimers on the cell surface (19).
The conserved E2 domain, which is located near the middle of APP’s ectodomain, may contribute to the dimerization of the full-length molecule (Fig. S1). The atomic structures of the E2 domains of APP and APP homologs obtained from different crystal forms suggest a conserved mode of protein dimerization (23–25). The E2 domain also contains the high affinity heparin binding site of the full-length molecule (26), and heparin binding induces the formation of E2 dimers in solution (25). Despite efforts to model heparin binding based on crystal structures of the apo proteins and complexes with sucrose octasulfate (23, 27), the details of E2:heparin interaction and the mechanism of heparin-induced protein dimerization are not well understood. Here we report the crystal structure of a complex between the E2 domain of human APLP1 (NP_005157) and a heparin hexasaccharide. Based on the previously undescribed structural data and additional biochemical analysis of wild-type and mutant E2 proteins, we propose an asymmetric binding model for longer heparin chains, which explains their dimerization effect at low concentrations, and their apparent ability to disrupt protein dimer at high concentrations.
Results
Structural Determination.
The E2:heparin complex was generated by directly soaking a homogenous heparin hexasaccharide into preformed APLP1 crystals. Difference Fourier analysis showed a continuous stretch of positive density near the protein’s 2-fold symmetry axis (Fig. 1A). The difference density corresponds to a single heparin molecule. Four sugar residues could be modeled into the density: The prominent features for the two sulfate groups on N-sulfo-glucosamine-6-sulfate (SGN) and for the single sulfate group on iduronate-2-sulfate (IDS) helped distinguish the two types of sugar rings; the directionality of the polysaccharide chain could also be unequivocally determined (Fig. 1B). At the end of the modeled tetrasaccharide, the electron density for C4 and C5 of IDS-1 was poor. It was therefore unclear whether IDS-1 was a regular L-iduronic acid or a Δ4-uronic acid, which had a planar C4–C5 double bond and occurred only once at the nonreducing end of the hexasaccharide (the missing disaccharide unit could be on either side of the tetrasaccharide). In the structure presented here, IDS-1 was modeled as a regular L-iduronic acid.
Fig. 1.

The difference electron density for the heparin hexasaccharide soaked into preformed APLP1 crystals. (A) A single heparin molecule is bound near the 2-fold axis of the dimeric protein (shown as the Cα-trace). The A chain is shown in green and the B chain in orange. (B) A detailed view of the electron density that corresponds to the ordered portion of the hexasaccharide. The Fo-Fc map shown in A and B was calculated before heparin was included for refinement, and thus not biased by the model. The map was contoured at 3σ-levels. These figures and those in Figs. 2A, 3 B and C, and 4D were generated by program PyMOL (http://www.pymol.org).
The two glucosamine residues (SGN-2 and SGN-4) of the tetrasaccharide adopt the preferred 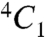 chair conformation (Fig. 1B). Unlike glucosamine residues, internal L-iduronic acids are usually flexible and can assume either the
chair conformation (Fig. 1B). Unlike glucosamine residues, internal L-iduronic acids are usually flexible and can assume either the 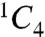 chair conformation or the
chair conformation or the 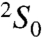 skew boat conformation. The features of the electron density indicate that IDS-3 adopts a chair conformation (Fig. 1B): In the 2.2-Å resolution map, three protrusions can be clearly seen originating from the planar shape of the sugar ring; these protrusions correspond to the axially positioned 2-O-sulfate and 3-hydroxyl groups, and the equatorially positioned C6 carboxylate group. IDS-3 is apparently locked into this conformation to maximize its interactions with the protein. The glycosydic linkages that connect the sugar residues have very similar ϕ/Ψ angular values as those observed for free heparins in solution, indicating that the binding to the E2 domain did not introduce any strains into the backbone of the polysaccharide (28).
skew boat conformation. The features of the electron density indicate that IDS-3 adopts a chair conformation (Fig. 1B): In the 2.2-Å resolution map, three protrusions can be clearly seen originating from the planar shape of the sugar ring; these protrusions correspond to the axially positioned 2-O-sulfate and 3-hydroxyl groups, and the equatorially positioned C6 carboxylate group. IDS-3 is apparently locked into this conformation to maximize its interactions with the protein. The glycosydic linkages that connect the sugar residues have very similar ϕ/Ψ angular values as those observed for free heparins in solution, indicating that the binding to the E2 domain did not introduce any strains into the backbone of the polysaccharide (28).
Heparin:Protein Interactions.
Within the APLP1:heparin complex the sugar is bound inside a deep groove between the two subdomains of one E2 protomer (the A chain) with its nonreducing (O4) end extending toward the 2-fold symmetry axis of the protein where it makes additional contacts with three basic residues from the second E2 protomer (the B chain). The tetrasaccharide forms a total of 20 hydrogen bonds with the A chain and 3 hydrogen bonds with the B chain. The binding of heparin causes little structural change in the protein.
Most interactions between heparin and E2 are mediated through the O- and N-sulfate groups on the negatively charged sugar (Fig. 2 A and B). The 2-O-sulfate group of IDS-1 is simultaneously hydrogen bonded to Lys-314, Arg-369, and His-433* (the asterisk denotes residues from the B chain). In SGN-2, the 2-N-sulfate group forms a direct hydrogen bond with His-430 and interacts with His-426 and Arg-429 via a water molecule. The 6-O-sulfate group of SGN-2 is hydrogen bonded to His-433, Lys-314*, and Arg-369*. In IDS-3, the 2-O-sulfate group forms a hydrogen bond with His-426. The same sulfate group also interacts with His-307, His-376, and Arg-429 through water-mediated hydrogen bonds. Besides the 2-O-sulfate, the ring oxygen and the ester oxygen on C2 of IDS-3 also appear to interact with His-376 via a water molecule. In SGN-4, the 2-N-sulfate group points away from the binding groove and is not involved in any interactions with the protein. The 6-O-sulfate group of SGN-4 is hydrogen bonded to Lys-422. It also forms a hydrogen bond with a water molecule that is positioned near the N terminus of the αB helix. Therefore, the dipole moment of the helix may also contribute to the binding of the sulfate group.
Fig. 2.

Heparin is bound inside a conserved groove between E2’s two subdomains. (A) A detailed view of the interactions between APLP1 and heparin. Shown as stick models are the oligosaccharide (yellow), protein side chains that interact with it (green, A chain; orange, B chain), and part of the protein’s main chain near the N terminus of the αB helix. The helices are shown as ribbons. Two turns of αB (between His-307 and Lys-314) were omitted to show His-376 behind the helix. Water molecules that mediate hydrogen bonds between protein and heparin are shown as small red spheres. Hydrogen bonds are shown as dashed lines. The carbon atoms of each sugar are labeled in blue from 1 to 6. Resides and helices from the B chain are indicated by the asterisk. (B) A schematic diagram of E2:heparin interactions. The hydrogen bonds are represented by the dashed lines, and their distances are shown. The oligosaccharide is shown in gray, and amino acids are shown in green (A chain) and orange (B chain). B was generated by LIGPLOT (35).
The heparin binding site revealed by the crystal structure is completely conserved in APP and APLP2, the other two mammalian members of the family (Fig. S1B). A total of nine residues, four from the N-terminal subdomain (His-307, Lys-314, Arg-369, and His-376) and five from the C-terminal subdomain (Lys-422, His-426, Arg-429, His-430, and His-433), contribute to the binding of the tetrasaccharide. Mutation of these residues affects heparin binding in various degrees (25, 27). A complete mapping of the region by mutagenesis has identified two pockets that are most important in ligand binding (27). The first pocket consists of His-376, Lys-422, and Arg-429. In the crystal structure of the complex with tetrasaccharide, these residues interact exclusively with the disaccharide unit (IDS-3, SGN-4) that is bound inside the intersubdomain groove. The second pocket consists of Lys-314 and its symmetry mate Lys-314*. In the crystal structure, Lys-314 and Lys-314* form hydrogen bonds with the disaccharide unit (IDS-1, SGN-2) that is bound at the interfacial site. It is interesting to note that the footprint of the tetrasaccharide covers four histidines (His-307, His-376, His-426, and His-433) that are absolutely conserved from the worm homolog to the human proteins. At low pH, the protonation of these histidines is expected to increase the binding affinity between the E2 domain and the negatively charged sugar (23).
Binding Stoichiometry.
The 2∶1 protein:heparin binding stoichiometry revealed by the crystal structure was not expected. To determine the binding stoichiometry in solution with longer heparin chains, and to obtain other thermodynamic parameters, we conducted isothermal titration calorimetry (ITC) experiment using a heparin preparation that has an average molecular weight of 5 kDa, which corresponds roughly to a chain length of 15 sugar residues. As illustrated in Fig. 3A, the binding reaction is exothermic. Fitting the titration curve to a single type of binding site generated a binding stoichiometry of 1.7∶1, which is close to the 2∶1 stoichiometry observed in the crystal. The dissociation constant for the (E2)2:heparin complex was calculated to be approximately 0.8 μM, indicating that the binding of heparin to the E2 domain is approximately six times tighter than its binding to the E1 domain (29), which harbors the other heparin binding site of the full-length molecule.
Fig. 3.

The binding stoichiometry. (A) Binding isotherm for the interaction between E2 and heparin. (Top) The downward pointing peaks represent the heat released upon injection of protein into the heparin solution. The integrated peak area is plotted against the protein/heparin molar ratio (Lower). The line represents the best fit of the ITC data. From this analysis, the following thermodynamic parameters were obtained: n, 1.71( ± 0.091); Kd, 0.787( ± 0.124) μM; ΔH, -39.75( ± 2.96) kCal/mol; ΔS, -103 cal/(mol·K). The injection of heparin into protein solution yielded similar results. B and C show the molecular surface of the two heparin binding sites joined at the 2-fold axis (color-coded by electrostatic potential). In B, heparin is omitted to reveal the inner shape of the binding sites. The 2-fold axis, roughly perpendicular to the paper, is indicated by the filled oval at the center. In C, heparin is shown as the space-filling model (yellow).
The unique arrangement of protein protomers within the E2 dimer and the geometric shape of the heparin binding site in each protomer provide a possible explanation for the observed binding stoichiometry. The E2 domain dimerizes in such a way that the binding site for heparin in one protein protomer is right across the 2-fold symmetry axis from the binding site in the second protomer (Fig. 3B). The binding groove within each protein protomer, wide enough for just one heparin molecule to fit deeply inside, guides the polysaccharide toward the 2-fold axis (Fig. 3C). Although it is not yet clear whether the sugar chain ends there, or can extend further to occupy the second binding site (see below), it is certain that a heparin molecule similarly bound to the second protein protomer would create steric clashes with the first heparin’s nonreducing end.
Heparin Binding and E2 Dimerization.
In solution, the E2 domain exists in equilibrium between monomers and dimers (25). We suggested previously that heparin promotes E2 dimerization by binding more tightly to the dimers. The thermodynamic coupling between the binding of ligand and protein dimerization predicts that weakening the dimeric interface should also reduce the protein’s overall affinity for heparin. To test the relevance of the E2 dimer observed in the crystal, we mutated a pair of conserved leucine residues (Leu-428 and Leu-453) that are found at the dimeric interface (25). Leu-428 and Leu-453 do not interact directly with heparin. As shown in Fig. 4A, the double mutant (L428A/L453A) elutes from the heparin column at a lower salt concentration (84%) than the wild-type protein, indicating that it indeed has a lower affinity for heparin.
Fig. 4.

Heparin binding and E2 dimerization. (A) L428A/L453A (green) elutes at a lower salt concentration from the heparin column than WT protein (blue). The dotted line represents the salt gradient. (B) Heparin tetrasaccharide (green) and hexasaccharide (blue) quench the protein’s fluorescence slightly. (C) Titration of the fluorescence intensities of wild-type and mutant proteins. The number in the parentheses indicates the salt concentration (percentage of the wild-type value) at which the protein is eluted from a heparin column (27). In C and E, the 5 kDa heparin was used. (D) A model of the 2∶1 complex between E2 and heparin. The A chain is shown in green and the B chain in orange. A segment of the ligand that forms the primary contact with the protein is shown in white stick model. The segment forming the secondary contract is represented by the white dots. The white arrows point from the reducing end (O1) to the nonreducing end (O4) of heparin. The colored arrows point from the protein’s N-terminal subdomain to the C-terminal subdomain. (E) The fluorescence spectra of the protein at different heparin concentrations (0 μM, black; 5 μM, blue; 10 μM, red; 100 μM, green; 600 μM, purple).
In the absence of heparin, the equilibrium favors monomers probably because interactions between the monomers are weak. The positively charged residues clustered at the dimeric interface also generate electrostatic repulsions (Fig. 3B). The crystal structure shows that the negatively charged heparin molecule functions as an electrostatic tether: It strengthens the interface by forming hydrogen bonds with both protein protomers and reduces the electrostatic repulsion between protomers (Fig. 2). The crystal structure also predicts that heparin chains as short as a tetrasaccharide should be sufficient to induce some degree of dimerization. To test this, we measured the fluorescence of the protein in the presence of short heparin oligosaccharides. Previously we found that E2 dimerization causes a decrease of the protein’s fluorescence intensity (25). As shown in Fig. 4B, the disaccharide produced no effect, whereas both the tetrasaccharide and hexasaccharide reduced the fluorescence, indicating that they can induce protein dimerization. It is, however, important to note that the level of fluorescence quenching induced by the short oligosaccharides is much lower than that induced by longer heparin chains. This latter observation suggests that longer heparins may form additional contacts with the protein, which can also contribute to the stabilization of the E2 dimer.
Most mutations that affect heparin binding also reduce dimerization (Fig. S2). In Fig. 4C, the change of protein’s fluorescence intensity is plotted against the concentration of heparin. As expected, the titration curves for those mutants where key heparin binding residues have been modified (K422A, K314A, R429A, H376A, H307A, and R369A) are flatter than that for the wild-type protein, indicating that a higher heparin concentration is required to achieve half maximum fluorescence quenching, which is consistent with their lower affinities for the ligand (27). What is surprising in the titration experiment is that many mutants also appear to quench to a lesser degree than the wild-type protein, even at high heparin concentrations where the titration curves have started to plateau, indicating less protein dimerization. Based on the crystal structure with the hexasaccharide, one might predict that only K314A and R369A should affect the maximum level of protein dimerization because, after the binding site in the A chain is fully occupied, the interactions of heparin with Lys-314* and Arg-369* from the B chain contribute directly to the stabilization of the dimer, whereas interactions with the others (e.g., Lys-422) do not (Fig. 2). Based on this finding and the result of the previous experiment, we propose a bipartite binding model where the longer heparin chain interacts with residues in the heparin binding sites of both protein protomers (Fig. 4C). Because heparin does not have an internal 2-fold symmetry, its binding to the E2 dimer has to be asymmetrical: Within the binding site of one protein protomer (primary contact), the nonreducing end (O4) of the polysaccharide points toward the protein’s 2-fold axis, in a fashion similar to what we observed in the crystal structure; within the other binding site (secondary contact), the polysaccharide’s nonreducing end points away from the symmetry axis (dotted line in Fig. 4D). The fact that the hexasaccharide binds to the protein with a well-defined orientation in the crystal suggests that the binding mode revealed by the crystal structure is preferred over the binding mode where the sugar is bound in the opposite orientation (Fig. S3). The E2 dimer is thus different from many other heparin binding proteins in its asymmetric interaction with the ligand (30, 31). Finally, it is interesting that some mutants (e.g., H426A) show enhanced fluorescence quenching (Fig. S2), indicating that not all interactions between heparin and E2 contribute positively to dimerization.
The possibility that the secondary contact is weaker suggests that, at high ligand concentrations, a new heparin molecule may compete for the binding site by occupying it in the preferred orientation and disrupt the protein dimer by clashing with the first heparin molecule (Fig. S3). As shown in Fig. 4E, the fluorescence of the protein dropped at low heparin concentrations (blue, 5 μM; red, 10 μM). After reaching a minimum, the fluorescence became less sensitive to the change of heparin concentration, indicating that a maximum level of protein dimerization has been achieved. When the concentration of heparin was raised higher (green, 100 μM; purple, 600 μM), a significant rebound of the fluorescence intensity was observed. This second change of fluorescence reflects a lower-affinity binding event, and its opposite trend suggests that protein dimers are breaking apart when the second binding takes place, which is consistent with the prediction made above.
Discussion
The centrally located E2 domain possesses several biochemical and structural properties suitable for homophilic binding. The isolated E2 domain can reversibly dimerize in solution (25). The protein dimerizes in a conserved and antiparallel fashion (24, 25). Heparin binding promotes E2 dimerization (25). Now adding to these properties we have shown that heparin binds to a highly conserved site in the E2 domain and that heparin strengthens the dimeric interface by engaging in electrostatic interactions with conserved basic residues from both protein protomers. The 2∶1 binding stoichiometry between E2 and heparin is the same as that observed for sAPP and heparin (22).
Dimerization at the E2 domain may affect the ability of the domain to interact with other proteins. It was reported that APP can bind to collagen, a component of the connective tissue (32). The region that was thought to contribute to collagen binding is buried at the E2 dimer interface and inaccessible from the solution. The E2 domain of APP was also found to interact with F-spondin, a secreted neuronal protein, which inhibits APP processing (33). Because F-spondin also binds to the APLPs, it must recognize an exposed region that is conserved in the three proteins. There are only two such regions on the protein surface: One is involved in heparin binding, as shown here, and the other is involved in dimerization (24). Therefore, the interaction between APP and F-spondin could also be affected by heparin-induced E2 dimerization. The growth-promoting activities of sAPP have been attributed to an RERMS sequence motif within the E2 domain (12, 34). This sequence motif is partially buried at the dimer interface.
Heparin binding also induces the dimerization of the E1 domain (29). Although the crystal structure of E1 has been solved, the structure of the heparin complex is not yet known (29). The contribution of E2 to the dimerization of APP and APLPs has been questioned by some studies that seem to suggest that the dimerization is primarily mediated through the E1 domain (17, 29). This is difficult to reconcile with our results at the structural level: E1 and E2 are separated by an unstructured domain of 100 amino acids; dimerization at the E1 domain is therefore unlikely to hinder E2 dimerization, even though the latter occurs in an antiparallel fashion. Anchoring the molecule in the membrane is also unlikely to affect E2’s ability to dimerize because E2 is separated from the transmembrane domain by another unstructured domain of about 130 amino acids.
Materials and Methods
Reagents.
The heparin hexasaccharide, tetrasaccharide, and disaccharide were purchased from Iduron. The 5 KDa heparin (sc-203075) was purchased from Santa Cruz Biotechnology.
Crystallization and Structural Determination.
The recombinant E2 domain of APLP1 (construct APLP1285–494) was prepared and crystallized as previously described (27). Briefly, for crystallization, a 10 mg/mL protein solution was mixed with the well solution, which contains 25% PEG 3,350, 0.1 M bis-tris pH 6.5, 0.2 M Li2SO4, and 25 mM ATP in a 72-well microbatch plate (Hampton Research); after streak-seeding, rod-shaped crystals usually appeared within 24 h. The hexasaccharide was dissolved in an artificial crystallization mother liquor containing 25% PEG 3,350, 0.1 M bis-tris pH 6.5 (final concentrations 2 and 5 mM). Soaking in either 2 or 5 mM hexasaccharide solutions caused deformation of most crystals within 2 h. However, a small number of crystals survived the 2-mM soak overnight and remained visually intact. They were cryopreserved by stepwise exchanging into an artificial mother liquor supplemented with 25% glycerol. The cryopreserved crystals were flash-cooled in liquid nitrogen. X-ray diffraction data were collected from beamline X25 at the National Synchrotron Light Source (NSLS) and processed by HKL2000 (36). A previously determined apo-structure (27), with water and sulfate ions removed, was used as the initial model for rigid body refinement by the Crystallography and NMR System (CNS) program (37). The crystallographic asymmetric unit contains an E2 dimer (chain A and chain B). We have also shown previously that the two subdomains within each protein protomer are flexible (25). Therefore, the orientations and positions of the four protein subdomains were independently refined, which lowered Rfree from 47% to 37%. Subsequent rounds of positional and b-factor refinement and model adjustment using O (38) further lowered Rfree to 33%. It was at this stage that a tetrasaccharide and three sulfate ions were modeled into the difference Fourier map, which lowered Rfree by another 2%. Automatic water picking and further refinement were carried out using CNS and REFMAC5 (39). The final model has an Rfree value of 26.7% and reasonable geometry (Table 1).
Table 1.
Crystallographic statistics
| Data collection | APLP1:heparin |
| Cell dimensions, Å | a = 74.2, b = 81.9, c = 90.8 |
| Wavelength, Å | 1.071 |
| Resolution, Å* | 40.0–2.2 (2.28–2.20) |
| Observed reflections | 196,757 |
| Unique reflections | 27,708 |
| Redundancy | 7.1 |
| Completeness, %* | 99.9 (100.0) |
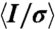 * *
|
11.2 |
| Rmerge*,† | 0.073 (0.506) |
| Refinement | |
| Resolution, Å | 40.0–2.2 |
| Rwork/Rfree‡ | 0.218/0.267 |
| Number of atoms | |
| Protein | 3,019 |
| Heparin | 71 |
| Sulfate ion | 15 |
| Water | 275 |
| B factors | |
| Protein | 48 |
| Heparin | 69 |
| Sulfate ion | 108 |
| Water | 59 |
| rms deviations | |
| Bond lengths, Å | 0.009 |
| Bond angle, ° | 1.13 |
APLP1 was crystallized in space group P212121.
*Highest resolution shell is shown in parentheses.
†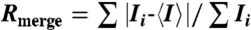 .
.
‡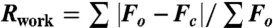 . Rfree is the cross-validation R factor for the test set of reflections (10% of the total) omitted in model refinement.
. Rfree is the cross-validation R factor for the test set of reflections (10% of the total) omitted in model refinement.
Isothermal Titration Calorimetry.
ITC measurements were carried out on a VP-ITC microcalorimeter (GE Healthcare MicroCal) in the Biophysics Resource of the W. M. Keck Biotechnology Research Laboratory at Yale University. The protein solution was dialyzed overnight against a buffer containing 10 mM sodium phosphate and 10 mM sodium acetate (pH 7.5) at 4 °C. The protein concentration was accurately determined by amino acid analysis. A stock solution of the 5 kDa heparin was prepared in water and diluted by about 1,000-fold with the dialysate. The ITC experiment was performed with 1.3 μM heparin in the cell and 31 μM protein in the syringe. There were 20 injections in total; the first injection was 3 μL and the remaining 19 were 15 μL each, with 240-s spacing between injections. The thermogram was analyzed with the Microcal ORIGIN 7.0 software supplied with the instrument.
Heparin Binding.
The effect of the mutations on heparin binding was empirically determined by applying the protein to a 1-mL Hi-Trap heparin HP column (GE Healthcare) and eluting it with a linear NaCl gradient (0 to 2 M). Mutants with reduced binding affinities elute at lower salt concentrations. Before loading, the protein was first dialyzed against the chromatographic buffer, which contained 10 mM sodium phosphate and 10 mM sodium acetate (pH 7.5) (25).
Fluorescence Spectroscopy.
Protein fluorescence was measured on a spectrofluorometer from Photon Technology International. The protein was excited at 295 nm and the emission spectra were collected from 300 to 450 nm at 1-nm intervals with a speed of 1 nm/s. Three scans were acquired and averaged. The protein concentration was fixed at 5 μM. Blank spectra were collected from the buffer and subtracted from the data.
Supplementary Material
Acknowledgments.
We thank A. Heroux and A. Soares at NSLS X25 for assistance during X-ray diffraction data collection. We also thank J. Schlessinger for allowing us to use the spectrofluorometer in his laboratory, and Ewa Folta-Stogniew for assistance with the ITC experiment. Financial support for the beamlines comes principally from US Department of Energy, and from the National Institutes of Health. This work was supported by NIH Grant GM077547 (to Y.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinate and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3QMK).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1103407108/-/DCSupplemental.
References
- 1.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bettens K, Sleegers K, Van Broeckhoven C. Current status on Alzheimer disease molecular genetics: From past, to present, to future. Hum Mol Genet. 2010;19:R4–R11. doi: 10.1093/hmg/ddq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hornsten A, et al. APL-1, a C elegans protein related to human Amyloid Precursor Protein, is essential for viability. Proc Natl Acad Sci USA. 2007;104:1971–1976. doi: 10.1073/pnas.0603997104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Koch CS, et al. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18:661–669. doi: 10.1016/s0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- 5.Heber S, et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herms J, et al. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23:4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang P, et al. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-like protein 2. J Neurosci. 2005;25:1219–1225. doi: 10.1523/JNEUROSCI.4660-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, et al. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheraland central synaptogenesis. J Neurosci. 2009;29:10788–10801. doi: 10.1523/JNEUROSCI.2132-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, et al. Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc Natl Acad Sci USA. 2010;107:17362–17367. doi: 10.1073/pnas.1012568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saitoh T, et al. Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell. 1989;58:615–622. doi: 10.1016/0092-8674(89)90096-2. [DOI] [PubMed] [Google Scholar]
- 11.Milward EA, et al. The amyloid protein precursor of Alzheimer’s disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9:129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- 12.Jin LW, et al. Peptides containing the RERMS sequence of amyloid beta/A4 protein precursor bind cell surface and promote neurite extension. J Neurosci. 1994;14:5461–5470. doi: 10.1523/JNEUROSCI.14-09-05461.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattson MP, et al. Evidence forexcitoprotective and intraneuronal calcium-regulating roles for secreted forms ofthe beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 14.Pietrzik CU, et al. From differentiation to proliferation: The secretory amyloid precursor protein as a local mediator of growth in thyroid epithelial cells. Proc Natl Acad Sci USA. 1998;95:1770–1775. doi: 10.1073/pnas.95.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Scheuermann S, et al. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J Biol Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- 17.Soba P, et al. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munter LM, et al. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gralle M, Botelho MG, Wouters FS. Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J Biol Chem. 2009;284:15016–15025. doi: 10.1074/jbc.M808755200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaden D, et al. Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J Cell Sci. 2009;122:368–377. doi: 10.1242/jcs.034058. [DOI] [PubMed] [Google Scholar]
- 21.Eggert S, Midthune B, Cottrell B, Koo EH. Induced dimerization of the amyloid precursor protein leads to decreased amyloid-beta protein production. J Biol Chem. 2009;284:28943–28952. doi: 10.1074/jbc.M109.038646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gralle M, et al. Solution conformation and heparin-induced dimerization of the full-length extracellular domain of the human amyloid precursor protein. J Mol Biol. 2006;357:493–508. doi: 10.1016/j.jmb.2005.12.053. [DOI] [PubMed] [Google Scholar]
- 23.Hoopes JT, et al. Structural characterization of the E2 domain of APL-1, a Caenorhabditis elegans homolog of human amyloid precursor protein, and its heparin binding site. J Biol Chem. 2010;285:2165–2173. doi: 10.1074/jbc.M109.018432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 25.Lee S, et al. The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry. 2011;50:5453–5464. doi: 10.1021/bi101846x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mok SS, et al. Expression and analysis of heparin-binding regions of the amyloid precursor protein of Alzheimer’s disease. FEBS Lett. 1997;415:303–307. doi: 10.1016/s0014-5793(97)01146-0. [DOI] [PubMed] [Google Scholar]
- 27.Xue Y, Lee S, Wang Y, Ha Y. The crystal structure of the E2 domain of APP-like protein 1 in complex with sucrose octasulfate. J Biol Chem. 2011;286:29748–29757. doi: 10.1074/jbc.M111.219659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulloy B, et al. The effect of variation of substitution on the solution conformation of heparin: A spectroscopic and molecular modelling study. Carbohydr Res. 1994;255:1–26. doi: 10.1016/s0008-6215(00)90968-6. [DOI] [PubMed] [Google Scholar]
- 29.Dahms SO, et al. Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc Natl Acad Sci USA. 2010;107:5381–5386. doi: 10.1073/pnas.0911326107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiGabriele AD, et al. Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature. 1998;393:812–817. doi: 10.1038/31741. [DOI] [PubMed] [Google Scholar]
- 31.Schlessinger J, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 32.Beher D, Hesse L, Masters CL, Multhaup G. Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J Biol Chem. 1996;271:1613–1620. doi: 10.1074/jbc.271.3.1613. [DOI] [PubMed] [Google Scholar]
- 33.Ho A, Südhof TC. Binding of F-spondin to amyloid-beta precursor protein: A candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci USA. 2004;101:2548–2553. doi: 10.1073/pnas.0308655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ninomiya H, Roch JM, Sundsmo MP, Otero DA, Saitoh T. Amino acid sequence RERMS represents the active domain of amyloid beta/A4 protein precursor that promotes fibroblast growth. J Cell Biol. 1993;121:879–886. doi: 10.1083/jcb.121.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1996;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallog Sect D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 38.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 39.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.