

Abstract
The mechanisms governing atlastin-mediated membrane fusion are unknown. Here we demonstrate that a three-helix bundle (3HB) within the middle domain is required for oligomerization. Mutation of core hydrophobic residues within these helices inactivates atlastin function by preventing membrane tethering and the subsequent fusion. GTP binding induces a conformational change that reorients the GTPase domain relative to the 3HB to permit self-association, but the ability to hydrolyze GTP is required for full fusion, indicating that nucleotide binding and hydrolysis play distinct roles. Oligomerization of atlastin stimulates its ability to hydrolyze GTP, and the energy released drives lipid bilayer merger. Mutations that prevent atlastin self-association also abolish oligomerization-dependent stimulation of GTPase activity. Furthermore, increasing the distance of atlastin complex formation from the membrane inhibits fusion, suggesting that this distance is crucial for atlastin to promote fusion.
Keywords: Drosophila, endoplasmic reticulum
The endoplasmic reticulum (ER) forms an elaborate network that spreads throughout the cell. The ER is a dynamic organelle, continuously undergoing membrane fusion. One of the primary functions of the ER is folding and glycosylation of secreted proteins, as well as the distribution of resident membrane proteins within the secretory pathway. Vesicular traffic exiting and entering the ER requires heterotypic membrane fusion, which uses the SNARE protein family and their associated chaperones [soluble N-ethylmaleimide-sensitive factor attachment protein receptors] for membrane merger (1). In contrast to vesicular transport, the establishment and maintenance of the ER network requires homotypic membrane fusion (2, 3). Membrane fusion occurs through an initial tethering step, which locks apposing membranes together, followed by lipid bilayer merger. Drosophila atlastin forms transoligomeric complexes between adjacent ER membranes and promotes liposome fusion in vitro, and its overexpression induces ER fusion in vivo, indicating that this GTPase is responsible for mediating ER homotypic fusion (4).
The atlastins constitute a family of very closely related, integral membrane GTPases. They are distant members of the dynamin family of GTPases and are localized on the ER membrane. Mammals have three atlastins, and mutations in ATL1 are responsible for one of the most frequent and earliest-onset forms of pure hereditary spastic paraplegia (5, 6). Human atlastins interact with the ER tubule-shaping proteins reticulons and DP1 and have been proposed to play a role in the formation of an interconnected tubular network, indirectly implicating these GTPases in the fusion of ER membranes (7). Structurally atlastins resemble mitofusins, a class of GTPases also belonging to the dynamin family essential for the homotypic fusion of mitochondria membranes (8, 9), although the ability of mitofusins to directly induce lipid bilayer merger has yet to be demonstrated (10).
Despite the identification of the dual tethering and fusion-promoting role of Drosophila atlastin, it is not known how atlastin mechanistically brings about membrane fusion. The specific role(s) of GTP binding and/or hydrolysis during atlastin-mediated ER membrane fusion have not been determined. Recent structural work has provided new information regarding a potential fusion mechanism (11, 12). The structures of the cytosolic portion of human atlastin-1 reveal a GTPase domain similar to that of GBP1 and a three-helix bundle (3HB), connected by a linker region. One of the solved structures is proposed to correspond to a prefusion state with atlastin-1 molecules, interacting through their GTPase domains to form a dimer. A second dimer, thought to correspond to a postfusion state generated after GTP hydrolysis, displays a 90° rotation of the 3HBs relative to the interacting GTPase domains.
Here we uncover key features underlying atlastin ability to promote membrane fusion that reveal unique mechanistic insights into its function. Drosophila atlastin contains a 3HB middle domain, akin to that reported for human atlastin-1, that is necessary for oligomerization. Mutations disrupting the structure of the helices within the 3HB inactivate atlastin by preventing tethering and the subsequent fusion of ER membranes. Atlastin undergoes oligomerization-dependent stimulation of GTPase activity, and mutations of essential hydrophobic residues within the 3HB prevent this effect. We show uncoupling between GTP binding, which alone is sufficient to promote atlastin self-assembly, and GTP hydrolysis required for full fusion. Furthermore, we find that increasing the distance of the middle domain from the membrane by insertion of a linker upstream of the transmembrane anchor abolishes atlastin's fusogenic properties, demonstrating that atlastin complex formation adjacent to the membrane is necessary to induce lipid bilayer merger.
Results
To investigate the structural basis of atlastin-mediated fusion, we sought to identify the domains of atlastin responsible for homooligomerization using a molecular modeling approach. Atlastin contains a tandem transmembrane domain with a long N-terminal region that includes the GTPase domain (1–422) and a short C-terminal tail, both protruding into the cytoplasm. Initial homology modeling revealed that residues 383–419 share structural similarity to helical coiled-coil proteins, including a myosin heavy chain/GCN4 bZIP domain chimera [Protein Data Bank (PDB) code: 3BAS], the bZIP domain of GCN4 (13) (PDB code: 2DGC; 32% identity), and the heptad repeat region of mitofusin (PDB code: 1T3J; 10% identity). Recent reports that the atlastin-1 middle domain folds into a 3HB and the observation that the 383–419 helix corresponds to the most C-terminal of the three helices prompted us to model atlastin on the basis of the structure of atlastin-1 (Fig. 1A). Our analysis clearly indicates that the N-terminal cytosolic region of atlastin is highly likely to adopt a conformation similar to that observed for atlastin-1. In particular, the middle region contains the predicted α-helix, and its sequence is compatible with folding as a 3HB.
Fig. 1.

3HB domain is involved in homooligomerization. (A) 3D model of the 1–422 fragment of Drosophila atlastin, based on the atlastin-1 structure. (B) Coimmunoprecipitation of atlastin deletion constructs. Schematic representation of atlastin deletion constructs. HeLa cells were simultaneously transfected with HA and myc tagged versions of each deletion construct. Lysates were immunoprecipitated using anti-myc and probed with anti-HA antibody. (C) Microsomes from HeLa cells expressing atl(328-541)-HA or atl(328-541)-myc were mixed, immunoprecipitated with anti-myc antibodies, and analyzed by Western blotting. L, lysate; S, supernatant; P, pellet.
Middle Domain Is Involved in Oligomerization and Tethering.
To identify the region responsible for mediating the physical interaction between atlastin molecules, we generated a series of deletion constructs. These constructs are atl(1-328) encompassing the GTP binding domain, atl(328-422) corresponding to the 3HB, atl(1-422), atl(423-541) including the two transmembrane domains and the C-terminal tail, and atl(328-541) (Fig. 1B). Immunofluorescence analysis of transfected HeLa cells showed that protein fragments containing the transmembrane domains display a typical ER localization without affecting ER structure, whereas all of the others were characterized by a diffuse cytoplasmic distribution (Fig. S1). To establish the ability of these proteins to self-associate, differentially tagged deletion constructs were cotransfected in HeLa cells, and lysates prepared from these cells were subjected to immunoprecipitation using anti-myc antibodies. The resulting immunoprecipitates were analyzed by Western blot using both anti-myc and anti-HA antibodies. Only atl(328-541) and atl(1-422) oligomerized (Fig. 1B). In control experiments neither atl(328-541)-HA nor atl(1-422)-HA were precipitated by anti-myc antibodies (Fig. S2A). Although atl(1-422) seems to oligomerize more weakly than atl(328-541), GTP-dependent dimerization of the N-terminal cytosolic region has been reported both for atlastin-1 and atlastin using different techniques (11, 12, 14). Consistent with our data, atl(1-328) is unable to self-associate when examined by analytical ultracentrifugation (14). These coimmunoprecipitation experiments suggest that the region 328–422, common to atl(328-541) and atl(1-422), but not the GTPase domain, is required for stable oligomerization. The fact that atl(328-422) does not oligomerize may be due to incorrect folding of this domain when removed from the context of the protein.
To establish whether oligomerization through the 3HB region leads to membrane tethering, we performed a vesicle coimmunoprecipitation assay using microsomes prepared from HeLa cells separately transfected with atl(328-541)-HA or atl(328-541)-myc. These vesicles were mixed and immunoprecipitated with anti-myc antibodies. The pellet thus obtained was positive for the anti-HA signal, indicating that the two populations of vesicles become linked by membrane-bound atl(328-541) (Fig. 1C and Fig. S2B). These results demonstrate that the 3HB region has the ability to support oligomerization and membrane tethering.
Mutation of Core Hydrophobic Residues Within the Helices of the 3HB Region Inactivates Atlastin.
To obtain further evidence of the importance of the 3HB domain for atlastin function, we designed mutations predicted to disrupt its structure by targeting critical hydrophobic residues within the three helices. We reasoned that if these helices are crucial for atlastin oligomerization, mutation of core hydrophobic residues would result in the loss of atlastin self-association properties and potentially in protein inactivation. We therefore introduced proline substitutions, known to have a severely destabilizing effect on α-helix conformation, at positions 346 in helix α7, 374 in α8, and 396 and 404 in α9 of the 3HB and examined the consequences on atlastin function.
To allow for a rapid analysis of the effects induced by atlastin variants, we established the effects of atlastin expression in human cell lines. In Drosophila, overexpression of atlastin causes the formation of expanded ER and the absence of morphologically normal Golgi, with dispersal of Golgi proteins to the ER (4). In contrast to controls (Fig. S3), transfected HeLa cells overexpressing Drosophila atlastin displayed ER spots, likely owing to enlargement caused by excessive membrane fusion, and a dispersed Golgi apparatus (Fig. 2A), indicating that spotty ER and redistribution of Golgi markers can be used as a read-out for atlastin function in mammalian cells. HeLa cell transfection with individual proline mutants results in normal ER and Golgi morphology, indicating that mutant atlastin is inactive (Fig. 2B and Fig. S4). All transfected proteins expressed at comparable levels, indicating that the observed effects are not the result of overexpression-induced toxicity (Fig. S5). We also generated transgenic Drosophila for the expression of atl(F404P). Ubiquitous expression of transgenic atlastin is embryonic lethal, and eye-specific expression gives rise to a small eye (4). In contrast, ubiquitous expression of atl(F404P) allowed normal adult survival and had no phenotypic consequences in the eye (Fig. S6), suggesting that this mutant is nonfunctional also in vivo.
Fig. 2.

F404P substitution in the α9 helix of the 3HB inhibits atlastin function. (A) Transfection of wild-type atlastin in HeLa cells results in the formation of enlarged ER and redistribution of Golgi proteins. (B) HeLa cells transfected with atl(F404P) show normal ER and Golgi morphology, indicating that this mutant is no longer capable of fusing ER membranes. PDI and GM130 were used as ER and Golgi markers, respectively. (Scale bars in A and B, 10 μm.) (C) Anti-myc immunoprecipitates from HeLa cells cotransfected with atl(F404P)-HA and atl(F404P)-myc lack atl(F404P)-HA. (D) A lysate from HeLa cells transfected with HA-tagged atl(F404P) was incubated with GTP-agarose and bound proteins analyzed by immunoblotting. L, lysate; S, supernatant; P, pellet. (E) atl(F404P) is unable to fuse proteoliposomes in vitro. In all fusion experiments the percentages represent endpoint fusion at 60 min after subtraction of the background without nucleotide. Histograms are an average of at least three experiments. Error bars represent SEM.
To test whether loss of atlastin fusogenic activity depends on the inability of the mutant protein to oligomerize, we simultaneously expressed in HeLa cells HA- and myc-tagged proline mutants and immunoprecipitated the lysates from these cells with anti-myc antibody. Western blot analysis of these immunoprecipitates demonstrated that mutant atlastin completely loses its ability to oligomerize (Fig. 2C and Fig. S4). Together these analyses suggest that atlastin contains a 3HB and that packing of 3HBs from distinct atlastin molecules is required for oligomerization.
Because all proline substitutions in the 3HB helices had identical oligomerization properties and functional behavior in cell culture, we chose to test the effects of one mutation on GTPase activity and the ability of atlastin to fuse proteoliposome (4). We found that the F404P mutation bound GTP efficiently (Fig. 2D) but displayed reduced GTPase activity (Fig. 3A) and entirely eliminated atlastin fusogenic activity in vitro (Fig. 2E). Because other large GTPases show oligomerization-dependent stimulation of GTP hydrolysis (15), this finding raised the possibility that the reduced GTPase activity of mutant atlastin could be caused by the inability to oligomerize. Because the oligomeric state of atlastin in detergent solution may not reflect accurately the organization of the protein complex inserted into the membrane, we assayed the GTPase activity of wild-type and mutant atlastin after liposome reconstitution. Consistent with our hypothesis, reconstituted atlastin showed a threefold increase in the ability to hydrolyze GTP (Fig. 3B). In contrast, atl(F404P) showed no increase of GTPase activity and after reconstitution the rate of GTP hydrolysis was essentially unchanged (Fig. 3B). These results suggest that wild-type atlastin undergoes oligomerization-dependent stimulation of GTPase activity and that replacement of crucial hydrophobic residues in the middle domain significantly distant from the GTPase active site prevents this stimulation. Thus, destabilization of the 3HB eliminates atlastin function owing to the impairment of its oligomerization ability, demonstrating that this region is essential for atlastin-mediated fusion of ER membranes.
Fig. 3.

GTPase activity of wild-type and mutant atlastin. (A) GTPase activity of recombinant wild-type and mutant atlastin in detergent. GTPase activity is represented at turnover number (μM phosphate per min per μM protein) for each protein. (B) GTPase activity of wild-type and F404P atlastin upon reconstitution. GTPase activity for wild-type atlastin in Triton X-100 (D), proteoliposome (L), or detergent solubilized liposomes (SL) is compared with the same conditions for atl(F404P). Error bars represent SEM.
GTP Binding Promotes Atlastin Oligomerization, Whereas GTP Hydrolysis Is Required for Fusion.
We have previously reported that atl(K51A), carrying a substitution in the P-loop, is unable to self-assemble and thereby loses its capacity to mediate membrane tethering and fusion (4). However, it is unclear whether nucleotide binding or hydrolysis or both are required for oligomerization because the K51A mutation prevents nucleotide binding as well as hydrolysis. We therefore asked whether GTP binding and hydrolysis might have distinct roles in atlastin-mediated fusion by examining the ability of the nonhydrolyzable analog GTPγS to support independently the two steps, tethering and lipid bilayer merger. Coimmunoprecipitation of lysates containing only endogenous GTP from HeLa cells transfected with atlastin-HA and atlastin-myc showed that atlastin-HA was coimmunoprecipitated by anti-myc antibodies (Fig. 4A). However, the addition of 1 mM GTP to the reaction resulted in a threefold increase in complex formation between atl-myc and atl-HA (Fig. 4 A and B). A similar increase was also observed after addition of GTPγS, indicating that tethering is equally robust also in the absence of hydrolysis (Fig. 4 A and B). In contrast, GTPγS is unable to promote the fusion of atl liposomes and efficiently and rapidly inhibited an ongoing fusion reaction (Fig. 4C). Therefore, GTP binding alone is sufficient to promote atlastin self-assembly, indicating that the roles of nucleotide binding and hydrolysis can be uncoupled.
Fig. 4.

GTPγS enhances atlastin self-association ability but inhibits ongoing liposome fusion. (A) Coimmunoprecipitation of transfected atlastin-HA and atlastin-myc was performed in the absence of exogenous nucleotide or presence of 1 mM GTP or GTPγS. (B) Quantification of the data from five independent coimmunoprecipitation experiments shows that the presence of GTP and GTPγS increases atlastin complex formation. (C) Addition of 0.5 mM GTPγS to an ongoing liposome fusion reaction rapidly blocks fusion. Error bars represent SEM.
To investigate this further, we replaced the arginine in position 48 with alanine in the GTP-binding domain of atlastin. An identical mutation, which results in charge neutralization, in GBP1 inhibits GTP hydrolysis without loss of GTP binding ability (16). Because arginine 48 is absent from all other GTPase families but conserved between the atlastins and GBP1, we postulated that atl(R48A) would be GTPase-deficient but retain unaltered GTP-binding ability. On the basis of our homology model of the Drosophila variant, the arginine at position 48 is not located inside the GTP binding cleft and does not seem to be directly involved in the stabilization of the GTP–atlastin complex, suggesting that it might play a cooperative role during phosphate hydrolysis. This hypothesis was confirmed by the ability of transfected atl(R48A)-myc to bind GTP-agarose efficiently. In a parallel experiment, atlastin carrying a K51A mutation did not bind to GTP-agarose (Fig. 5A). Additionally, the analysis of recombinant GST-atl (1-422) established that wild type and R48A have similar affinity for Mant-GTPγS (kd = 2.15 ± 0.39 for wild type and 4.07 ± 1.51 for R48A), whereas K51A binds Mant-GTPγS with a much weaker affinity (kd = 102.28 ± 7.44) (Fig. 5B). As predicted, full-length GST-atl(R48A) showed a reduction of the GTPase activity similar to that of other fusion-deficient mutants (Fig. 3A). As a consequence, GST-atl(R48A) fusogenic ability was eliminated both in a liposome assay (Fig. 5C) and in transfected HeLa cells where this mutant protein, expressed at levels comparable to wild-type atlastin (Fig. S5), displayed a reticular distribution and, unlike wild-type atlastin, did not induce ER fusion or the associated redistribution of Golgi (Fig. 5D). However, because atl(R48A) binds GTP normally, we predicted that its ability to form a complex would remain intact, and coimmunoprecipitation of differentially tagged atl(R48A) confirmed this prediction (Fig. 5E). These results provide strong evidence that GTP binding is necessary and sufficient to permit atlastin oligomerization. A plausible explanation for this observation is that binding of GTP induces a conformational rearrangement that facilitates the physical interaction between atlastin molecules, possibly by reorienting the middle domain and allowing assembly of 3HBs from distinct atlastin molecules.
Fig. 5.

Atl(R48A) binds GTP and retains its oligomerization properties but does not support fusion. (A) Lysates prepared from HeLa cells transfected with HA-tagged atl(R48A) or atl(K51A) were incubated with GTP-agarose, and bound proteins were analyzed by immunoblotting. (B) Increasing concentrations of atl(1-422), atl(R48A), and atl(K51A) were bound to 1 μM Mant-GTPγS. The change in Mant fluorescence upon protein binding is plotted vs. concentration of protein. (C) Atl(R48A) is inactive in a liposome fusion assay. Error bars represent SEM. (D) HeLa cells transfected with atl(R48A) display normal ER and Golgi morphology. PDI and GM130 were used as ER and Golgi markers, respectively. (Scale bar, 10 μm.) (E) Coimmunoprecipitation of differentially tagged atl(R48A) demonstrates unchanged self-association properties. L, lysate; S, supernatant; P, pellet.
Distance of Atlastin Complex Formation from the Membrane Is Critical for Fusogenic Activity.
For membranes to achieve the close apposition required to undergo fusion, the tethering complex must be able to promote membrane–membrane proximity. The 3HB middle domain is positioned four to five amino acid residues N-terminally to the transmembrane anchor. A consequence of this structural organization is that formation of transoligomers between atlastin 3HBs would bring apposed membranes into very close proximity to allow the atlastin complex to force membrane fusion. We thus examined the effect of progressively increasing the length of the juxtamembrane region of atlastin with a flexible linker containing the sequence gly-gly-ser repeated three times (17) (Fig. 6A). This sequence moves complex formation farther away from the lipid bilayer interface. We generated constructs for the expression in mammalian cells of atlastin containing one or three tandem copies of the linker, atl(1xlinker) and atl(3xlinker). Transfection of either construct did not cause alteration of ER localization or ER and Golgi morphology (Fig. 6B and Fig. S7A), suggesting that linker-containing atlastin sorted accurately but had lost its fusogenic properties. This result was further substantiated by the observation that unlike expression of wild-type atlastin, in vivo expression of transgenic atl(3xlinker) in flies resulted in viability and normal eye morphology (Fig. S6). Moreover, atl(1xlinker) was essentially unable to support liposome fusion in vitro (Fig. 6C), despite its normal GTPase activity (Fig. 3A), confirming that this mutant specifically lacks fusogenicity. To determine whether loss of fusogenicity may be due to the inability to oligomerize, we performed coimmunoprecipitation using differentially tagged linker constructs. These experiments demonstrated that complex formation ability was retained by atlastin with linker even in the most severe case (3xlinker) (Fig. 6D and Fig. S7B), suggesting that linker insertion does not prevent correct protein folding. Thus, a small increase in the separation between the 3HB and transmembrane domains induces protein inactivation without inhibiting atlastin oligomerization properties. Furthermore, limited fusion occurred when atl(1xlinker) proteoliposomes were assayed against wild-type atlastin proteoliposomes (Fig. S8), presumably because in this combination the presence of the single linker decreases the intermembrane distance, allowing bilayer merger to partly take place. These results provide evidence that the distance of atlastin complex formation from the membrane interface is an essential determinant of atlastin's fusogenic properties.
Fig. 6.

Increasing the distance between the 3HB and the transmembrane domain causes inactivation of atlastin without inhibiting its oligomerization ability. (A) Sequence of full-length atlastin is represented, and the juxtamembrane region is magnified. The distance between the 3HB and the transmembrane domains was increased by introducing one or three copies of a linker containing the sequence gly-gly-ser repeated three times. (B) ER and Golgi morphology is unperturbed in HeLa cells transfected with atl(1xlinker). PDI and GM130 were used as ER and Golgi markers, respectively. (Scale bar, 10 μm.) (C) Atl(1xlinker) lacks fusogenic activity. Error bars represent SEM. (D) atl(1xlinker)-HA is present in myc immunoprecipitates from HeLa cells cotransfected with atl(1xlinker)-myc and atl(1xlinker)-HA, indicating that this mutant protein oligomerizes normally. L, lysate; S, supernatant; P, pellet.
Discussion
In this study, we provide insights into Drosophila atlastin-mediated fusion. We identify the 3HB middle domain as the region mediating self-association. Packing of 3HBs from atlastin molecules localized on opposing membranes would result in the formation of a transcomplex responsible for membrane tethering, bringing the membranes into close enough proximity for fusion to occur. In agreement with this prediction, mutation of core hydrophobic residues within any of 3HB helices prevents atlastin dimerization, leading to the inability to fuse ER membranes in vivo and in vitro. Interaction between 3HB domains is controlled by a conformational change of the protein induced by GTP binding. In fact, oligomerization efficiently takes place in the presence of GTP or the nonhydrolyzable analog GTPγS but is prevented when GTP binding is abolished by a K51A substitution (4). In contrast, atl(R48A), a hydrolysis-specific mutant that does not support membrane fusion but maintains GTP-binding ability, allows efficient complex formation, reinforcing the hypothesis that GTP binding and GTP hydrolysis play distinct and separable roles. In contrast to tethering, full membrane fusion has a strict requirement for GTP hydrolysis, and an ongoing fusion reaction is potently and rapidly inhibited by addition of GTPγS.
The basal GTPase activity of dynamin-1 as well as that of other dynamin family members is increased upon oligomer formation (9). Likewise, after reconstitution atlastin self-assembly results in stimulation of GTPase activity. However, this stimulation is approximately threefold in contrast to a 10- to 100-fold increase observed for dynamin-related proteins. This difference may depend on the oligomerization properties of these proteins. In fact, whereas dynamin assembles into large oligomeric complexes, atlastin oligomerization is limited to dimerization (11, 12, 14, 18).
An essential determinant of atlastin fusogenic ability is the distance of complex formation from the membrane. Increasing this distance results in the uncoupling of atlastin tethering and fusogenic abilities. Indeed, atlastin containing a 10-aa linker inserted between the α9 helix and the membrane anchor is unable to fuse membranes either in HeLa cells or liposomes. However, this insertion mutant oligomerizes as efficiently as the wild-type protein, demonstrating that the length of the juxtamembrane region must be very short to allow the two membranes to come in close enough apposition for the bilayers to fuse. Attenuation but not abrogation of fusion when the assay is performed with one liposome population containing wild-type atlastin and the other atl(1xlinker) corroborates this hypothesis. Insertion of a similar linker between the transmembrane domain and the coiled-coil domain of SNAREs also decreases fusion efficiency (17), implying that the distance of complex formation from the membrane is a general critical factor for protein-mediated fusion.
Our hypothesis that stable atlastin dimerization requires the 3HB domain differs from the interpretation of the structural data on atlastin-1, which suggest that dimerization occurs through the GTPase domain. We have shown that, unlike atlastin fragments containing the middle domain, the isolated GTPase domain does not dimerize even in the presence of GTP in vitro (ref. 14 and this work). Further, atl(1-422), but not atl(1-328), acts as an inhibitor of membrane fusion in vitro and in cell culture (14). Presumably owing to the inability to oligomerize, atl(1-328) does not hydrolyze GTP, yet inclusion of the 3HB switches the protein to an efficient GTPase, likely because of newly acquired oligomerization ability (14). In vitro solution experiments found that both atlastin-1 and atlastin only dimerize in the presence of nonhydrolyzable GTP (11, 12, 14), yet all reported atlastin-1 dimer structures are GDP-bound (11, 12), making it unclear which functional states these crystals represent. Additionally, mutations within the proposed GTPase domain dimer interface of atlastin-1 do not completely eliminate dimer formation, with the exception of the unique R77E (R48 in atlastin) (11, 12). This effect of the R77E mutation is in contrast with our results for atlastin and published data for GBP1, the closest atlastin relative and the only other GTPase containing an equivalent arginine residue, whose dimerization is unaffected by substitution of this arginine with alanine (16). Interestingly, R48A replacement in both atlastin and GBP1 reduces GTPase activity without changing GTP affinity. Unlike mutation of residues at the proposed GTPase domain interface, single proline substitutions of core hydrophobic residues within any helix of the 3HB abolish dimerization, underscoring the importance of 3HB packing in this process. These considerations underscore the need for a GTP-bound, full-length atlastin structure. Alternatively, species-specific differences in protein structure may account for the observed discrepancies because atlastin-1 mediated proteoliposome fusion has yet to be demonstrated.
Our findings reveal important mechanistic insights into the functional properties of atlastin and suggest a model for atlastin-mediated homotypic fusion of ER membranes. Upon nucleotide binding, atlastin inserted within the ER membrane undergoes a conformational change that reorients the 3HB, making it available for interaction with the 3HB from a similarly primed atlastin molecule. Formation of a transcomplex induced by assembly of the 3HBs pulls the two membranes into very close apposition. The observations that in vitro fusion of atlastin liposomes is prevented in the presence of GTPγS and that the hydrolysis-specific R48A mutant is fusion-deficient show that GTP hydrolysis is required for bilayer merger and is not used to power a downstream event, such as disassembly of the atlastin complex. 3HB-mediated oligomerization promotes the interaction between GTPase domains, which in turn stimulates atlastin basal GTPase activity. The energy released after GTP hydrolysis is transduced to the lipid bilayers, resulting in their destabilization. The combination of close proximity and membrane destabilization then drives the fusion reaction. Release of the nucleotide could lead to complex disassembly, and another cycle would be prompted by binding of a new GTP molecule.
Our studies support a mode of membrane fusion that uses GTP binding to drive a conformational rearrangement that promotes membrane tethering and the chemical energy of GTP hydrolysis to merge opposing phospholipid bilayers. If confirmed, this observation will establish a new paradigm in membrane fusion.
Experimental Procedures
Molecular Modeling.
Databases.
The amino acid sequences of Drosophila atlastin and human atlastin-1 were retrieved from the protein sequence database at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). The 3D structures were obtained from the PDB (19).
Software and hardware.
Geneious Pro (version 5.4.4) and Molecular Operating Environment (MOE version 2010.10) suite were used for display and manipulation of sequences. MOE was also used for visualization and/or rendering, for homology modeling of the 3D structures, and for the stereochemical quality of the generated models. All software was run on a 16 CPU (Intel Core2 Quad CPU 2.40 GHz) Linux cluster. Default values were used for all of the parameters, unless specified otherwise.
Sequence alignment.
Sequence alignments were performed using Blom62 similarity matrix implemented by Geneious.
Homology modeling.
The homology model of the 1–422 domain of Drosophila atlastin was assembled according to the X-ray structures of the cytosolic segment of human atlastin-1 (PDB code: 3QNU). An ensemble of 55 model structures was generated. These were ranked by analysis of their packing score and contact energy, using the “Homology Modeling” tool implemented by MOE. The analysis of homology models stereochemistry was carried out using the “Protein Geometry” tools implemented by MOE.
Molecular Biology.
The atlastin full-length cDNA and the deletion constructs were amplified by PCR and cloned in the pCDNA3.1 vector in-frame with a myc or HA tag. Mutagenesis to generate point mutants was performed with the QuikChange kit (Stratagene). To generate linker insertion mutants a unique SacII site was introduced in the juxtamembrane region of pCDNA3.1-atlastin using the following primers: CAGCACGGACACCCGCGGTGTACTTC and GAAGTACACCGCGGGTGTCCGTGCTG. The annealed double-stranded oligo GGS-f (CGGTGGATCCGGTGGTTCCGGAGGTTCCGC) and GGS-r (CGGCCACCTAGGCCACCAAGGCCTCCAAGG) was cloned into the artificial SacII site in pCDNA3.1-atlastin. atl(F404P) and atlastin 3xlinker cDNAs were subcloned in the pUAST vector for P-element mediated transformation.
Drosophila Genetics.
Fly culture and transgenesis were performed according to standard procedures. Several transgenic lines for each construct were generated and tested. Drosophila strains used were GMR-Gal4 and tubulin-Gal4.
HeLa Cell Transfection, Immunocytochemistry, Immunoprecipitation, and Vesicle Immunoprecipitation Assay.
These experiments were carried out using standard protocols. Immunoprecipitation in the presence of 1 mM GTP or GTPγS was preceded by the addition of nucleotides to the cell lysate and incubation at room temperature for 1 h. To quantitate the effect of GTP and GTPγS on the ability of atlastin to oligomerize, the ratio between HA and myc signal intensity was calculated for each sample. This ratio was then normalized to the control sample (no addition of exogenous nucleotides). The final data represent the average of five experiments. The vesicle immunoprecipitation assay was performed as previously described (4). The following antibodies were used: mouse anti-myc (Cell Signaling), mouse anti-HA (Cell Signaling), mouse anti-PDI (BD Biosciences), rabbit anti-myc (Sigma Aldrich), mouse anti-GM130 (BD Biosciences), and mouse anti-tubulin (Sigma Aldrich). Secondary antibodies for immunofluorescence (Cy3 conjugate from Jackson Laboratories, Alexa Fluor 488 conjugates from Invitrogen) were used at 1:1,000. Anti-mouse HRP conjugate (Dako) was used at 1:10,000. Confocal images were acquired with a Nikon C1 confocal microscope.
In Vitro Fusion Assay and GTPase Activity Measurement.
All protein expression and purification, liposome fusion, and GTPase activity measurements were performed as previously described (4). Inhibition of liposome fusion was performed by adding 0.5 mM GTPγS to the ongoing fusion reaction.
GTP Binding.
HeLa cells expressing HA-tagged atlastin were lysed in buffer containing 50 mM Tris, 150 mM NaCl2, 10 mM MgCl2, and 0.1% TritonX-100. Cleared lysates were incubated with 100 μL of GTP-agarose beads (Innova Biosciences) overnight at 4 °C. The resin was washed three times with lysis buffer, and the proteins were resolved by SDS/PAGE and analyzed by immunoblotting using anti-HA antibodies. Bacterially expressed GST fusion proteins atl(1-422), atl R48A (1-422), and atl K51A (1-422) were purified as previously described (4). Atlastin protein, buffer [25 mM Hepes-KOH (pH 7.4), 100 mM KCl, 10% glycerol, and 2 mM 2-mercaptoethanol], 5 mM Mg2+, and 1 μM Mant-GTPγS [2′/3′-O-(N-Methyl-anthraniloyl)-guanosine-5′-(γ-thio)-triphosphate; Jena Biosciences] were mixed in a black 96-well plate and Mant fluorescence (λEx = 366 nm, λEm = 435 nm) measured at 25 °C in a TECAN Infinite M200 fluorimeter every 2 min for 20 min and averaged. The absolute fluorescence intensities were plotted vs. protein concentration and curve fitted in KaleidaGraph (Synergy Software) to determine an equilibrium Kd according to the following equation (20):
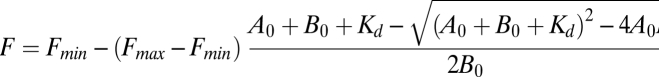 |
where Fmin is the fluorescence minimum, Fmax is the fluorescence maximum, A0 is the concentration of protein in μM, B0 is the concentration of Mant-GTPγS in μM, and Kd is the equilibrium binding constant in μM. Values for Fmin and Fmax from the wild-type protein titration were used in all three curve fits. Average Kd is (μM) ± SEM; n = 4 for wild type and K51A; n = 3 for R48A. kd measurements were carried out with at least two independent preparations of each protein.
Supplementary Material
Acknowledgments
We thank Gia Voeltz for kindly providing the GFP-Sec61β construct, Massimo Bellanda and Stefano Mammi for their preliminary analysis of the 3HB domain, and Nicoletta D'Elia for technical support. Work in the A.D. laboratory is supported by grants from the Italian Ministry of Health, the Association Française contre les Myopathies (Grant AFM 14482), and the Fondazione Telethon (Grant S00059). Work in the J.A.M. laboratory is supported by National Institutes of Health Grant GM071832 and the G. Harold and Leila Y. Mathers Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1106421108/-/DCSupplemental.
References
- 1.Cai H, Reinisch K, Ferro-Novick S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev Cell. 2007;12:671–682. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Anderson DJ, Hetzer MW. Nuclear envelope formation by chromatin-mediated reorganization of the endoplasmic reticulum. Nat Cell Biol. 2007;9:1160–1166. doi: 10.1038/ncb1636. [DOI] [PubMed] [Google Scholar]
- 3.Dreier L, Rapoport TA. In vitro formation of the endoplasmic reticulum occurs independently of microtubules by a controlled fusion reaction. J Cell Biol. 2000;148:883–898. doi: 10.1083/jcb.148.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orso G, et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- 5.Rismanchi N, Soderblom C, Stadler J, Zhu PP, Blackstone C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum Mol Genet. 2008;17:1591–1604. doi: 10.1093/hmg/ddn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao X, et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- 7.Hu J, et al. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoppins S, Nunnari J. The molecular mechanism of mitochondrial fusion. Biochim Biophys Acta. 2009;1793:20–26. doi: 10.1016/j.bbamcr.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Koshiba T, et al. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 11.Bian X, et al. Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc Natl Acad Sci USA. 2011;108:3976–3981. doi: 10.1073/pnas.1101643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrnes LJ, Sondermann H. Structural basis for the nucleotide-dependent dimerization of the large G protein atlastin-1/SPG3A. Proc Natl Acad Sci USA. 2011;108:2216–2221. doi: 10.1073/pnas.1012792108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keller W, König P, Richmond TJ. Crystal structure of a bZIP/DNA complex at 2.2 A: Determinants of DNA specific recognition. J Mol Biol. 1995;254:657–667. doi: 10.1006/jmbi.1995.0645. [DOI] [PubMed] [Google Scholar]
- 14.Moss TJ, Andreazza C, Verma A, Daga A, McNew JA. Membrane fusion by the GTPase atlastin requires a conserved C-terminal cytoplasmic tail and dimerization through the middle domain. Proc Natl Acad Sci USA. 2011;108:11133–11138. doi: 10.1073/pnas.1105056108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Praefcke GJ, McMahon HT. The dynamin superfamily: Universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 16.Praefcke GJ, et al. Identification of residues in the human guanylate-binding protein 1 critical for nucleotide binding and cooperative GTP hydrolysis. J Mol Biol. 2004;344:257–269. doi: 10.1016/j.jmb.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 17.McNew JA, Weber T, Engelman DM, Söllner TH, Rothman JE. The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol Cell. 1999;4:415–421. doi: 10.1016/s1097-2765(00)80343-3. [DOI] [PubMed] [Google Scholar]
- 18.Zhu PP, et al. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J Biol Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
- 19.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunzelmann S, Praefcke GJ, Herrmann C. Nucleotide binding and self-stimulated GTPase activity of human guanylate-binding protein 1 (hGBP1) Methods Enzymol. 2005;404:512–527. doi: 10.1016/S0076-6879(05)04045-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.