

Abstract
Spectral changes in the photocycle of the photoactive yellow protein (PYP) are investigated by using ab initio multiconfigurational second-order perturbation theory at the available structures experimentally determined. Using the dark ground-state crystal structure [Genick, U. K., Soltis, S. M., Kuhn, P., Canestrelli, I. L. & Getzoff, E. D. (1998) Nature (London) 392, 206–209], the ππ* transition to the lowest excited state is related to the typical blue-light absorption observed at 446 nm. The different nature of the second excited state (nπ*) is consistent with the alternative route detected at 395-nm excitation. The results suggest the low-temperature photoproduct PYPHL as the most plausible candidate for the assignment of the cryogenically trapped early intermediate (Genick et al.). We cannot establish, however, a successful correspondence between the theoretical spectrum for the nanosecond time-resolved x-ray structure [Perman, B., Šrajer, V., Ren, Z., Teng, T., Pradervand, C., et al. (1998) Science 279, 1946–1950] and any of the spectroscopic photoproducts known up to date. It is fully confirmed that the colorless light-activated intermediate recorded by millisecond time-resolved crystallography [Genick, U. K., Borgstahl, G. E. O., Ng, K., Ren, Z., Pradervand, C., et al. (1997) Science 275, 1471–1475] is protonated, nicely matching the spectroscopic features of the photoproduct PYPM. The overall contribution demonstrates that a combined analysis of high-level theoretical results and experimental data can be of great value to perform assignments of detected intermediates in a photocycle.
How photosensors absorb visible light to initiate signaling, which ultimately leads to a biological response, is one of the essential issues in photobiology. It is also worth mentioning that, apart from its own intrinsic importance, a detailed molecular knowledge of the mechanism of photosensing could be the source of inspiration to develop new devices of current technological interest. After absorption of a photon by the chromophoric group, the photoactive proteins undergo a series of dark reactions through a number of intermediates, resulting in the reformation of the initial state of the protein. The overall process is known as a photocycle. Changes in the position of the absorbance maximum and/or in the magnitude of the molar extinction coefficient usually are associated with such a photocycle. However, spectroscopic information for the intermediates is in certain cases difficult to extract from the recorded data. First, the mixture of different intermediates cannot be clearly separated in most of the usual experimental conditions. Second, the absorbance spectra of the species involved often show large overlap, both spectrally and temporally. As shall be illustrated here, in those cases, a combined analysis of theoretical results derived from quantum-chemical calculations and experimentally derived data can be of great value in advancing and/or complementing the spectroscopic behavior of the intermediates in a photocycle.
Photoactive yellow protein (PYP) is probably the most extensively examined photoreceptor. The low size of the protein and its water solubility have made investigations easy. PYP has been isolated from three halophilic phototrophic purple bacteria (1–3). Their negative phototactic response to intense blue light is attributed to this cytosolic protein. In the family of eubacterial blue-light photoreceptors, the PYP from Ectothiorhodospira halophila is the best-studied member. PYP has a unique chromophore, p-coumaric acid (4-hydroxycinnamic acid), covalently bound to the only cysteine residue in the polypeptide chain (Cys-69) via a thioester linkage (4). PYP undergoes a series of transformations induced by light that are remarkably similar in their photochemical properties to those of the sensory rhodopsins (5–7). In analogy with rhodopsins, the family of photoactive yellow proteins is called xanthopsins (8).
To clarify some aspects of the photocycle of PYP, a comprehensive ab initio study on its electronic spectra has been carried out. The results derived from the investigation are presented here. The high-resolution PYP structures available (9–11) allow quantum-chemical studies that can help to provide the prototype for comparison to other protein photocycles and signaling processes. The electronic states have been characterized by using the complete active space perturbation theory to second order (CASPT2) (12, 13), with the indirect interaction of the resulting states taken into account within the framework of its multistate extension (MS-CASPT2) (14). The successful performance of the theoretical approach in computing spectroscopic properties is well established (15–17). It yields quantitatively accurate results, that is, states are obtained in correct order with correct character and oscillator strengths. In most cases the excitation energies are within ± 0.2 eV of the experimental estimates (when comparison with experimental data is appropriate). Particularly connected to the present study are the investigations carried out on the trans-cis photoisomerization of retinal (18, 19), stilbene (20, 21), and styrene (22, 23), as well as the determination of the electronic spectra of anions (24, 25).
A number of key questions have been addressed in this theoretical investigation. What type of low-lying excited states has the chromophore of PYP? In particular, is the nπ* state placed above or below the lowest ππ* state? How do they contribute to the photocycle of PYP? Can the computed electronic spectra be used as fingerprints to identify a given intermediate? Finally, how does the protonation/deprotonation of the chromophore affect the electronic spectra? We believe that the answers to these questions can give further insight into the knowledge of PYP.
The Photocycle of the PYP: A Brief Outlook
Upon blue-light irradiation at 446 nm, PYP enters a photocycle, initiated by electronic excitation of the ground state (PYPG). Chronologically, two transient species were first detected sequentially by flash photolysis at room temperature, a red-shifted intermediate (PYPL), in the nanosecond time scale after irradiation, and a blue-shifted intermediate (PYPM) in the millisecond time scale (5–7) (see Scheme S1a). The term bleaching of protein is associated with the formation of the colorless PYPM, because its absorption maximum moves out of the visible range of electromagnetic radiation. Because of the relatively long lifetime, PYPM is assumed to be the signaling species (9, 26). The original conformation of the (dark) ground state (PYPG) is recovered with a rate constant of about 2–3 s−1 (9).
Scheme 1.

(a) Photoproducts observed at room temperature (7). (b) Species detected at low temperature in glycerol (27) (see text).
Additional intermediates in the early events of the photocycle have been detected by low-temperature spectroscopy. Imamoto et al. (27) have proposed two pathways from electronically excited PYPG, both leading to the formation of PYPL. Those authors have found that irradiation of PYPG at −190°C produced a photo-steady-state mixture composed of bathochromic (PYPB) and hypsochromic (PYPH) photoproducts (see Scheme S1b). Upon warming, they were thermally converted to the blue-shifted intermediates PYPBL and PYPHL. The PYPL intermediate thermally reverted to PYPG above −50°C, completing the photocycle. In the recovery process at low temperature PYPM has not been detected. This behavior has been ascribed to the effects of temperature and the presence of glycerol (27).
More recent studies using picosecond (28) and femtosecond (29)
spectroscopic techniques at room temperature also have revealed the
presence of two early photochemical intermediates
(Io and
I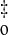 ) exhibiting absorption maxima around
510 nm, close to the spectroscopic feature of
PYPB at low temperature (27). In addition,
Devanathan et al. (29) have detected an alternative route
for PYP excitation and photochemistry initiated by excitation at 395
nm, on the blue edge of the observed absorption band. They have
suggested that it presumably involves a different excited state of the
chromophore. As shall be seen below, their suggestion is fully
supported by the present findings.
) exhibiting absorption maxima around
510 nm, close to the spectroscopic feature of
PYPB at low temperature (27). In addition,
Devanathan et al. (29) have detected an alternative route
for PYP excitation and photochemistry initiated by excitation at 395
nm, on the blue edge of the observed absorption band. They have
suggested that it presumably involves a different excited state of the
chromophore. As shall be seen below, their suggestion is fully
supported by the present findings.
Model Compound and Geometries
From the structural standpoint, the photocycle of PYP involves trans-cis isomerization of the yellow anionic chromophore (30). The crystal structure obtained in the absence of light, which corresponds to PYPG (9–11), has the chromophore buried in a hydrophobic pocket with a trans conformation. The negative charge of the dark form is stabilized by a network of hydrogen bonds.
An early protein photocycle intermediate has been cryogenically trapped (11). The structure at 0.85-Å resolution has revealed that the 4-hydroxy-cinnamoyl anion chromophore isomerizes by flipping its thioester linkage with the protein. Hence, collisions resulting from large-scale movement of the aromatic ring are avoided during the initial light reaction. The protein reaches a distorted geometry, with a perpendicularly twisted ethenic bond, and the stored energy is used to drive the PYP light cycle. The observed light-activated structure has been tentatively assigned by Genick et al. (11) to the early intermediate denoted PYPBL by Imamoto et al. (27) (Scheme S1b). As discussed below, a more plausible candidate can be inferred from the present spectroscopic results.
A structure determined by nanosecond time-resolved x-ray crystallography (10) has been assigned to the spectroscopically observed intermediate PYPL, which develops in solution within 1 ns after electronic excitation (5–7). It should be noted, however, that the assignment performed by Perman et al. (10) is not confirmed by the present contribution. In the crystal structure, the chromophore has cis conformation and remains in its binding pocket. The center of its aromatic moiety does not move, although the ring rotates in its plane. The trans-to-cis photoisomerization process is accompanied by the specific formation of new hydrogen bonds that replace those broken upon excitation of the chromophore. The reverse photoreaction (PYPL→PYPG) also occurs but it is much less efficient (31).
The colorless long-lived PYPM intermediate has been determined by millisecond time-resolved crystallography (9), with the chromophore in a cis conformation. During the bleaching of the protein an arginine gateway opens, allowing solvent exposure and protonation of the phenolic oxygen to occur. Nevertheless, that the amino acid residue Glu-46 acts as the actual proton donor to the chromophore has been seen with IR techniques (32, 33). The cycle is completed when the PYPG structure is recovered by means of a protein-mediated thermal process (34).
The selected model for the ab initio computation is the methyl thioester of the chromophore, namely, CH3—S—CO—CH⩵CH—(C6H4)—O−. The 0.85-Å resolution data have been used (11) for dark ground state and the early protein intermediate, denoted as structures I and II hereafter. Taking the coordinates from the crystal structures developed in the nanosecond (10) and millisecond (9) time scales, structures III and IV have been built. Two additional molecules have been made by protonating (III.P) and deprotonating (IV.DP) the phenolic oxygen from structures III and IV, respectively. Requests for explicit coordinates can be addressed to the authors.
Computational Details
Generally, contracted basis sets of atomic natural orbital type have been used for expanding the molecular orbitals (MOs) (35, 36). From the S(17s12p5d)/C,O(14s9p4d)/H(8s4p) primitive sets, the contraction scheme S[4s3p1d]/C,O[3s2p1d]/H[2s1p] has been used. The reference wave functions and the (MOs) are obtained from average complete active space self-consistent field (CASSCF) calculations (37). The active space comprises 12 electrons in 10 orbitals, including the relevant MOs to describe the low-lying electronic excited states. In a subsequent step, the remaining electron correlation effects have been taken into account by using MS-CASPT2. The CASPT2 method (12, 13) involves a second-order perturbation treatment on a multiconfigurational CASSCF wave function. At the MS-CASPT2 level (14), the considered states are allowed to interact under the influence of dynamic correlation.
Perturbation-modified CAS reference functions (the model states) (14), i.e., linear combinations of all CASs involved in the MS-CASPT2 calculation, have been used to compute the corresponding transition dipole moments according to the CAS state interaction (CASSI) protocol (38, 39).
All calculations were performed on a Cray-SGI Origin 2000 computer at the University of Valencia by using the MOLCAS-4 quantum-chemical package (40).
Results and Discussion
We have focused on certain aspects of the PYP photocycle where the theoretical study of the electronic excited states may yield relevant information. First, the initial step of the process is itself an electronic excitation by blue light. Thus, using structure I, characterization of the involved excited states and computation of the vertical transition energies and the corresponding oscillator strengths have been basic objectives of the investigation. Second, the accurate performance and predictive character of the method used makes it possible for a reliable assignment of the cryogenically trapped early intermediate (11), structure II. Third, PYPL is the only intermediate, together with the ground-state structure PYPG, which has been observed in both low- and room-temperature spectroscopic studies. It seems therefore appropriate to undertake a theoretical spectroscopic study of structure III. Finally, the influence of protonation/deprotonation of the chromophore during the bleaching process is investigated through structures IV and IV.DP, related to PYPM (9).
The computed vertical transitions and related oscillator strengths are compiled in Table 1. For the sake of comparison, selected experimental data are also included.
Table 1.
Computed excitation energies and oscillator strengths (f)
| State | MS-CASPT2 | f | Experimental data |
|---|---|---|---|
| Structure I | |||
| S1 | 2.58 eV | 0.567 | 446 nm (2.78 eV)* |
| S2 | 2.95 eV | <10−3 | 395 nm (3.14 eV)† |
| S3 | 3.64 eV | 0.070 | |
| Structure II | |||
| S1 | 1.24 eV | 0.161 | |
| S2 | 2.59 eV | 0.115 | 447 nm (2.77 eV)‡ |
| S3 | 2.91 eV | 0.013 | |
| Structure III | |||
| S1 | 1.29 eV | 0.122 | |
| S2 | 1.70 eV | 0.101 | |
| S3 | 1.86 eV | 0.008 | |
| S4 | ≈2.9 eV | <10−3 | |
| Structure III.P | |||
| S1 | 1.34 eV | 0.001 | |
| S2 | 1.94 eV | 0.008 | |
| S3 | 3.02 eV | 0.013 | |
| Structure IV.DP | |||
| S1 | 2.03 eV | 0.427 | |
| S2 | 3.06 eV | 0.001 | |
| S3 | 3.47 eV | 0.056 | |
| Structure IV | |||
| S1 | 3.58 eV | 0.105 | 355 nm (3.49 eV)* |
| S2 | 3.71 eV | 0.018 | |
| S3 | 4.23 eV | 0.214 | |
Electronic Spectrum of the Ground-State Structure.
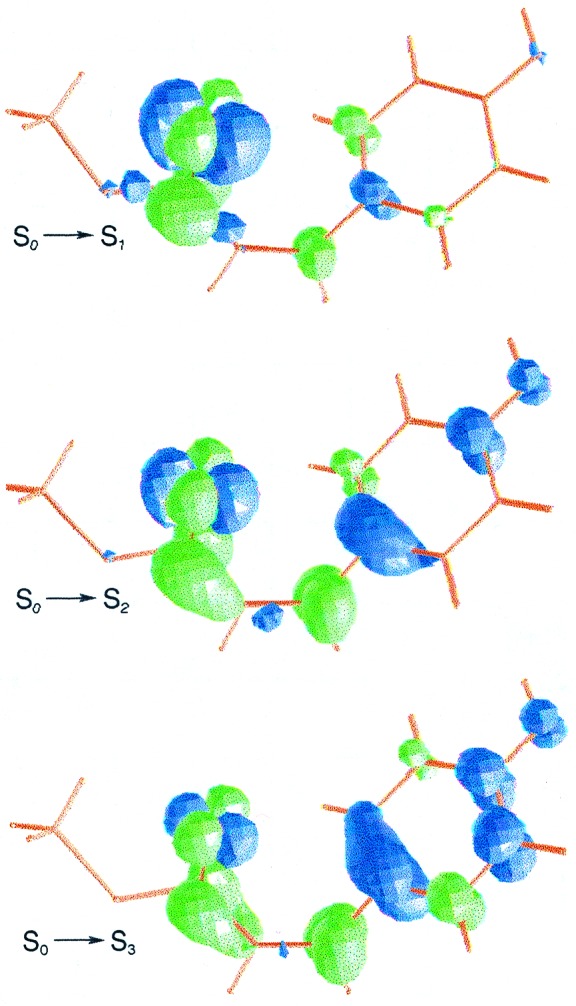
The computed excitation energies for structure I and the available experimental data for PYPG can be unambiguously related within 0.2 eV. That shift can be ascribed to the inherent differences between the theoretical and experimental investigations (limitations of the methods, possible effects due to the geometry, surrounding protein, solvent, etc.). Transition to the lowest singlet excited-state S1 appears at 2.58 eV with a relatively high oscillator strength, in agreement with the observed absorbance maxima (1, 6, 7). The present finding supports the only previous theoretical result reported for PYP, as far as we know, at the semiempirical level (41). The electronic promotion induced by light has been characterized by means of the differential electron density between the ground and the excited state. As shown in Fig. 1, the lowest excited state has a clear ππ* nature: an overall electronic shift from the phenolate oxygen to the carbonyl group takes place (electron density comes from darker to lighter areas).
Figure 1.
Computed differential electron density for the vertical electronic transitions of structure I.
The second singlet excited state, S2, placed at 2.95 eV, has nπ* character and is predicted at the high-energy side of the intense absorption band with an exceedingly weak intensity (compare Table 1). As can be seen in Fig. 1, the lone-pair orbital mainly belongs to the phenolate oxygen. In addition, the ππ* electronic transition to S3 is found to be localized within the phenolate group. The S3 state is computed at 3.64 eV above the ground state and the corresponding oscillator strength associated with the transition is small (0.07). No direct experimental evidence is available for the nπ* and the highest ππ* states. It is worth mentioning, however, that an alternative route for PYP photochemistry has been observed from 395-nm excitation (3.14 eV) (29). Based on the MS-CASPT2 results, it can be related to the second excited state vertically computed. Therefore, the distinct behavior observed at 446-nm and 395-nm excitation can be ascribed to the different nature of the two low-lying excited states, S1 and S2, ππ* and nπ*, respectively.
The agreement between theoretical results and experimental data for PYPG seems to point out that the interaction between protein and chromophore, which has been neglected here, is similar in both ground and excited states. Indeed, the effect of mutants on excitation energies has been at most recorded to be 0.1 eV, for instance, when Glu-46 is replaced by Gln (34). On the other hand, in a recent theoretical study of the phenolate anion in the environment of PYP, He et al. (42) have shown that the considered hydrogen bonds do not significantly affect the excitation energies of the protein-bound phenolate anion.
Identification of the Early Intermediate.
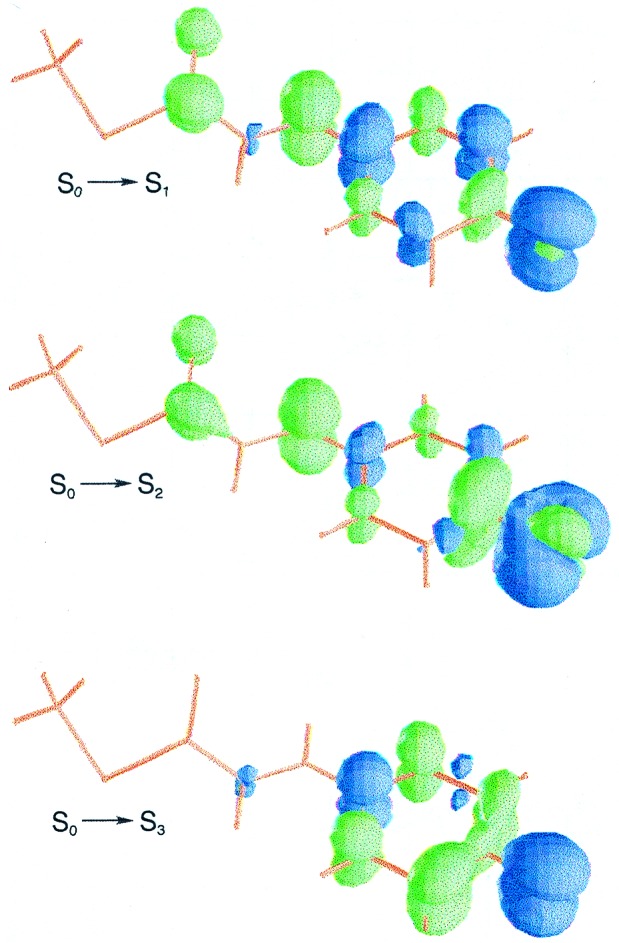
The chromophore of the cryotrapped intermediate (11) has the two lowest electronic transitions at 1.24 and 2.59 eV with similar oscillator strengths (compare Table 1). A relatively weaker electronic transition is also predicted at 2.91 eV. As shown in Fig. 2, electron density is clearly provided by the π orbital of the carbonyl group in the S0→S1 transition. For the vertical excitations to S2 and S3 the nπ* one-electron promotion (n corresponding to the lone pair orbital of the carbonyl group) also plays a relevant role.
Figure 2.
Computed differential electron density for the vertical electronic transitions of structure II.
There are four possible candidates, in principle, to be related to the cryotrapped intermediate. The photoproducts (PYPB, PYPH, PYPBL, and PYPHL) have been spectroscopically detected at low temperature. As shown in Scheme S1, within the PYP photocycle two possible pathways from electronically excited PYPG converging to PYPL can occur, involving two transients each. The mixture of photoproducts can be either (PYPB, PYPH) or (PYPBL, PYPHL) depending on temperature conditions (27). Such considerations, together with preliminary spectroscopic studies of PYP crystals, led Genick et al. (11) to conclude that the observed light-activated structure corresponds to the early intermediate termed PYPBL. Nevertheless, even taking into account a similar theoretical-experimental shift as it occurs for dark structure I, the observed band maximum for PYPBL (27) (3.10 eV) is too high (around 0.5 eV) with respect to the computed electronic transition S0→S2, predicted with significant intensity at 2.59 eV. The present results support instead that PYPHL (27), with a band maximum at 2.77 eV, corresponds to the actual cryotrapped structure in the early events of the photocycle (11). Moreover, in agreement with the energy difference between the recorded band maxima of PYPG and PYPHL (see Scheme S1), the computed electronic transitions around the 446-nm region for structures I and II are found to be within 0.01 eV. The formation of the 400-nm shoulder on the PYPG spectrum from light-activated PYP crystals (11) is also consistent with the weak S0→S3 electronic transition computed at 2.91 eV for the chromophore of the early intermediate. Furthermore, the theoretical results suggest that the monitoring of the early intermediate could be achieved by scanning the energy region 800–1,000 nm, where no absorption overlap with the dark ground-state structure occurs.
Nanosecond Time-Resolved X-Ray Structure.
Despite the fact that both the PYPL intermediate in solution (1, 6, 7, 27) and the crystal structure reported by Perman et al. (10) develop in the nanosecond time scale, we cannot establish a relationship between the electronic spectrum described theoretically for structure III and the recorded feature for PYPL (≈2.7 eV).
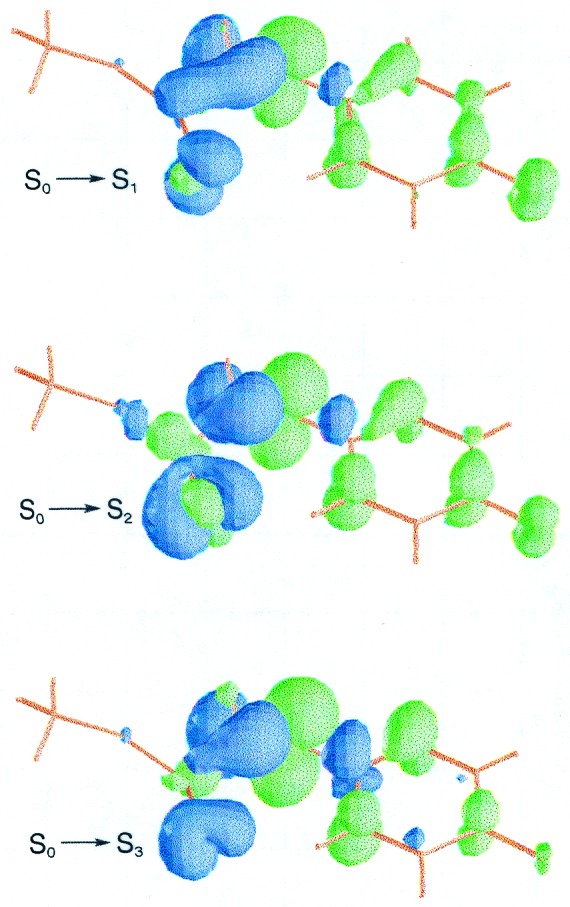
The crystal structure of the chromophore (10) offers two differential characteristics that markedly determine its spectral properties. The thioester linkage is stretched to a linear geometry and the carbonyl group is bent into a near perpendicular conformation with respect to the plane defined by the surrounding atoms. As can be seen in Fig. 3, the S0→S1 and S0→S3 transitions of the anionic system are described mainly by electronic promotions from the sulfur lone pairs to the π* molecular orbital of the carbonyl group. The S1 and S3 excited states are placed at 1.29 and 1.86 eV, respectively. The S2 state lies at 1.70 eV, with the corresponding electronic transition involving an electronic shift from the phenolate fragment to the carbonyl group. In addition, the weak S0→S4 transition has been estimated around 2.9 eV. Therefore, the main features for structure III are predicted below 1.7 eV, well separated from the typical absorption band of PYPL.
Figure 3.
Computed differential electron density for the vertical electronic transitions of structure III.
The possibility that the chromophore could actually be protonated at this stage also has been examined through structure III.DP. The electronic transitions to S1, S2, and S3 are computed at 1.34, 1.94, and 3.02 eV, respectively. They are slightly higher in energy than the corresponding promotions to S1, S3, and S4 of structure III, having a similar nature. Smaller oscillator strengths are found, however, for the protonated structure (III.P). Thus, it might be an intermediate especially difficult to detect by standard absorption spectroscopy.
We conclude that the nanosecond time-resolved crystal structure cannot be related to the PYPL intermediate. A time-resolved spectroscopical study toward lower energy regions might yield new light on the species involved in the photocycle.
Protonation Effects in the Bleaching Process.
The main feature in the theoretical spectrum of the cis anionic form (IV.DP) lies in the visible range, as it occurs in the trans structure I. Transition from the ground state to the lowest excited state is computed at 2.03 eV for structure IV.DP, around 0.5 eV red-shifted with respect to structure I. The S2 and S3 excited states are placed at 3.06 and 3.47 eV, respectively. The nature, energy ordering, and oscillator strengths of the three electronic transitions is similar in both trans and cis anionic models.
The low-lying excited states of the neutral protonated system (IV) are computed out of the visible range. The lowest singlet excited state has a local nπ* character within the carbonyl group (see Fig. 4). Accordingly, it is predicted with a relatively low intensity. The S1 state is placed at 3.58 eV, consistent with the recorded band maximum of the spectrum (3.49 eV7). The agreement with experiment is surprisingly good, especially taking into account that PYPM exists in solution as a family of multiple conformers that exchange on a millisecond time scale (43). The vertical excitation to S2 is predicted as a weak transition on the blue edge of the lowest-energy band. The main feature of the theoretical spectrum in the studied range is computed at 4.23 eV. To summarize, theory confirms that protonation of the phenolic oxygen actually moves the absorption spectra out of the visible range.
Figure 4.
Computed differential electron density for the vertical electronic transitions of structure IV.
Final Remarks
Whether the photoproducts spectroscopically detected up to date can be unambiguously related to the recorded light-activated x-ray structures has been the major underlying reason for this study. By combining the computational evidence and the existing spectroscopic data we conclude that the assignments previously proposed for the early intermediates should be revised. This issue could be clarified by carrying out spectroscopic measurements in the IR/visible borderline. In this energy region theory predicts no absorption overlap with the spectra of the dark ground-state structure. The lowest excited state plays a fundamental role in the PYP photocycle.
Quantum-chemical calculations effectively helped in assigning spectra and guiding experiments in photobiology long ago. The current contribution brings an additional example that demonstrates a constructive interplay between experiment and theory, providing insights into the electronic states that cannot be easily derived from the recorded spectra alone. We believe that the protocol followed here could be successfully applied to related challenging issues in the realm of photobiology.
Acknowledgments
We thank Björn O. Roos for suggesting the topic 3 years ago. More recently, Massimo Olivucci reminded us of it. This research has been supported by Dirección General de Enseñanza Superior Project PB97–1377 of Spain and by the European Commission through Training and Mobility of Researchers Network Contract ERB FMRX-CT96–0079 (Quantum Chemistry for the Excited State). V.M. also thanks the Generalitat Valenciana for a personal grant.
Abbreviations
- PYP
photoactive yellow protein
- CASPT2
complete active space perturbation theory to second order
- MS-CASPT2
multistate CASPT2
References
- 1.Meyer T E. Biochim Biophys Acta. 1985;806:175–183. doi: 10.1016/0005-2728(85)90094-5. [DOI] [PubMed] [Google Scholar]
- 2.Meyer T E, Tollin G, Causgrove T P, Cheng P, Blankenship R E. Biophys J. 1991;59:988–991. doi: 10.1016/S0006-3495(91)82313-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koh M, van Driessche G, Samyn B, Hoff W D, Meyer T E, Cusanovich M A, van Beeumen J J. Biochemistry. 1996;35:2526–2534. doi: 10.1021/bi951494t. [DOI] [PubMed] [Google Scholar]
- 4.Baca M, Borgstahl G E O, Boissinot M, Burke P M, Williams D R, Slater K A, Getzoff E D. Biochemistry. 1994;33:14369–14377. doi: 10.1021/bi00252a001. [DOI] [PubMed] [Google Scholar]
- 5.Meyer T E, Yakali E, Cusanovich M A, Tollin G. Biochemistry. 1987;26:418–423. doi: 10.1021/bi00376a012. [DOI] [PubMed] [Google Scholar]
- 6.Meyer T E, Tollin G, Hazzard J H, Cusanovich M A. Biophys J. 1989;56:559–564. doi: 10.1016/S0006-3495(89)82703-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoff W D, van Stokkum I H M, van Ramesdonk H J, van Brederode M E, Brouwer A M, Fitch J C, Meyer T E, van Grondelle R, Hellingwerf K J. Biophys J. 1994;67:1691–1705. doi: 10.1016/S0006-3495(94)80643-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kort R, Hoff W D, van West M, Kroon A R, Hoffer S M, Vlieg K H, Crielaard W, van Beeumen J J, Hellingwerf K J. EMBO J. 1996;15:3209–3218. [PMC free article] [PubMed] [Google Scholar]
- 9.Genick U K, Borgstahl G E O, Ng K, Ren Z, Pradervand C, Burke P M, Šrajer V, Teng T, Schildkamp W, McRee D E, et al. Science. 1997;275:1471–1475. doi: 10.1126/science.275.5305.1471. [DOI] [PubMed] [Google Scholar]
- 10.Perman B, Šrajer V, Ren Z, Teng T, Pradervand C, Ursby T, Bourgeois D, Schotte F, Wulff M, Kort R, et al. Science. 1998;279:1946–1950. doi: 10.1126/science.279.5358.1946. [DOI] [PubMed] [Google Scholar]
- 11.Genick U K, Soltis S M, Kuhn P, Canestrelli I L, Getzoff E D. Nature (London) 1998;392:206–209. doi: 10.1038/32462. [DOI] [PubMed] [Google Scholar]
- 12.Andersson K, Malmqvist P-Å, Roos B O, Sadlej A J, Wolinski K. J Phys Chem. 1990;94:5483–5488. [Google Scholar]
- 13.Andersson K, Malmqvist P-Å, Roos B O. J Chem Phys. 1992;96:1218–1226. [Google Scholar]
- 14.Finley J, Malmqvist P-Å, Roos B O, Serrano-Andrés L. Chem Phys Lett. 1998;288:299–306. [Google Scholar]
- 15.Roos B O, Fülscher M P, Malmqvist P-Å, Merchán M, Serrano-Andrés L. In: Quantum Mechanical Electronic Structure Calculations with Chemical Accuracy. Langhoff S R, editor. Dordrecht, The Netherlands: Kluwer; 1995. pp. 357–431. [Google Scholar]
- 16.Roos B O, Andersson K, Fülscher M P, Malmqvist P-Å, Serrano-Andrés L, Pierloot K, Merchán M. In: Advances in Chemical Physics: New Methods in Computational Quantum Mechanics. Prigogine I, Rice S A, editors. XCIII. New York: Wiley; 1996. pp. 219–331. [Google Scholar]
- 17.Merchán M, Serrano-Andrés L, Fülscher M P, Roos B O. In: Recent Advances in Multireference Methods. Hirao K, editor. Vol. 4. New York: World Scientific; 1999. pp. 161–195. [Google Scholar]
- 18.Merchán M, González-Luque R. J Chem Phys. 1997;106:1112–1122. [Google Scholar]
- 19.González-Luque R, Garavelli M, Bernardi F, Merchán M, Robb M A, Olivucci M. Proc Natl Acad Sci USA. 2000;97:9379–9384. doi: 10.1073/pnas.97.17.9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molina V, Merchán M, Roos B O. J Phys Chem A. 1997;101:3478–3487. [Google Scholar]
- 21.Molina V, Merchán M, Roos B O. Spectrochim Acta. 1999;55A:433–446. [Google Scholar]
- 22.Molina V, Smith B R, Merchán M. Chem Phys Lett. 1999;309:486–494. [Google Scholar]
- 23.Molina V, Merchán M, Roos B O, Malmqvist P-A. Phys Chem Chem Phys. 2000;2:2211–2217. [Google Scholar]
- 24.Rubio M, Merchán M, Ortí E, Roos B O. J Phys Chem. 1995;99:14980–14987. [Google Scholar]
- 25.Pou-Amérigo R, Serrano-Andrés L, Merchán M, Ortí E, Forsberg N. J Am Chem Soc. 2000;122:6067–6077. [Google Scholar]
- 26.Hellingwerf K J, Hoff W D, Crielaard W. Mol Microbiol. 1996;21:683–693. doi: 10.1046/j.1365-2958.1996.411402.x. [DOI] [PubMed] [Google Scholar]
- 27.Imamoto Y, Kataoka M, Tokunaga F. Biochemistry. 1996;35:14047–14053. doi: 10.1021/bi961342d. [DOI] [PubMed] [Google Scholar]
- 28.Ujj L, Devanathan S, Meyer T E, Cusanovich M A, Tollin G, Atkinson G H. Biophys J. 1998;75:406–412. doi: 10.1016/S0006-3495(98)77525-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devanathan S, Pacheco A, Ujj L, Cusanovich M, Tollin G, Lin S, Woodbury N. Biophys J. 1999;77:1017–1023. doi: 10.1016/S0006-3495(99)76952-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Changenet P, Zhang H, van der Meer M J, Hellingwerf K J, Glasbeck M. Chem Phys Lett. 1998;282:276–282. [Google Scholar]
- 31.Gensch T, Hellingwerf K J, Braslavsky S E, Schaffner K. J Phys Chem A. 1998;102:5398–5405. [Google Scholar]
- 32.Xie A, Hoff W D, Kroon A R, Hellingwerf K J. Biochemistry. 1996;35:14671–14678. doi: 10.1021/bi9623035. [DOI] [PubMed] [Google Scholar]
- 33.Imamoto Y, Mihara K, Hisatomi O, Kataoka M, Tokunaga F, Bojgova N, Yoshihara K. J Biol Chem. 1997;272:12905–12908. doi: 10.1074/jbc.272.20.12905. [DOI] [PubMed] [Google Scholar]
- 34.Genick U K, Devanathan S, Meyer T E, Canestrelli I L, Williams E, Cusanovich M A, Tollin G, Getzoff E D. Biochemistry. 1997;36:8–14. doi: 10.1021/bi9622884. [DOI] [PubMed] [Google Scholar]
- 35.Widmark P-O, Malmqvist P-Å, Roos B O. Theor Chim Acta. 1990;77:291–306. [Google Scholar]
- 36.Widmark P-O, Persson B J, Roos B O. Theor Chim Acta. 1991;79:419–432. [Google Scholar]
- 37.Roos B O. In: Advances in Chemical Physics, Ab Initio Methods in Quantum Chemistry II. Lawley K P, editor. Chichester, U.K.: Wiley; 1987. pp. 399–445. [Google Scholar]
- 38.Malmqvist P-Å. Int J Quant Chem. 1986;30:479–494. [Google Scholar]
- 39.Malmqvist P-Å, Roos B O. Chem Phys Lett. 1989;155:189–194. [Google Scholar]
- 40.Andersson K, Blomberg M R A, Fülscher M P, Karlstöm G, Lindh R, Malmqvist P-Å, Neogrády P, Olsen J, Roos B O, Sadlej A J, et al. molcas. Lund, Sweden: Univ. of Lund; 1997. , Version 4.0. [Google Scholar]
- 41.Yamato T, Niimura N, Go N. Proteins Struct Funct Genet. 1998;32:268–275. doi: 10.1002/(sici)1097-0134(19980815)32:3<268::aid-prot2>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 42.He Z, Martin C H, Birge R, Freed K F. J Phys Chem A. 2000;104:2939–2952. [Google Scholar]
- 43.Rubinstenn G, Vuister G W, Mulder F A A, Düx P E, Boelens R, Hellingwerf K J, Kaptein R. Nat Struct Biol. 1998;5:568–570. doi: 10.1038/823. [DOI] [PubMed] [Google Scholar]