

Abstract
Rationale
Calcium/calmodulin dependent protein kinase II (CaMKII) is a key mediator of intracellular signaling in the heart. However, the tools currently available for assessing dynamic changes in CaMKII localization and activation in living myocytes are limited.
Objective
Here we use Camui, a novel FRET based biosensor in which full length CaMKII is flanked by CFP and YFP, to measure CaMKII activation state in living rabbit myocytes.
Methods and Results
We show that Camui and mutant variants which lack the sites of CaMKII autophosphorylation (T286A) and oxidative regulation (CM280/1VV) serve as useful biosensors for CaMKIIδ activation state. Camui (WT or mutant) was expressed in isolated adult cardiac myocytes and localization and CaMKII activation state were determined using confocal microscopy. Camui, like CaMKIIδ is concentrated at the z-lines, with low baseline activation state. Camui activation increased directly with pacing frequency, but the maximal effect was blunted with the T286A, consistent with frequency dependent phosphorylation of CaMKII at T286 mainly at high frequency and amplitude Ca transients. Camui was also activated by four neurohormonal agonists. Angiotensin II and endothelin-1 activated Camui, largely via an oxidation-dependent mechanism, while isoproterenol and phenylephrine mediated mechanisms had a significant autophosphorylation-dependent component.
Conclusions
Camui is a novel, non-destructive tool that allows spatio-temporally resolved measurement of CaMKII activation state in physiologically functioning myocytes. This represents a first step in using Camui to elucidate key mechanistic details of CaMKII signaling in live hearts and myocytes.
Keywords: CaMKII, biosensor, FRET
INTRODUCTION
The multifunctional calcium/calmodulin (Ca2+/CaM) dependent protein kinase II (CaMKII) translates numerous intracellular signals into downstream physiological effects. This plasticity of function derives from the unique structural features of CaMKII. Recent crystal structures show that the holoenzyme assembles as a dodecamer comprised of two stacked rings.1 Individual monomers feature three domains, a C-terminal association domain that directs dodecamer assembly, an N-terminal catalytic domain that interacts with substrates and performs kinase function, and a central regulatory domain that modulates the activity of CaMKII. In resting conditions, the regulatory domain forms a tight association with the catalytic domain, preventing substrate binding. When [Ca2+] is elevated, Ca2+/CaM binds to the regulatory domain of CaMKII and induces a conformational shift that disrupts association with the catalytic domain, resulting in a shift from an autoinhibited to an active state.1,2
In prolonged conditions of elevated Ca2+, intersubunit autophosphorylation occurs at the T286 residue (or T287, the numbering is isoform specific). Phosphorylation at T286 prevents the reassociation of the regulatory and catalytic domains,3,4 resulting in autonomous CaMKII activity that persists in the absence of Ca2+/CaM. The ability to shift from Ca2+-dependent to Ca2+-independent states allows CaMKII to transform shifts in the frequency or amplitude of Ca2+ transients into critical cellular outcomes,2 but how this signal integration occurs in cardiac myocytes is not understood.
An additional mechanism of autonomous CaMKII activation by reactive oxygen species (ROS) has been reported recently.5 After initial activation by Ca2+/CaM, M280/281 residues are subject to oxidation that blocks autoinhibition in a similar manner to T286 autophosphorylation. CaMKII is thus able to translate changes in redox potential from both acute and chronic stimuli into downstream physiological effects.6 But again, how this integrative signaling dovetails with other CaMKII activating signals in cardiac myocytes is poorly understood.
Hayashi and colleagues developed Camui, a fluorescence resonance energy transfer (FRET) biosensor based on the full length sequence of CaMKII, to monitor the activation state of the kinase in neurons.7 Fluorescent proteins are added to the C- and N-terminal ends of the protein, allowing robust FRET in the compactly folded autoinhibited state. Activation of the kinase results in a conformational shift that is detected as a reduction in fluorescence transfer (Fig. 1A). Expression of Camui in cells allows CaMKII activation state to be monitored both temporally and spatially.
Figure 1.
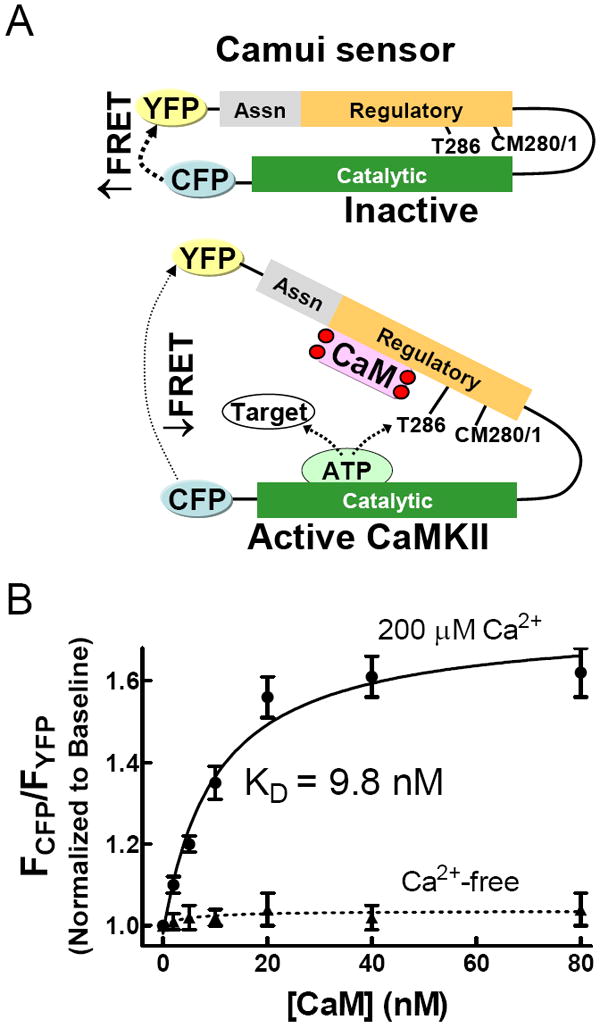
Camui is a FRET biosensor based on the structure of CaMKII. (A) Binding of Ca2+/CaM results in a conformational shift that increases the distance between the N-terminal catalytic domain and the C-terminal regulatory/association domains, reducing FRET between the CFP/YFP pair. (B) Titration of Camui with CaM in the presence of saturating Ca2+ shows the dynamic range of the sensor.
Activation of CaMKII plays a key role in cardiac pathophsyiology. CaMKII expression and activity are increased during heart failure,8 while genetic inhibition of cardiac CaMKII protects against the transition to structural heart disease.9 CaMKII also contributes to arrhythmogenesis via direct effects on ryanodine receptors,10-12 sodium channels,13,14 and CaV1.2.15,16 ROS-dependent CaMKII activation is a critical mediator of angiotensin II induced myocyte apoptosis and plays an important role in structural remodeling after myocardial infarction.5 CaMKII is widely considered to be a potential therapeutic target in prevention of heart disease,17 yet the detailed signaling mechanisms and dynamic modulation of CaMKII function in intact cardiomyocytes are still largely unknown.
Here we develop Camui variants into adenoviral vectors to examine pathways of CaMKII activation by Ca2+ transients and neurohormonal agonists. Camui is a novel, non-destructive tool that allows the online measurement of CaMKII activity in physiologically functioning adult myocytes. We show here that changes in FRET associated with a conformational shifts of Camui are effective measures of CaMKII activation in living mocytes. In addition, we use Camui to demonstrate that CaMKII activity increases with enhanced pacing frequency (and elevated [Ca]o), while a mutant Camui variant lacking the T286 site has a significantly blunted response to pacing frequency. Finally, stimulation of cardiac myocytes expressing WT or mutant variants of Camui reveal that angiotensin II (AngII) and endothelin-1 (ET-1) activate CaMKII by a primarily oxidation-dependent pathway, while isoproterenol (Iso) and phenylephrine (PE) activate CaMKII by a Ca2+ and autophosphorylation dependent pathway. This study positions Camui as an important, emerging tool for examining CaMKII activation in cellular physiology and pathophysiology.
METHODS
Construction of Adenoviral Vectors Encoding Biosensors
The Camui construct7 was incorporated in adenoviruses using the AdEasy™ adenoviral vector system (Qbiogene, Inc., Carlsbad, CA) to ensure high infection efficiency in the terminally differentiated adult ventricular myocytes. Mutant variants of Camui (T286A and MM280/281VV) were generated using the commercially available QuickChange site directed mutagenesis kit (Stratagene), and likewise incorporated into adenovirus.
HEK293 Cell Transfection
HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) with 5% fetal bovine serum and penicillin/streptomycin for 24 h and then transiently transfected with expression plasmids encoding Camui using a mammalian transfection kit (Stratagene). Cells were cultured for an additional 36 hours post transfection. Camui expression was checked by fluorescence microscopy prior to experiments.
In Vitro Fluorescence and CaMKII activity assays
Fluorescence measurements were performed using a MS SpectraMax plate reader spectrophotometer (Molecular Devices). Excitation and emission slits were set at 4 nm. Excitation wavelength of 440 nm was used, and dual photon counting emission detectors were set at 477 nm (FCFP) and 527 nm (FYFP), respectively. The cytosolic fraction of the transfected HEK cells was diluted in Ca2+-free buffer containing 50 mM Tris-HCl buffer (pH 7.5), 5 mM MgCl2, and protease inhibitors. Camui fluorescence was measured in the presence of 10 μM CaM and 200 μM Ca2+. For some experiments, 1 mM EGTA was used to chelate Ca2+. Autonomous CaMKII activity was measured in the presence of 1 mM EGTA, 100 μM ATP and/or 1 μM H2O2. Incubation time was five minutes to allow achievement of steady state. CaMKII kinase activity was confirmed by measuring incorporation of 32P-ATP into an artificial substrate, syntide-2, as previously described.5
Myocyte Isolation and Adenoviral Infection
All protocols involving animals were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the University of California, Davis Institutional Animal Care and Use Committee. Adult rabbit ventricular myocytes were isolated as previously described.18 Myocytes were seeded on laminin-coated coverslips in serum-free PC-1 medium (Lonza) supplemented with penicillin/streptomycin. Myocytes were infected for 2 hours at multiplicity of infection of 10–100 with adenovirus encoding Camui, followed by replacement with fresh medium. Infected cells were kept in culture for 36 hours with one final replacement of fresh medium 1 hr before experiments.
Confocal Microscopy Imaging
Cover slips were mounted on the stage of an inverted microscope (Zeiss, LSM5 Pascal) equipped with a 40× 1.4 NA water immersion objective lens. Argon laser excitation wavelengths were 458 nm for CFP and 514 nm for YFP. CFP emission fluorescence was measured by confocal microscopy at 485 ± 15 nm, while YFP emitted fluorescence was measured at ≥535 nm. For some experiments, cells were field stimulated at 0.25, 0.5, and 1 Hz. Some cells were treated with 1 μM angiotensin II, isoproterenol, phenylephrine, or endothelin-1. All myocyte experiments were performed in Tyrode solution containing 1 mM Ca2+ unless otherwise noted. For each experimental condition, a minimum of 30 cells (3 rabbits, 10 cells from each) was analyzed. Image-J software was used for image analysis.
Statistics
Pooled data are represented as the means ± S.E. Statistical comparisons were made using repeated two-way analysis of variance and paired Student’s t test where applicable. p < 0.05 was considered significant.
RESULTS
Camui design and dynamic range
Currently available tools for measuring the extent of CaMKII activation are not sufficient to separate phosphorylation-dependent and redox-dependent activity. To achieve this goal, we are developing new tools based on the FRET-based biosensor Camui, which takes advantage of conformational changes in CaMKII during activation. Full length CaMKIIα protein is flanked by two fluorescent proteins (K26R/N164H CFP and “Venus” F46L YFP).7 In the absence of Ca2+/CaM binding (inactive state), the proximity of CFP and YFP produces FRET (Figure 1A). Upon activation of the kinase, the distance between CFP and YFP increases due to the conformational change in CaMKII, decreasing FRET. This decrease in FRET is observable as an increase in donor (CFP) fluorescence (FCFP) accompanied by a concomitant decrease in acceptor fluorescence (FYFP). Throughout this manuscript we represent Camui FRET change as the FCFP/FYFP fluorescence ratio, such that FCFP/FYFP increases when CaMKII activation state increases.
To determine the dynamic range of our Camui sensors, we titrated expressed Camui (from the soluble fraction lysates of HEK cells) with CaM in the presence of a saturating concentration of Ca2+ (200 μM). Control experiments were performed in Ca2+ chelating conditions (1 mM EGTA). FCFP/FYFP ratio reached a maximum 60% increase at a CaM concentration of 20 μM (Figure 1B) with a KD of about 10 nM. Importantly, there was no significant change in FRET in Ca2+-free conditions, indicating that fluorescence change in the presence of Ca2+ is due to CaM association with CaMKII and not to changes in background fluorescence.
Changes in Camui fluorescence report changes in kinase conformation and activity
Addition of Ca2+/CaM to lysates from HEK cells expressing Camui resulted in a significant change in FCFP/FYFP compared to baseline, indicating a change in protein conformation (Fig. 2A; first two sets of bars). This effect could be fully reversed by the addition of EGTA, as Ca2+ removal allows the kinase to return to an autoinhibited state (Fig. 2A; third set of bars). However, treatment with Ca2+/CaM in conditions favoring either autophosphorylation (+ATP) or oxidation (+H2O2) resulted in a sustained shift in FCFP/FYFP even after the addition of EGTA, thus demonstrating that CaMKII has undergone autonomous activation. These results match prior observations that autophosphorylation and oxidation preserve CaMKII activity by preventing re-association of the regulatory and catalytic domains.3,5 Moreover, our results exclude the possibility that Camui functions only as a biosensor for CaM binding, because autophosphorylation/redox dependent activation of CaMKII (and FRET change) is sustained after Ca2+ is sequestered and CaM dissociates from the kinase. Interestingly, treatment with both ATP and H2O2 in the presence of Ca2+/CaM resulted in an additive effect on the shift in Camui fluorescence even after subsequent removal of Ca2+/CaM, suggesting that the two autonomous mechanisms are distinct but potentially additive.
Figure 2.
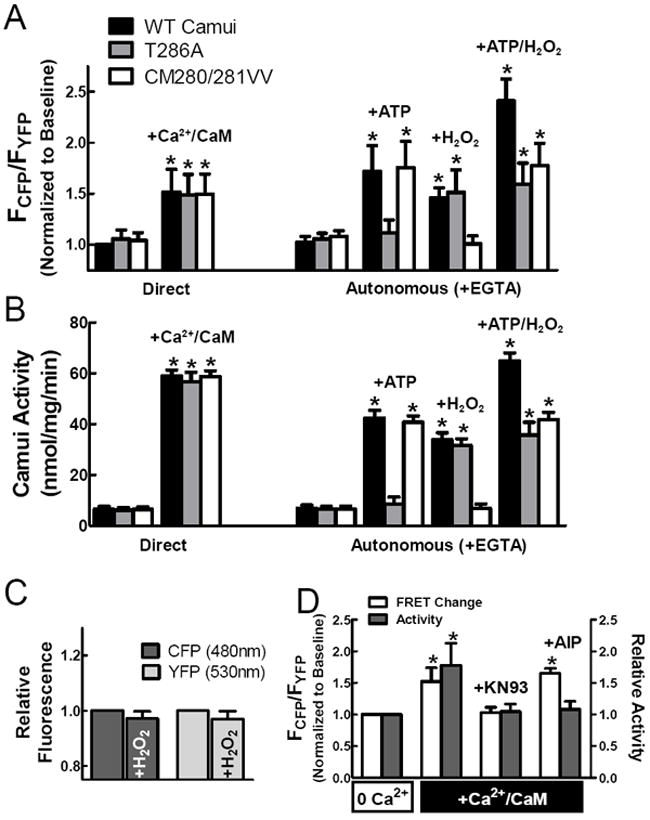
Camui detects activation of CaMKII. (A) Camui FRET changes reflect activation of CaMKII either directly through Ca2+/CaM binding or autonomously by phosphorylation or oxidation. (B) Traditional kinase assays confirm our findings with Camui. (C) Oxidative stress does not affect CFP or YFP fluorescence. (D) KN-93, but not AIP, blocks the change in FRET associated with Camui activation.
To further investigate the role of post-translational modifications on CaMKII activity, we designed Camui variants lacking either the autophosphorylation site (T286A) or the oxidation site (CM280/281VV) and repeated the previous experiments (Fig. 2A). While the mutant forms of Camui showed a normal response to Ca2+/CaM, the T286A mutant did not exhibit an autonomous shift in fluorescence under autophosphorylation conditions (+ATP). However, the T286 mutant did maintain a conformational shift in conditions favoring oxidation (H2O2). Conversely, the CM280/281VV mutant showed WT-like shift in fluorescence under autophosphorylation conditions, but not after H2O2 exposure. These observations further demonstrate the importance of the T286 and M280/281 sites in modulating CaMKII activity, and further that autophosphorylation and oxidation are independent but complementary events.
We also performed traditional kinase assays to confirm that the shift in fluorescence observed for Camui is associated with a concomitant increase in kinase activity. Total kinase activity was measured as a function of incorporation of 32P into an artificial CaMKII substrate (syntide-2). We observed a significant increase in kinase activity for each Camui variant in the presence of Ca2+/CaM (Fig. 2B). Conditions favoring autophosphorylation preserved kinase activation levels for the WT and CM280/281VV Camui, but not for the T286A mutant, while treatment with H2O2 resulted in autonomous activity for the WT and T286A, but not the CM280/281VV Camui variant. These activity results corroborate our changes in FRET and demonstrate the efficacy of shifts in Camui fluorescence as a proxy for CaMKII activation state.
The Camui construct is based on full length CaMKIIα but the predominant isoform expressed in cardiac myocytes is CaMKIIδ. We performed additional activity assays using purified WT CaMKIIδ, as well as autophosphorylation and oxidation resistant mutants from a baculovirus system,5 to determine whether Camui activity mimics that of endogenous cardiac CaMKIIδ (Supplemental Fig. IA). In every condition tested, CaMKIIδ activity closely paralleled that observed with Camui in Fig 2B. We also plotted change in Camui FRET against the autonomous kinase activity observed for both the sensor and purified kinase (Supplemental Fig. IB). This plot highlights two key results: (1) Camui FRET varies directly with autonomous kinase activity of both CaMKIIδ and Camui, and (2) autonomous CaMKII activation by autophosphorylation and oxidation appear to be additive as measured by both change in FRET and kinase activity. Interestingly, direct activation of CaMKII by Ca2+/CaM yields greater kinase activity than would be predicted by the observed change in Camui FRET. This is consistent with studies demonstrating that autonomous activation of CaMKII yields lower (but sustained) kinase activity compared to the direct CaM-induced mechanism.3,5
To test whether the change in Camui fluorescence after H2O2 treatment was due to altered fluorophore fluorescence (as has been observed in a circularly permuted form of YFP)19 control cells were transfected with viruses encoding either CFP or YFP and treated as before. No significant redox-dependent change in FCFP or FYFP were observed (Fig. 2C). This, along with the lack of effect of H2O2 on the CM280/281VV Camui mutant indicates that the changes observed with H2O2 are due to ROS-dependent CaMKII activation involving CM280/281, and thus reflect the in vivo activation mechanism of the kinase.
We also tested the effects of two potent CaMKII inhibitors, KN93 and autocamtide-2 related inhibitory peptide (AIP), on Camui FRET in activating conditions (+Ca2+/CaM). The addition of 1 μM KN-93 was sufficient to block FRET change associated with activation of Camui, while 1 μM AIP had no effect in similar conditions (Fig. 2D, white bars). However, both inhibitors were confirmed to block Camui activity at these concentrations (Fig. 2D, gray bars). We infer that KN-93 inhibits CaMKII activity by blocking the conformational shift associated with Ca2+/CaM binding, while AIP permits the conformation change but inhibits CaMKII activity through a separate mechanism, possibly by binding to the catalytic domain (mimicking the regulatory domain) or hindering substrate binding. This observation suggests that Camui, a potential tool for screening CaMKII inhibitors, can distinguish between inhibitory mechanisms that rely on blocking CaMKII conformational shift versus those that rely on substrate exclusion.
Localization of Camui in cardiomyocytes
Isolated rabbit cardiomyocytes were treated with a replication-deficient adenoviral construct encoding Camui. Images were taken 36 hours after exposure to the virus (Fig. 3A). We observed a significant increase in total fluorescence at both the 477 nm and 527 nm emission wavelengths, indicating the expression of CFP and YFP, respectively. Line scan imaging shows periodic sarcomeric localization of Camui with highest expression at the z-lines, consistent with past observations of CaMKII localization using immunofluorescent imaging.5 Cells expressing Camui were loaded with di-8-ANEPPS, a fluorescent dye that strongly associates with t-tubules, to further examine Camui localization. After YFP bleaching, CFP and di-8-ANEPPS fluorescence were measured using line scan imaging (Supplemental Fig. II). Matching patterns of peak fluorescence for the two fluorophores further confirms the enrichment of Camui at the z-line. Ratiometric images were also generated by measuring FCFP/FYFP for cells expressing Camui, allowing us to determine whether Camui activation varies by subcellular localization (Fig. 3B). We found that the basal Camui signal was slightly greater in the perinuclear space and cytoplasm than in the nucleus at rest.
Figure 3.
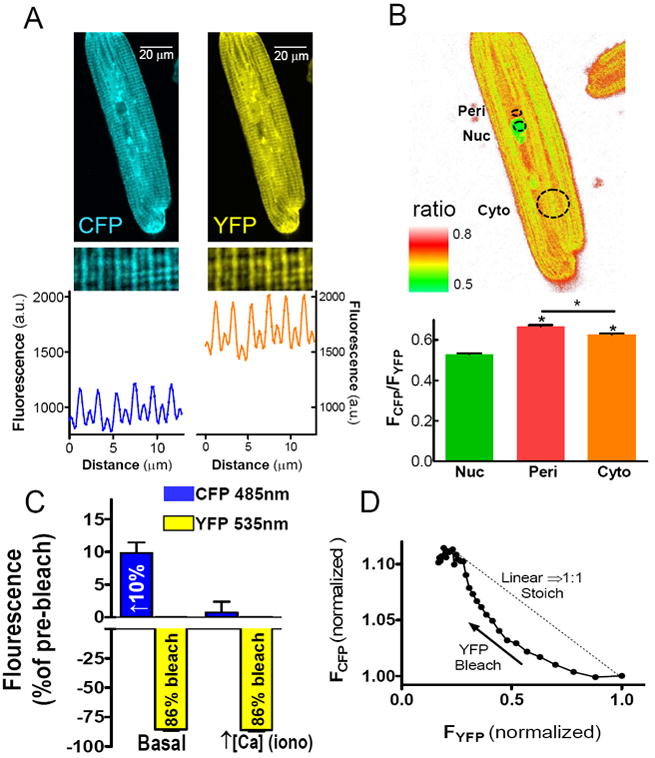
Localization of Camui in cardiomyocytes. (A) Fluorescent confocal microscopy of CFP and YFP emission signals from myocytes expressing Camui. A sample line scan confirms Camui association with myofilaments. (B) A sample ratio plot shows localized differences in Camui activation in the nucleus (Nuc), perinuclear space (Peri) and cytoplasm (Cyto) at baseline conditions. (C) Selective photobleach of the YFP acceptor results in increased donor fluorescence, which is ablated by activation of CaMKII via increased [Ca2+]I. (D) CFP fluorescence is not enhanced by YFP photobleach in a 1:1 stoichiometric manner.
To directly assess the FRET efficiency of Camui in myocytes, we performed selective high intensity acceptor (YFP) photobleach, and measured the resultant increase in donor (CFP) fluorescence. This confirms that true FRET is occurring and FCFP (485 nm) increased by about 10% upon 86% bleach of YFP (3C). This is consistent with a FRET efficiency of about 12%. Addition of ionomycin, which increases intracellular [Ca2+] and activates Camui, virtually eliminated the increase in FCFP seen upon YFP bleach. These results confirm that FRET occurs in Camui and is nearly abolished by activation of the sensor.
CaMKII tagged with GFP on the C-terminal association domain has been shown not to interfere with homo- or hetero-multimerization of CaMKII monomers.20 To test whether Camui participates in multimerization we monitored the relationship between the rise in FCFP and bleach of YFP (Fig. 3D). A linear relationship would imply a 1:1 or monomeric Camui. In contrast, the curved relationship observed indicates that even when one YFP is bleached, the CFP on the same molecule can FRET with an alternate YFP, or in other words Camui can multimerize and produce intermolecular FRET.21 We interpret this finding as further evidence that the addition of CFP and YFP do not disrupt multimerization of Camui into a CaMKII-like holoenzyme. While the acceptor photobleach method is a sensitive, direct measurement of FRET efficiency, it is a terminal experimental protocol. To perform continuous, dynamic measurements in myocytes, we used continuous measurements of FCFP/FYFP.
Steady state CaMKII activity is increased in paced cells
The canonical mechanism of CaMKII activation is in response to Ca2+ transients.2 We utilized Camui to examine CaMKII activation resulting from excitation-contraction coupling. Rabbit cardiomyocytes were paced at different frequencies in Tyrode’s solution containing 1 or 2 mM Ca2+ and expressing either WT or T286A Camui. WT Camui detected increased CaMKII activation as a direct function of pacing frequency (Fig. 4, top panel). Additionally, steady state CaMKII activation was enhanced at each stimulation frequency by increased [Ca]o. Notably, in resting cardiomyocytes Camui seems to be very close to the deactivated level because inclusion of KN-93 and [Ca2+]o= 0 did not decrease the basal FCFP/FYFP signal appreciably (Fig. 4, inset).
Figure 4.
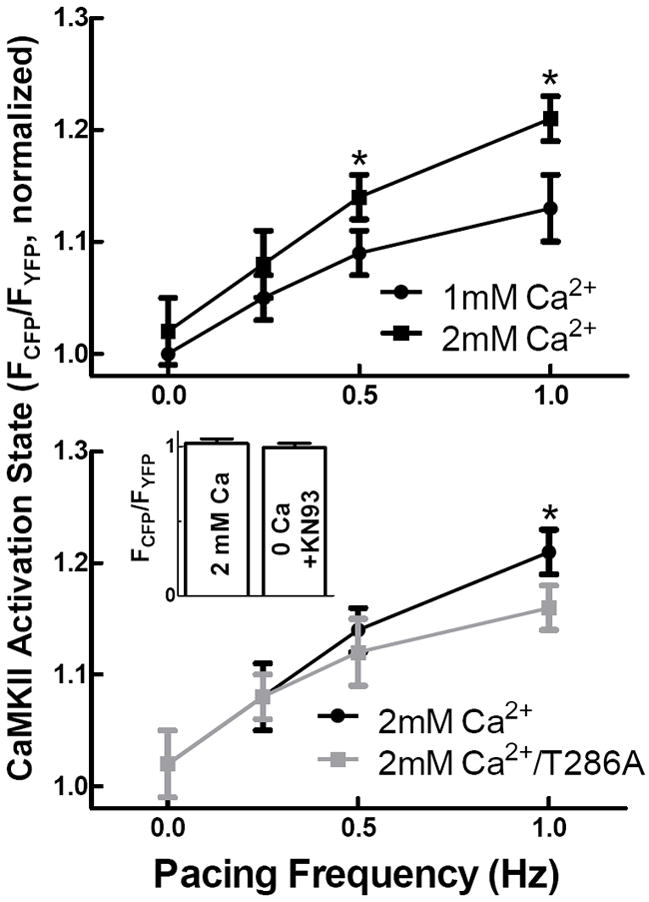
CaMKII activity is dependent on pacing frequency as measured by Camui. CaMKII activity increases with pacing frequency and [Ca2+]o. Mutation of the T286 phosphorylation site reduces CaMKII activation at a pacing frequency of 1 Hz. (Inset) FCFP/FYFP for baseline measurements was indistinguishable from Ca2+/CaMKII buffered conditions.
Interestingly, steady state CaMKII activity levels measured by the WT and T286A mutant were indistinguishable at the slowest pacing frequencies (0.25 and 0.5 Hz), but activation of the T286A mutant was significantly reduced at a pacing frequency of 1 Hz compared to WT (Fig. 4, bottom panel). Our results demonstrate that CaMKII activation state in cardiomyocytes is subject to acute regulation by the frequency and intensity of excitation-contraction coupling. Further, autophosphorylation at T286 contributes to CaMKII activation at higher frequency and [Ca2+].
Neurohormonal stimulation of rabbit myocytes enhances CaMKII activity
A number of additional intracellular signaling pathways are known to modulate CaMKII activity. These include neurohormonal stimulation, as has been observed with AngII and Iso.5 β-adrenergic dependent CaMKII activation also occurs via cAMP/Epac22 and cAMP/Ca2+-independent pathways.11,23 To determine the effects of neurohormonal stimulation on CaMKII activation, we treated isolated rabbit cardiomyocytes expressing Camui with four different agonists known to play critical roles in cardiac signaling. Cells were exposed to agonist for 40 min (Fig. 5A). Point measurements were used rather than continuous monitoring to limit fluorophore bleaching. All four agents caused a significant CaMKII activation (p<0.001 vs. control) after 20 min as measured by Camui FRET. AngII and ET-1 had very rapid effects on CaMKII activity, with a pronounced increase in activity within 5 min of exposure. PE showed an intermediate time course of CaMKII activation, while Iso was slowest, with no significant change until 10 min after exposure. Interestingly, although Iso caused the slowest initially response, the eventual peak intensity of CaMKII activation was the greatest. Note that all of these agonists activate CaMKII in quiescent myocytes at least as well as high frequency Ca2+ transients.
Figure 5.
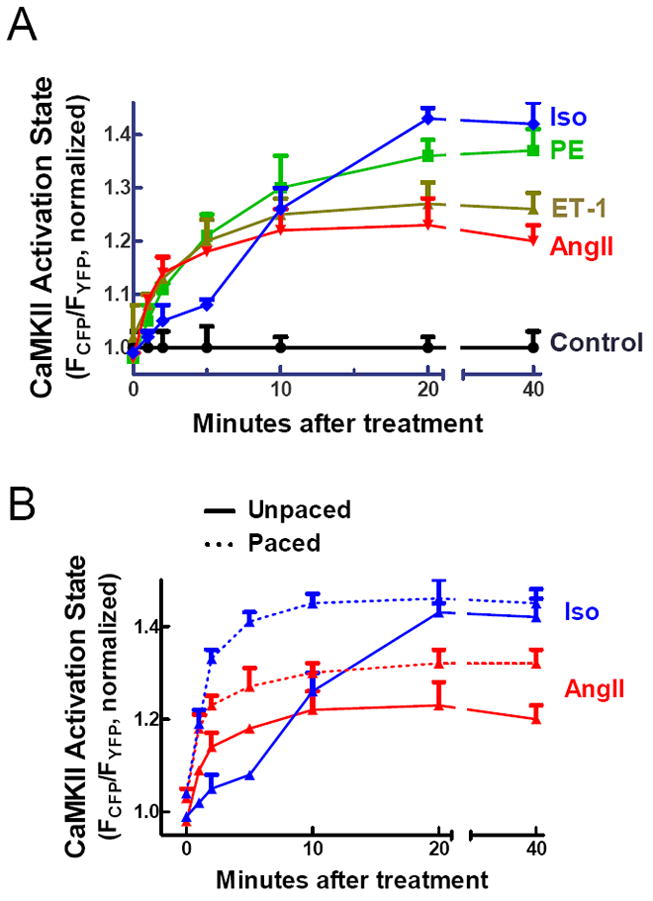
CaMKII activation by common cardiac agonists. (A) All four neurohormonal agonists cause significant activation (p<0.001 vs. control at 20 min) of CaMKII in isolated rabbit cardiomyocytes as measured by Camui. (B) 0.5 Hz pacing increases CaMKII activation for the time period 1-10 minutes after Iso treatment.
The slow response of CaMKII to Iso stimulation was somewhat surprising. We hypothesized that the effects of Iso would be accelerated by Ca2+ transients. So we repeated the experiments using AngII and Iso, but this time in paced (0.5 Hz) rabbit cardiomyoctyes expressing Camui. Pacing significantly altered the kinetics of CaMKII activation in response to Iso (p<0.01 for paced vs. unpaced cells at 1-10 min), consistent with our hypothesis, but had more modest effects on the timecourse with AngII (Figure 5B). Additionally, the increased availability of Ca2+ in the paced cells resulted in higher peak CaMKII activity for both agonists.
To further elucidate the mechanism of Iso-induced Camui activation, we pretreated cells for 10 min with either 10 μM thapsigargin, a potent and selective SERCA inhibitor, or 1 μM okadaic acid, a phosphatase inhibitor. Pretreatment with thapsigargin blocked Camui activation for the 1-20 minute period after Iso treatment (Supplemental Fig. IIIA). Conversely, okadaic accelerated the Iso-induced Camui activation (p<0.05 vs. –OA for 5 and 10 min time points) and significantly increased peak steady state Camui activation (Supplemental Fig. IIIB).
Mutant Camui variants elucidate activation mechanisms of CaMKII
All four agonists used caused CaMKII activation. To further elucidate the mechanisms for activation associated with each, we repeated the stimulation experiments in cells expressing mutant variants of Camui lacking either the T286 phosphorylation or CM280/281 oxidation sites (Fig. 6A). CaMKII activation by AngII and ET-1 was unchanged in the T286A mutant, but the CM280/281VV mutant showed a severely blunted response to both agonists. Peak CaMKII was reduced in the oxidation resistant mutant, and in the case of AngII, activity had returned to baseline levels within 20 min. These observations demonstrate that AngII and ET-1 modulate CaMKII activity by a pathway that importantly includes redox-dependent CaMKII activation for >50% of the maximal effect and up to 90% for AngII after 20 min (Fig. 6B).
Figure 6.
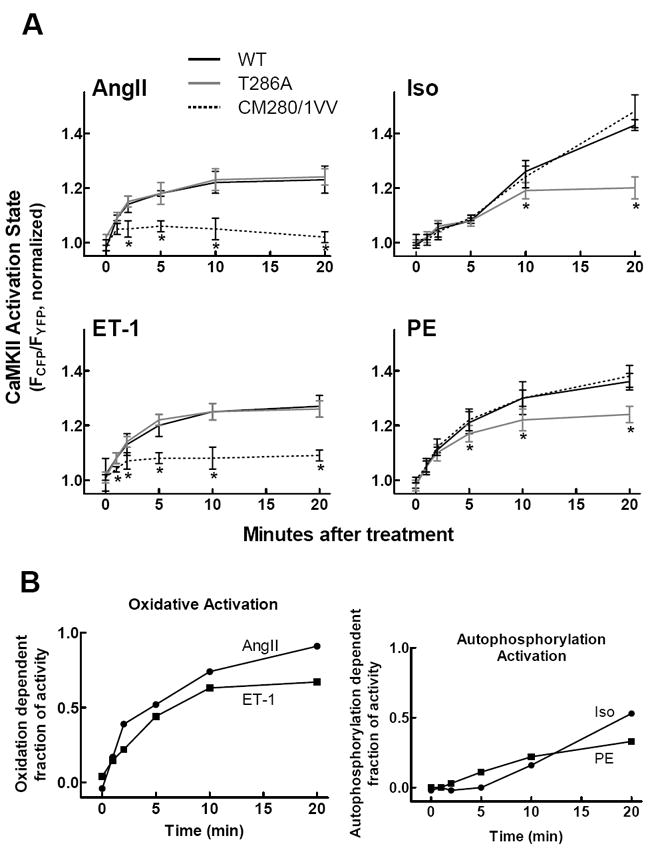
Time course of CaMKII activation after agonist treatment for the wild type, T286A, and CM280/281VV Camui variants. (A) Mutation of the redox-sensitive sites significantly reduces AngII and ET-1 mediated kinase activation, while mutation of the T286 phosphorylation site reduces Iso and PE mediated activation. (B) Difference curves for AngII/ET-1 oxidative activation and Iso/PE autophosphorylation activation.
For Iso and PE, we observed no significant difference in CaMKII activation with the WT and CM280/281VV Camui variants. However, peak CaMKII activity in response to Iso and PE was reduced significantly in the phosphorylation-resistant mutant. Thus, in contrast to the oxidation-dependent mechanism of CaMKII activation by AngII and ET-1, we conclude that modulation of CaMKII activity by Iso and PE is driven significantly by T286 phosphorylation.
As an additional test of the role of oxidation in CaMKII activation by AngII and Iso, we pre-treated cells expressing WT Camui with Trolox, a cell-permeable vitamin E derivative, to scavenge ROS. Peak CaMKII activation was unchanged in the presence of Trolox for cells treated with Iso (Fig. 7). However, Trolox completely prevented the AngII-mediated CaMKII activation as measured by Camui. These observations recapitulate our previous findings that AngII stimulates CaMKII activation by a redox-dependent pathway.
Figure 7.
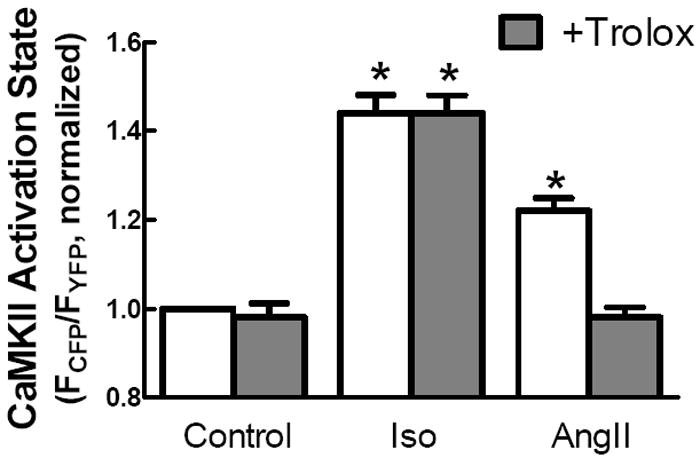
The cell permeable anti-oxidant Trolox ablates AngII dependent activation of Camui but has no significant effect on Iso dependent CaMKII activity.
DISCUSSION
The regular Ca2+ transients that occur at each heartbeat and are modulated in amplitude and frequency during physiological conditions are expected to modify CaMKII activity in myocytes. Until now, the only methods to assess CaMKII activation state in myocytes have been destructive (e.g. measures of target phosphorylation by phospho-antibodies), and direct activity measurements in cell lysates may not mimic cellular conditions. Measurements in cardiac myocytes with FRET-based Ca2+-CaM sensors24 and computational modeling25,26 have been steps toward enhanced understanding. The discovery that CaMKII can be activated by oxidation at the CM280/281 site,5 combined with the long known connection between T286 phosphorylation and frequency dependent CaMKII activation, further complicates our understanding of dynamic CaMKII activity regulation in the cellular environment. Probing for post-translational modifications of Camui using traditional immunoblot methods can provide a snapshot of CaMKII activation state after the fact.
However, the development of biosensors like Camui provides tools for monitoring the spatial and temporal status of CaMKII activation real-time in living myocytes. Indeed, our in vitro experiments demonstrate that the FRET change of Camui is a good proxy for CaMKII activity. This method offers several advantages over traditional methods of determining CaMKII activity. Immunoblot of T286 phosphorylation has often been used as an indication of CaMKII activation, yet this method ignores canonical Ca2+/CaM and oxidation-dependent activity. Likewise, blotting for phosphorylation of a downstream CaMKII target, such as phospholamban, may account for only limited pools of CaMKII activity while missing others (e.g. nuclear). Camui detects changes in CaMKII activation state dynamically, both with respect to the localization of the protein and the time of an activating signal.
Here we show that Camui is a powerful tool for detecting subtle differences in CaMKII activity within specific subcellular domains. Camui expression appears to be greatest at the z-line of the myofilament, suggesting close proximity of CaMKII and myocyte t-tubules. Our experiments demonstrate a critical advantage of Camui: the ability to track CaMKII activation spatially within living myocytes. Camui will prove invaluable in future studies focused on the subcellular mechanisms that drive CaMKII signaling pathways in the heart.
Camui FCFP/FYFP is a good reporter of CaMKII activation by Ca2+/CaM, autophosphorylation and oxidation, by sensing opening the regulatory-catalytic domain, but there are limitations. First, one cannot assume that the FCFP/FYFP signal is a linear readout of CaMKII activity. While the autonomous activity via autophosphorylation or oxidation is a fairly linear function of Camui FCFP/FYFP over a wide range, direct Ca2+/CaM activation produces a higher activity for a given FCFP/FYFP signal (Supplementary Fig IB). This is consistent with prior work showing higher maximum CaMKII activity via Ca2+/CaM than with either autonomous pathway individually.3,5 It is worth noting that the two autonomous modes are roughly additive in both activity and FCFP/FYFP signal and when combined the CaMKII activity reaches roughly the same level as upon maximal Ca2+/CaM activation.
Second, the increase in Camui FRET is not simple bimolecular FRET, because in the basal state CFP on the catalytic end of CaMKII can FRET with more than one YFP on the association domain (Fig 8B). This result is not surprising based on prior work with GFP-tagged CaMKII21 and recent structural evidence for interplay between CaMKII subunits during activation.27 This could contribute to some signal FCFP/FYFP nonlinearity as catalytic domains become activated (Fig 8B), but as we show changes in FCFP/FYFP signal are a good proxy for CaMKII activity. Moreover, the fact that Camui is based on full-length CaMKII and that it multimerizes may provide the advantage that it behaves much like endogenous CaMKII, making the FCFP/FYFP structural signal of particular value (even if it is not a precise readout of enzymatic activity). Camui thus also differs from FRET-based BsCaM sensors that we have used to monitor Ca2+/CaM binding to peptide CaM targets of different affinity as a readout of dynamic [Ca2+/CaM] signals in cardiac myocytes.24 These two types of sensors complement each other. While BsCaM data provides information about Ca2+/CaM dynamics that could activates CaMKII and other CaM targets like calcineurin, Camui reports on the actual impact of CaM as well as other factors (e.g. autophosphorylation) on CaMKII structure and activity. In this regard, the dynamics of T286A Camui probably gives a truer readout than BsCaM of the dynamics of CaM binding to real cellular CaMKII.
Figure 8.
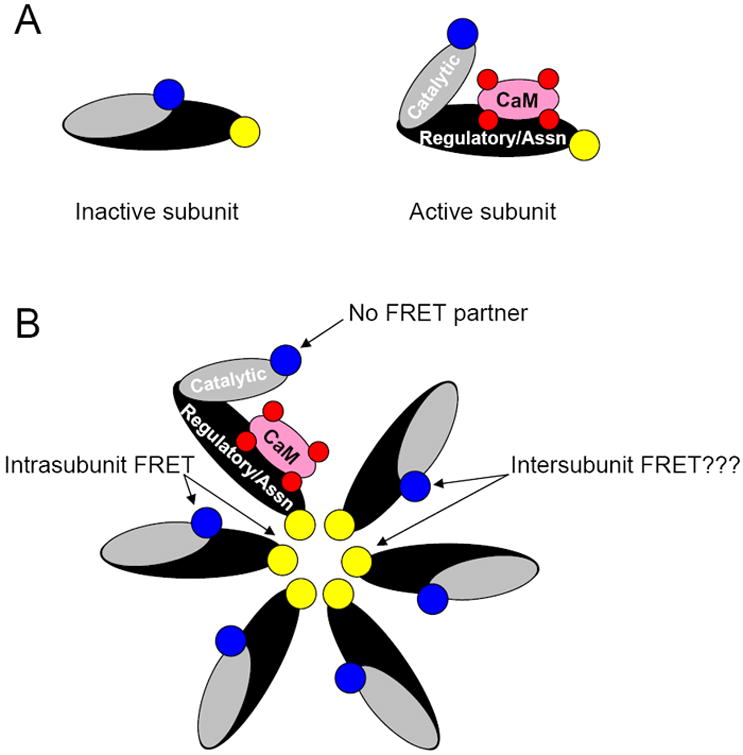
Models of the Camui sensor for both individual subunits and an assembled holoenzyme. (A) FRET is expected to occur between the fluorophore pair of an inactive subunit. The conformational shift associated with CaMKII activation (ex. via CaM binding) should largely abolish fluorescence transfer. (B) The geometry of the holoenzyme suggests the possibility that intersubunit FRET may occur between fluorophores from adjacent subunits.
In addition, the specific roles of CaMKII phosphorylation and oxidation can be examined using mutant forms of Camui lacking the target sites necessary for these modifications. This feature of Camui allows us to explore the mechanisms that translate upstream physiological signaling to downstream CaMKII activation. For example, our data demonstrate that AngII and ET-1 mediate CaMKII activity substantially through a redox dependent pathway involving oxidation at amino acids 280-281 on CaMKII. These findings are in good agreement with a previously published study on activation of CaMKII by AngII.5 Interestingly, while other studies have established a link between ET-1 signaling and CaMKII activity,28-30 our data is the first to show that ET-1 regulates CaMKII in large part by oxidation and autonomous activation. In contrast to AngII and ET-1, we show here that PE and Iso activate CaMKII through a pathway that is largely dependent on Ca2+ transient signaling and to a variable degree T286 phosphorylation, as demonstrated by reduced CaMKII activation of the T286A Camui mutant.
Remarkably, the kinetics for CaMKII activation by Iso in quiescent myocytes is very slow compared to the other agonists we tested. Part of the initial Iso-dependent activation of CaMKII also seems to depend on a functional SR (Fig S3A) and we speculate that this could be due to a PKA-dependent increase in SR Ca uptake (secondary to phospholamban phosphorylation) and enhancement of local Ca sparks.31 Conversely, the Iso-induced CaMKII activation was accelerated by both pacing the myocytes or inclusion of phosphatase inhibitors. Thus, for Iso exposure CaMKII activation is a complex function of [Ca2+], PKA, phosphatase and probably additional pathways,11,22,23 and autophosphorylation of CaMKII is an important contributor as well. It is also noteworthy that simple stimulation frequency at 2 mM [Ca2+]o does not achieve CaMKII activation levels seen with some GPCR agonists.
Because all known mechanisms of CaMKII activation require initial binding of Ca2+/CaM to the autoinhibited kinase,4 CaMKII function is thought to be acutely sensitive to the intensity, duration, and frequency of Ca2+ transients. The extent to which this is the case is currently unknown, though much progress has been made utilizing models of excitation-contraction coupling.26 The type of data that we have obtained here may also fuel improvements in these mathematical models, enhancing their utility as well. We show here that Camui is a powerful tool for detecting changes in CaMKII activity, with respect to frequency, and Ca2+ transient amplitude, and interaction with GPCR signaling. A clear direction of future study is examining the kinetics of Camui signaling on a beat-to-beat basis, as well as further efforts to ascertain the extent of CaMKII activation in response to acute Ca2+ signaling and how it may synergize in detail with these GPCR-linked activation pathways. Such data would lend valuable insight for detailed tuning of models and our integrated understanding of CaMKII signaling in heart.
Increased CaMKII activity is a common feature of structural heart disease and sudden heart failure in patients and in animal models, while inhibition32,33 and knock-out34,35 of CaMKII reduces apoptosis and improves mortality36 in structural heart disease models. These findings have garnered attention for CaMKII as a potential therapeutic target for treating heart failure and arrhythmias.37 Camui represents a powerful tool to aid in the development of treatment strategies centered on CaMKII. Utilizing Camui in combination with models of structural heart disease would provide novel mechanistic insight into CaMKII regulation in the context of the failing heart. Additionally, the ability to monitor kinase activity in cells expressing Camui would benefit future efforts to design therapeutics targeted at cardiac CaMKII.
Supplementary Material
NOVELTY AND SIGNIFICANCE
What Is Known?
Calcium/calmodulin-dependent kinase II (CaMKII) translates a broad range of upstream signaling mechanisms to downstream physiological effects in the heart.
Activation of CaMKII is a critical step in the transition to arrhythmia and heart failure.
CaMKII activity is regulated by several mechanisms, including calcium transient frequency and redox potential.
What New Information Does This Article Contribute?
We present a novel method for dynamic real-time monitoring of CaMKII activity in intact cardiac myocytes using the fluorescent biosensor Camui.
Camui allows spatial and temporal resolution of CaMKII activation state in living cells.
Signaling mechanisms known to enhance CaMKII activity do so through distinct molecular mechanisms.
Camui represents a critical tool in the translation of CaMKII research into clinical applications.
CaMKII has emerged as a critical mediator of cardiac physiology, particularly during the transition from healthy to failing myocardium. Thus, CaMKII is a potential target for future therapeutic approaches in the prevention of heart disease. However, the detailed mechanisms of CaMKII activation in cells are not fully understood. Here, we introduce a method for monitoring CaMKII activation state using the fluorescent biosensor Camui. This sensor can be used to measure CaMKII activity in intact cells, which is greatly advantageous compared to existing destructive methods, as it allows changes in CaMKII activity to be observed over time and within subcellular locations. We show here that CaMKII activity is influenced by both pacing rate and neurohormonal stimulation. Additionally, we use mutant forms of Camui lacking key regulatory sites to show that CaMKII activation state is determined by distinct molecular mechanisms that are specific to the activating stimulus. Camui will not only be an invaluable tool for future research on the mechanisms of CaMKII activation in the heart, but it will also provide new insight into clinical approaches aimed at the prevention of arrhythmia and sudden heart failure.
Acknowledgments
The authors would like to thank Dr. Y Hayashi (Massachusetts Institute of Technology, Cambridge, MA) for his generous gift of the Camui construct.
SOURCES OF FUNDING NIH T32 HL86350 (JRE), NIH R01 HL103933 and American Heart Association Scientist Development Grant 035312N (JB), and NIH P01 HL080101, R37 HL30077, and the Fondation Leducq Transatlantic CaMKII Alliance (DMB).
ABBREVIATIONS
- AIP
Autocamtide-2 related inhibitory peptide
- AngII
Angiotensin II
- CaM
Calmodulin
- CaMKII
Calcium/calmodulin dependent kinase II
- ET-1
Endothelin-1
- FRET
Fluorescence resonance energy transfer
- Iso
Isoproterenol
- PE
Phenylephrine
- ROS
Reactive oxygen species
Footnotes
DISCLOSURES None
References
- 1.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005:849–60. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schworer CM, Colbran RJ, Keefer JR, Soderling TR. Ca2+/calmodulin-dependent protein kinase II. Identification of a regulatory autophosphorylation site adjacent to the inhibitory and calmodulin-binding domains. J Biol Chem. 1988:13486–9. [PubMed] [Google Scholar]
- 4.Rellos P, Pike AC, Niesen FH, Salah E, Lee WH, von Delft F, Knapp S. Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol. 2010 doi: 10.1371/journal.pbio.1000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008:462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erickson JR, He BJ, Grumbach I, Anderson ME. CaMKII in the cardiovascular system: sensing redox states. Physiol Rev. 2011:889–915. doi: 10.1152/physrev.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwok S, Lee C, Sánchez SA, Hazlett TL, Gratton E, Hayashi Y. Genetically encoded probe for fluorescence lifetime imaging of CaMKII activity. Biochem Biophys Res Commun. 2008:519–25. doi: 10.1016/j.bbrc.2008.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003:912–9. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 9.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005:409–17. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 10.Currie S, Loughrey CM, Craig MA, Smith GL. Calcium/calmodulin-dependent protein kinase IIdelta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem J. 2004:357–66. doi: 10.1042/BJ20031043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005:1314–22. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 12.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010:2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006:3127–38. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010:3508–19. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, Grumbach IM, Luczak ED, Colbran RJ, Song LS, Hund TJ, Mohler PJ, Anderson ME. CaV1.2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci U S A. 2010:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bers DM. Beyond beta blockers. Nat Med. 2005:379–380. doi: 10.1038/nm0405-379. [DOI] [PubMed] [Google Scholar]
- 18.Bassani JW, Bassani RA, Bers DM. Calibration of indo-1 and resting intracellular [Ca]i in intact rabbit cardiac myocytes. Biophys J. 1995:1453–60. doi: 10.1016/S0006-3495(95)80318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008:279–90. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, Reddy LG, Bennett R, Silva ND, Jr, Jones LR, Thomas DD. A fluorescence energy transfer method for analyzing protein oligomeric structure: application to phospholamban. Biophys J. 1999:2587–99. doi: 10.1016/S0006-3495(99)77411-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lantsman K, Tombes RM. CaMK-II oligomerization potential determined using CFP/YFP FRET. Biochim Biophys Acta. 2005:45–54. doi: 10.1016/j.bbamcr.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Pereira L, Métrich M, Fernández-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Bénitah JP, Lezoualc’h F, Gómez AM. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J Physiol. 2007:685–94. doi: 10.1113/jphysiol.2007.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curran J, Hinton MJ, Ríos E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007:391–8. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 24.Song Q, Saucerman JJ, Bossuyt J, Bers DM. Differential integration of Ca2+-calmodulin signal in intact ventricular myocytes at low and high affinity Ca2+-calmodulin targets. J Biol Chem. 2008:31531–40. doi: 10.1074/jbc.M804902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hund TJ, Decker KF, Kanter E, Mohler PJ, Boyden PA, Schuessler RB, Yamada KA, Rudy Y. Role of activated CaMKII in abnormal calcium homeostasis and INa remodeling after myocardial infarction: insights from mathematical modeling. J Mol Cell Cardiol. 2008:420–8. doi: 10.1016/j.yjmcc.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saucerman JJ, Bers DM. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys J. 2008:4597–612. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chao LH, Pellicena P, Deindl S, Barclay LA, Schulman H, Kuriyan J. Intersubunit capture of regulatory segments is a component of cooperative CaMKII activation. Nat Struct Mol Biol. 2010:264–72. doi: 10.1038/nsmb.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006:675–82. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bossuyt J, Helmstadter K, Wu X, Clements-Jewery H, Haworth RS, Avkiran M, Martin JL, Pogwizd SM, Bers DM. Ca2+/calmodulin-dependent protein kinase IIdelta and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ Res. 2008:695–702. doi: 10.1161/CIRCRESAHA.107.169755. [DOI] [PubMed] [Google Scholar]
- 30.Komukai K, O-Uchi J, Morimoto S, Kawai M, Hongo K, Yoshimura M, Kurihara S. Role of Ca2+/calmodulin-dependent protein kinase II in the regulation of the cardiac L-type Ca2+ current during endothelin-1 stimulation. Am J Physiol Heart Circ Physiol. 2010:H1902–7. doi: 10.1152/ajpheart.01141.2009. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002:309–16. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 32.Zhu WZ, Wang SQ, Chakir K, Yang DM, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng HP, Xiao RP. Linkage of beta(1)-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP, Anderson ME. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol Heart Circ Physiol. 2006:H3065–75. doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- 34.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Heller Brown J. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009:1230–40. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009:2925–41. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khoo MS, Li J, Singh MV, Yang Y, Kannankeril P, Wu Y, Grueter CE, Guan X, Oddis CV, Zhang R, Mendes L, Ni G, Madu EC, Yang J, Bass M, Gomez RJ, Wadzinski BE, Olson EN, Colbran RJ, Anderson ME. Death, cardiac dysfunction, and arrhythmias are increased by calmodulin kinase II in calcineurin cardiomyopathy. Circulation. 2006:1352–9. doi: 10.1161/CIRCULATIONAHA.106.644583. [DOI] [PubMed] [Google Scholar]
- 37.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011 doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.