

Abstract
Background
The genus Pyrus belongs to the tribe Pyreae (the former subfamily Maloideae) of the family Rosaceae, and includes one of the most important commercial fruit crops, pear. The phylogeny of Pyrus has not been definitively reconstructed. In our previous efforts, the internal transcribed spacer region (ITS) revealed a poorly resolved phylogeny due to non-concerted evolution of nrDNA arrays. Therefore, introns of low copy nuclear genes (LCNG) are explored here for improved resolution. However, paralogs and lineage sorting are still two challenges for applying LCNGs in phylogenetic studies, and at least two independent nuclear loci should be compared. In this work the second intron of LEAFY and the alcohol dehydrogenase gene (Adh) were selected to investigate their molecular evolution and phylogenetic utility.
Results
DNA sequence analyses revealed a complex ortholog and paralog structure of Adh genes in Pyrus and Malus, the pears and apples. Comparisons between sequences from RT-PCR and genomic PCR indicate that some Adh homologs are putatively nonfunctional. A partial region of Adh1 was sequenced for 18 Pyrus species and three subparalogs representing Adh1-1 were identified. These led to poorly resolved phylogenies due to low sequence divergence and the inclusion of putative recombinants. For the second intron of LEAFY, multiple inparalogs were discovered for both LFY1int2 and LFY2int2. LFY1int2 is inadequate for phylogenetic analysis due to lineage sorting of two inparalogs. LFY2int2-N, however, showed a relatively high sequence divergence and led to the best-resolved phylogeny. This study documents the coexistence of outparalogs and inparalogs, and lineage sorting of these paralogs and orthologous copies. It reveals putative recombinants that can lead to incorrect phylogenetic inferences, and presents an improved phylogenetic resolution of Pyrus using LFY2int2-N.
Conclusions
Our study represents the first phylogenetic analyses based on LCNGs in Pyrus. Ancient and recent duplications lead to a complex structure of Adh outparalogs and inparalogs in Pyrus and Malus, resulting in neofunctionalization, nonfunctionalization and possible subfunctionalization. Among all investigated orthologs, LFY2int2-N is the best nuclear marker for phylogenetic reconstruction of Pyrus due to suitable sequence divergence and the absence of lineage sorting.
Background
The genus Pyrus L. belongs to the tribe Pyreae Baill. (the former subfamily Maloideae C. Weber) of the family Rosaceae [1] and is geographically divided into two groups: occidental pears and oriental pears [2]. The majority of oriental pears are native to China; a few are native to Japan and the Korean Peninsula. Chinese taxonomists agreed on 13 Pyrus species native to China, among which P. betulaefolia Bge. and P. calleryana Dcne. have retained characteristics believed to be ancestral for Pyrus [3] including the smallest fruit size and lowest carpel number. Based on morphological traits or crossing experiments, P. × bretschneideri Rehd., P. × serrulata Rehd., P. × sinkiangensis Yu, and P. × hopeiensis Yu are putative hybrids among other Pyrus species [4-6]. The circumscription of species, subspecies, and forms for occidental species remains controversial. It is believed that some cultivated pears frequently escaped from cultivation and became feral. These plants hybridize easily, both with cultivated and wild species, resulting in a number of intermediate forms and segregants [7]. Therefore, morphological characters are poor indicators of Pyrus phylogeny. Other data sources, like chemical characters, were used to distinguish some pear species [8], but these were plagued by low number of characters, polymorphisms, and environmental plasticity. During the last decade, molecular markers including RFLPs [9], RAPDs [10-12], genomic-SSRs [13,14], EST-SSRs [15,16] and AFLPs [10,17] have been applied in Pyrus. These data provided useful information on the origin of some cultivated pear groups, e.g. Chinese white pears (CWP), which are assigned to P. × bretschneideri. However, CWP are morphologically different from the so-called wild P. × bretschneideri in northern Hebei province [4]. They also show a close relationship to P. pyrifolia based on multiple molecular marker data and thus were treated as P. pyrifolia White Pear Group [11,14,17]. However, most of the studies have focused on the relationships of several oriental or occidental species or cultivar pear groups, and the phylogeny of the genus remains unresolved.
Plastid DNA sequence data and the internal transcribed spacer (ITS) of nuclear ribosomal DNA (nrDNA) have been used for plant phylogenetic reconstruction due to ease of amplification. In the Rosaceae, these data have been applied in phylogenetic studies at different taxonomic levels [18,19], but their utility is limited due to varied evolutionary rates [20-25]. Intra-individual ITS polymorphisms caused by incomplete concerted evolution of nrDNA arrays have been found in many Rosaceae [20,21,26,27]. Such polymorphisms provided evidence for hybrid origins of some species in Rosa [27], but they led to a poorly resolved phylogeny in Malus [21]. Similarly, our previous study in Pyrus [28] revealed a history of non-concerted evolution of ITS and a poorly resolved phylogenetic tree. Six non-coding regions of plastid DNA were found to be highly conserved in Pyrus [29], but they reflect only the maternal genealogies.
An alternative source of molecular sequence data, low-copy nuclear genes (LCNGs), has proven to be more phylogenetically informative than either ITS or plastid DNA [30]. These genes reflect biparental lineages and are less prone to homogenization [31-33]. Due to the accumulation of large number of gene sequences in GenBank, it is now possible to design taxa-specific primers. However, paralog and lineage sorting problems are still challenges to applying LCNGs to phylogenetic studies, since they may lead to topological incongruence similar to those caused by hybridization [31,34]. Gene duplication is a prominent feature of plant genome evolution, and duplicate segments account for 60% of the Arabidopsis thaliana genome [35]. In molecular phylogenetic studies, nuclear genes undergoing gene duplications or the birth-and-death process lead to problems in the identification of orthologs and paralogs and discordance between gene and species trees. Additionally, frequent gene duplications made the terms 'paralogy' and 'orthology' ambiguous. Thus the new terms 'inparalog' for paralogs that evolved after the ingroup speciation and 'outparalog' for those that evolved before ingroup speciation occurred [36]. Lineage sorting (or deep coalescence) results from random fixation of ancestral polymorphic alleles, which may induce similar topological incongruence to that of hybridization, and poses the most challenging problems for inter- and intra-specific phylogenetic inference [31,37]. However, lineage sorting is a random process, and fixation of ancestral alleles among species is rarely identical for two unlinked nuclear loci. Therefore, incongruence caused by hybridization and lineage sorting could be differentiated when comparing phylogenies based on multiple unlinked nuclear loci.
LCNGs that succeeded in other Rosaceae are potentially ideal nuclear markers for phylogenetic studies of Pyrus. The coding region of GBSSI has been successfully applied at intergeneric and higher levels [26,38], but the introns are too short and dispersed to be ideal gene regions for interspecific levels studies. Another gene region is the second intron of LEAFY, which is long enough and has been proven to be informative for studies at the interspecific level in Pyreae [39,40]. Complete coding sequences of two LEAFY loci have been isolated in Pyrus [41], and the corresponding genomic sequences in Malus species are available (DQ535885-AFL1, DQ535886-AFL2). Thus obtaining introns of LEAFY in the Pyrus taxa is possible.
Alcohol dehydrogenase gene (Adh) is one of the best-studied nuclear-encoded genes in plants. Two major ADH classes, class P with alcohol activity and class III with glutathione-dependent formaldehyde activity, have been identified in flowering dicot or monocot plants. The former is common for plants and the latter has been isolated in a few taxa including Pisum sativum (P80572) [42], Oryza sativa (U77637) and Araobidopsis thaliana (X82647) [43]. The Adh gene occurs in small gene families, and has proven to be a useful phylogenetic marker in the Poaceae and Paeoniaceae [44-46], but it is too complex a gene family or provided little phylogenetic resolution in other taxa such as the Gossypium and Carex [47,48]. Two distinct Adh loci (AF031900, AF031899) have been isolated in P. communis 'Packham's Triumph' [49], thus it is possible to isolate Adh genes among Pyrus species. However, the only genomic Adh sequence from a species of Rosaceae is that from Fragaria ananassa (X15588) in GenBank [50], and the exon/intron structure is unknown in Pyrus. The phylogenetic utility of Adh coding regions and the intron region have not been determined in any Rosaceae taxa.
Since no LCNG analysis had been applied to phylogenetic studies of Pyrus, we explored the utility of LEAFY and Adh. In this study, a comparison of genomic and RT-PCR-based approaches yielded an initial description of the composition and functionality of the Adh gene family in Pyrus. The phylogenetic utility of Adh gene regions and the second intron of LEAFY were determined after examining the sequence divergence, gene duplications, lineage sorting and recombination.
Malus taxa were once assigned to Pyrus, but Miller treated Malus Mill. as a separate genus in 1768 due to graft incompatibility between the two [51]. Malus taxa originated before Pyrus taxa according to the fossil occurrence [52], and are here used as outgroups (Table 1).
Table 1.
Plant taxa used in this study and subparalogs of LFY1int2, LFY2int2 and Adh1-1 recovered in each accession
| Accessions a | Species | Origin | Leaf source b | Subparalogs or copy typesc | ||
|---|---|---|---|---|---|---|
| LFY1int2 | LFY2int2 | Adh1-1 | ||||
| 'Korlaxiangli' | P. × sinkiangensis Yu | Xinjiang, China | CPGR | a, b | N | a, b, c |
| 'Cuiguan' | P. pyrifolia | Cross | Zhejiang University | / | / | / |
| 'Nanguoli' | P. ussuriensis | Liaoning Province, China | TU | a | N, Ins8 | a, b, c |
| 'Flemish Beauty' | P. communis | Belgium | ZZFI | / | / | / |
| 'Fuji' | M. domesitca | / | / | / | ||
| 'Chojuro' | P. pyrifolia Nakai | Kanagawa Pref. Japan | TU | a | N | a, b |
| 'Nijisseiki' | P. pyrifolia | Chiba Pref. Japan | TU | b | N | a, b, c |
| 'Yali' | P. pyrifolia White pear group | Hebei Province, China | TU | a, b | Del2, S | a |
| 'Dangshansuli' | P. pyrifolia White pear group | Anhui Province, China | ZZFI | a, b | N, Del2 | a, b, c |
| 'Yaguang' | P. ussuriensis Maxim. | Liaoning Province, China | CPGR | a | N, S | a, b |
| 'Jianbali' | P. ussuriensis | Liaoning Province, China | TU | a | N, S | a, b, c |
| P. pashia 1 | P. pashia D.Don | Yunnan Province, China | HRIYN | b | N | a, c |
| P. pashia 2 | P. pashia | Yunnan Province, China | HRIYN | b | N | a, b, c |
| P. dimorphophylla | P. dimorphophylla Makino | Mie Pref. Japan | TU | a, b | N, S | a, b |
| P. calleryana | P. calleryana Dcne. | South China | HRIYN | a, b | N, Del2 | a, c |
| P. fauriei | P. fauriei Schneid. | Korea | TU | a, b | S | a, b |
| P. betulaefolia | P. betulaefolia Bge. | Gansu Province, China | CPGR | b | N | a, c |
| P. × serrulata | P. × serrulata Rehd. | Hubei Province, China | CPGR | a, b | N | a, c |
| P. xerophila | P. xerophila Yu | Gansu Province, China | GPI | b | N | a, b |
| P. × hopeiensis | P. × hopeiensis Yu | Hebei Province, China | Hebei Province, China | a | N, Ins8 | a, b |
| P. × phaeocarpa | P. × phaeocarpa Rehd. | North China | CPGR | a | N | a, b, c |
| P. hondoensis | Nakai & Kikuchi | Middle Japan | TU | a | N, Ins8 | a, b, c |
| P. communis | P. communis L. | Europe | TU | b | 21-bp deletion | a, b, c |
| P. elaeagrifolia | P. elaeagrifolia Pall. | Turkey, Crimea, South East Europe | TU | b, c | N | a, c |
| P. amygdaliformis | P. amygdaliformis Vill. | Mediterranean area, South Europe | TU | b, c | N | a, c |
| P. cossonii | P. cossonii Rehder. | Algeria | TU | b | N | a, b |
| outgroup | ||||||
| M. sieboldii | M. sieboldii (Regel.) Rehd | Yunnan Province, China | HRIYN | / | N | a, b, c |
| M. rockii | M. rockii Schneid. | Yunnan Province, China | HRIYN | / | / | b, c |
| M. domestica subsp. chinensis | M. domestica subsp. chinensis Li Y. N.-(Nai) | North China | / | / | a, b, c | |
| M. neidzwetzkyana | M. neidzwetzkyana (Dieck) Langenf. | Xinjiang (Uygur Autonomous Region) | / | / | b, c | |
| 'Rall' | M. domestica Borkh | / | / | / | ||
a Accessions used for RT-PCR are in bold. b TU: Tottori University, Japan; CPGR: China Pear Germplasm Repository, Xingcheng, Liaoning Province; ZZFI: Zhengzhou Fruit Institute, Chinese Academy of Agriculture Science, Zhengzhou, Henan Province, China. GPI: Gansu Pomology Institute, Gansu Academy of Agricultural Science, Gansu Province, China; HRIYN: Horticultural Research Institute, Yunnan Academy of Agricultural Sciences, Kunming, Yunnan Province, China. c Different copy types of LFY1 and LFY2 identified by indels and phylogenetic analyses.
Results
Gene structure and paralog identification based on long Adh sequences
A total of 17 Adh1 and eight Adh2 long partial sequences were obtained by Genomic-PCR (G-PCR) using different primer sets (Table 2) in 'Cuiguan' (P. pyrifolia), 'Nanguoli' (P. ussuriensis), 'Korlaxiangli' (P. sinkiangensis), 'Flemish Beauty' (P. communis), M. rockii, M. domestica subsp. chinensis and 'Ralls' (M. domestica). All of these Adh genes encoded medium-chain ADH enzymes with 380 amino acid residues. After phylogenetic analyses, gene structure and sequence divergence comparisons, two paralogs representing Adh1 (Adh1-1, Adh1-2) and Adh2 (Adh2-1, Adh2-2) were identified.
Table 2.
Primers used for PCR amplification and sequencing in this study
| Target region | Primer sequence (5'-3') | |
|---|---|---|
| Adh | Long partial region | Adh1-F1:ATGTCTAATACTGCTGGTCA Adh1-F2: TGATGTTTACTTCTGGGAGG Adh1-R1: GATTGAATTGTGTTCTTTA Adh1-R2: TGTGGATTATGCAACGAAGA Adh2-F: TGTTGACTTCTGGGATGCCAA Adh2-R: ATGCTAACGATGCACCGCAA |
| Internal primer | Adh1-F5: AGGAGAATGCAAGGACTGCGCT Adh2-F5: CATTGCAAGTCTGAGGAAAG |
|
| reduced Adh1 | Adh1-F2 Adh1-R3: CAAAATGGTAGATAGGCTT |
|
| Specific primers | spAdh1-F: TCTACCATTTTGTTGGGACT spAdh1-R: AACGCTTCCTGTACATTCAA |
|
| spAdh2-F: GATTAATCACTTCCTCGGCA spAdh2-R: TAATATAGCCGGTGCACTCT |
||
| LEAFY | Long partial | LFY-F: TGTCGGAGGAGCCAGTGCAA LFY-R: GGCGTAGCAGTGCACATAGT |
| LFY1int2 | LFY1-F: TGGACGTTCATCAATAAAGA LFY1-R: AGTCGAACTAAATAGTTGAA |
|
| LFY2int2 | LFY2-F: GTGGGCCCATTTCCTGTAGT LFY2-R: GTTAAATCCGGTCAGATTAT |
|
| LFY2s | LFY2S-F: CTGTATTGACTATTTCTGTC | |
| MLFY2-F: CGTACGCTTATTTCTACTGCA LFY2-R |
||
| Actin | Partial | Pact-F: CCATCCAGGCTGTTCTCTC Pact-R: GCAAGGTCCAGACGAAGG |
As shown in Figure 1, Adh2-1 had a classical Adh gene structure with nine introns similar to Zea mays, Fragaria ananassa, and other characterized plant Adh genes [53]. Exon and intron codes described in this study were named following the classical gene structure to avoid confusion. Adh2-2 had lost intron 4, while both Adh1-1 and Adh1-2 had lost intron 7. Adh1-1 and Adh1-2 have the same gene structure, but the former was obtained by downstream primer Adh1-R1 located in the 3'UTR region, while the latter was obtained only by downstream Adh1-R2 located in the last exon (Figure 1). This indicates that the 3' UTR region of Adh1-2 may be divergent from that of Adh1-1. However, we were unable to obtain the 3'UTR region of Adh1-2. Adh1-2 obtained in 'Cuiguan' (P. pyrifolia) displayed a 20-bp deletion in exon 4. One of the three Adh1-2 clones in 'Korlaxiangli' (P. sinkiangensis) and one of the three Adh1-2 clones in 'Ralls' (M. domestica) displayed one or two 1-bp deletions, respectively, in exonic regions. A stop codon occurred in the exonic region in one of the two Adh2-2 sequences of M. rockii. These sequences were deemed putative pseudogenes and were removed from subsequent sequence analyses.
Figure 1.

Schematic diagram of Adh and LEAFY genes. Open boxes represent exons, and connecting lines represent introns. Combinations of neighboring exons show the loss of intron 4 in Adh2-2 and the loss of intron 7 in Adh1-1 and Adh1-2. Arrows indicate the locations and directions of primers used for PCR amplification. Rows a, b and c are different Adh genes from Malus and Pyrus obtained in this study; Row d is the genomic Adh sequence from Fragaria (X15588); Row e is Adh1-F from Zea (AF050457); Row f is LEAFY from Malus and Pyrus. For Rows b and c, the exon 1 and intron 1of Adh2-1 and Adh2-2 were not amplified by primers Adh2-F and Adh2-R, and the empty boxes indicate incomplete exon 2 and exon 10.
Among these four Adh paralogs (Adh1-1, Adh1-2, Adh2-1 and Adh2-2), only the intron regions of Adh1-1 and Adh1-2 could be aligned. Therefore, only the coding regions were used for nucleotide sequence divergence (NSD) and amino acid sequence divergence (ASD) comparisons. As shown in Table 3 NSD between Adh1 and Adh2 paralogs was as high as 0.3, while that within each paralog was lower than 0.02. NSD between Adh2-1 and Adh2-2 (0.19) was much greater than that between Adh1-1 and Adh1-2 (0.06). NSD of Adh1-1 and Adh1-2 between Pyrus and Malus were 0.027 and 0.035, respectively. All of these sequence divergence comparisons were consistent with the consequent phylogenetic inferences, indicating that our identification of paralogs was accurate. In most cases, NSD was greater than ASD between different homologs, while NSD was less than ASD within each homolog.
Table 3.
Sequence divergence (mean value) of the coding regions between and within Adh homologs (excluding putative pseudogenes)
| Pyrus Adh1-2 (3)c | Malus Adh1-2 (1) | Pyrus Adh1-1 (6) | Malus Adh1-1(4) | Malus Adh2-2 (3) | Pyrus Adh2-1 (4) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NSDa | ASDb | NSD | ASD | NSD | ASD | NSD | ASD | NSD | ASD | NSD | ASD | |
| Pyrus Adh1-2 | 0.015d | 0.021 | / | / | 0.016 | 0.019 | 0.016 | 0.027 | 0.017 | 0.017 | 0.015 | 0.024 |
| Malus Adh1-2 | 0.035 | 0.027 | ||||||||||
| Pyrus Adh1-1 | 0.060 | 0.047 | 0.062 | 0.046 | ||||||||
| Malus Adh1-1 | 0.058 | 0.050 | 0.062 | 0.050 | 0.027 | 0.039 | ||||||
| Malus Adh2-2 | 0.343 | 0.235 | 0.339 | 0.226 | 0.340 | 0.243 | 0.333 | 0.235 | ||||
| Pyrus Adh2-1 | 0.322 | 0.185 | 0.318 | 0.191 | 0.321 | 0.191 | 0.318 | 0.190 | 0.197 | 0.159 | ||
a Sequence divergence based on nucleotide sequence (NSD)
b Sequence divergence based on amino acid sequence (ASD)
c The numbers in the parentheses indicate the number of sequences, and both NSD and ASD in the table represent mean values.
d Sequence divergence within each homolog is highlighted in bold.
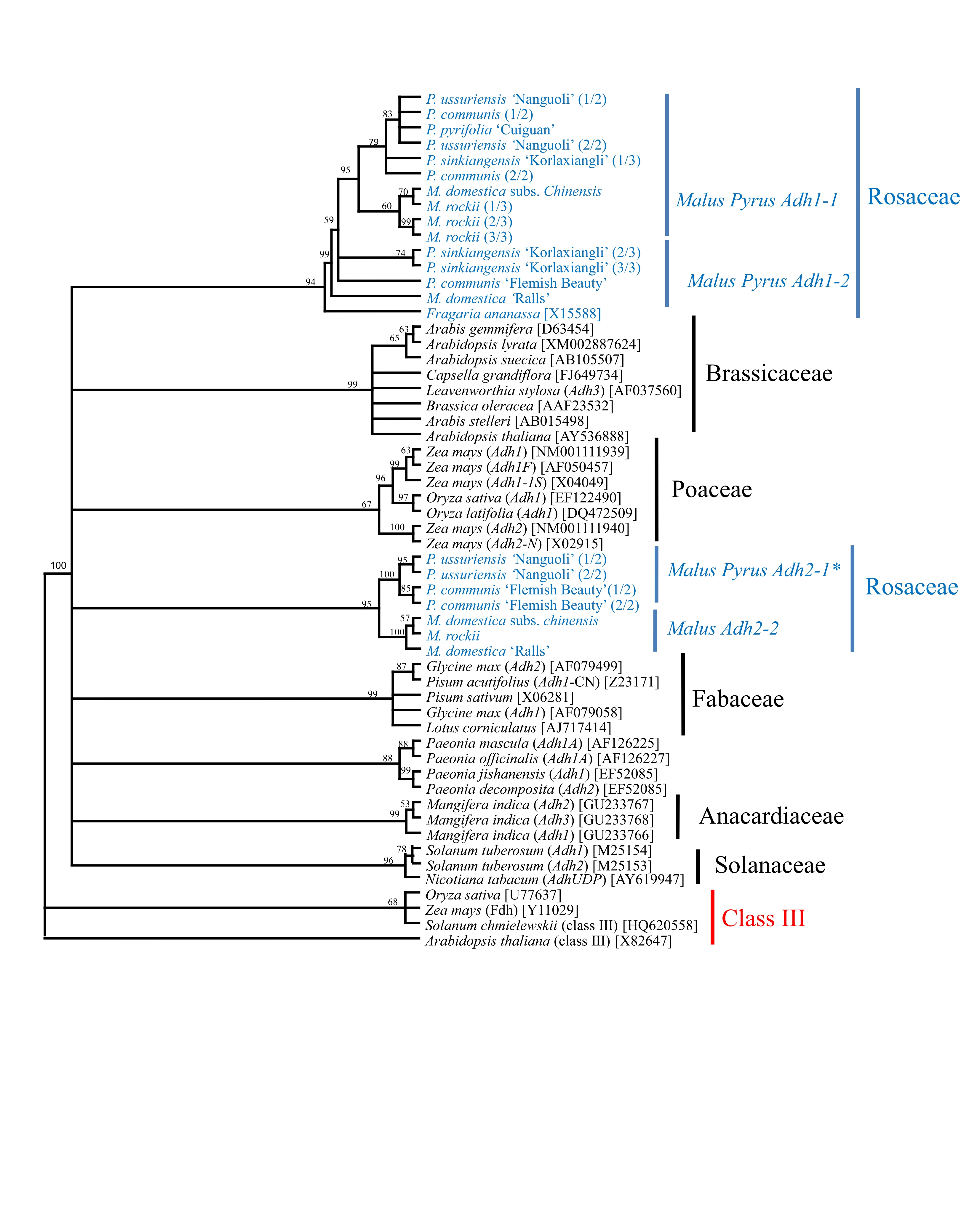
Both maximum parsimony (MP) (Additional file 1) and neighbor joining (NJ) (Figure 2) trees based on amino acid sequences of Adh genes from different plant taxa displayed similar topologies. Class III ADH formed clades separate from the putative class P ADH. The Adh genes from the Solanaceae, Brassicaceae, Anacardiaceae, and Fabaceae were monophyletic with high bootstrap values, suggesting that duplication events leading to these Adh genes occurred independently after diversification of these plant families. The Adh genes from Paeoniaceae and Poaceae also formed monophyletic clades; however, with low bootstrap values. Different Adh genes in Malus and Pyrus (Rosaceae) were not monophyletic. Adh1-1 and Adh1-2 in Malus and Pyrus formed two sister clades with Fragaria ananassa as their sister clade, suggesting that gene duplication leading to these two outparalogs occurred prior to diversification of Malus and Pyrus. Adh2-1 and Adh2-2 in Malus and Pyrus formed another clade. Due to lack of related sequences in other Rosaceae taxa, we cannot infer their origin. Among investigated families, Fabaceae is the most closely related to Rosaceae, but their Adh genes did not show a close relationship, indicating that the Adh genes have become highly diversified within each family.
Figure 2.

Neighbor-joining (NJ) tree based on amino acid sequences of Adh loci from diverse plant taxa. ADH sequences in Rosaceae are highlighted in blue. Numbers above the branches or near the branch nodes indicate bootstrap values (1000 replicates). GenBank accession numbers are in brackets. Multiple intra-individual clones for Adh1 (Adh1-1 and Adh1-2) and Adh2 (Adh2-1 and Adh2-2) are differentiated by the number in the parenthesis following the taxa name. *: Though Adh2-1 was not obtained by G-PCR in Malus, its transcription was detected by RT-PCR in 'Fuji' (M. domestica).
Phylogenetic analyses based on reduced Adh1 sequences
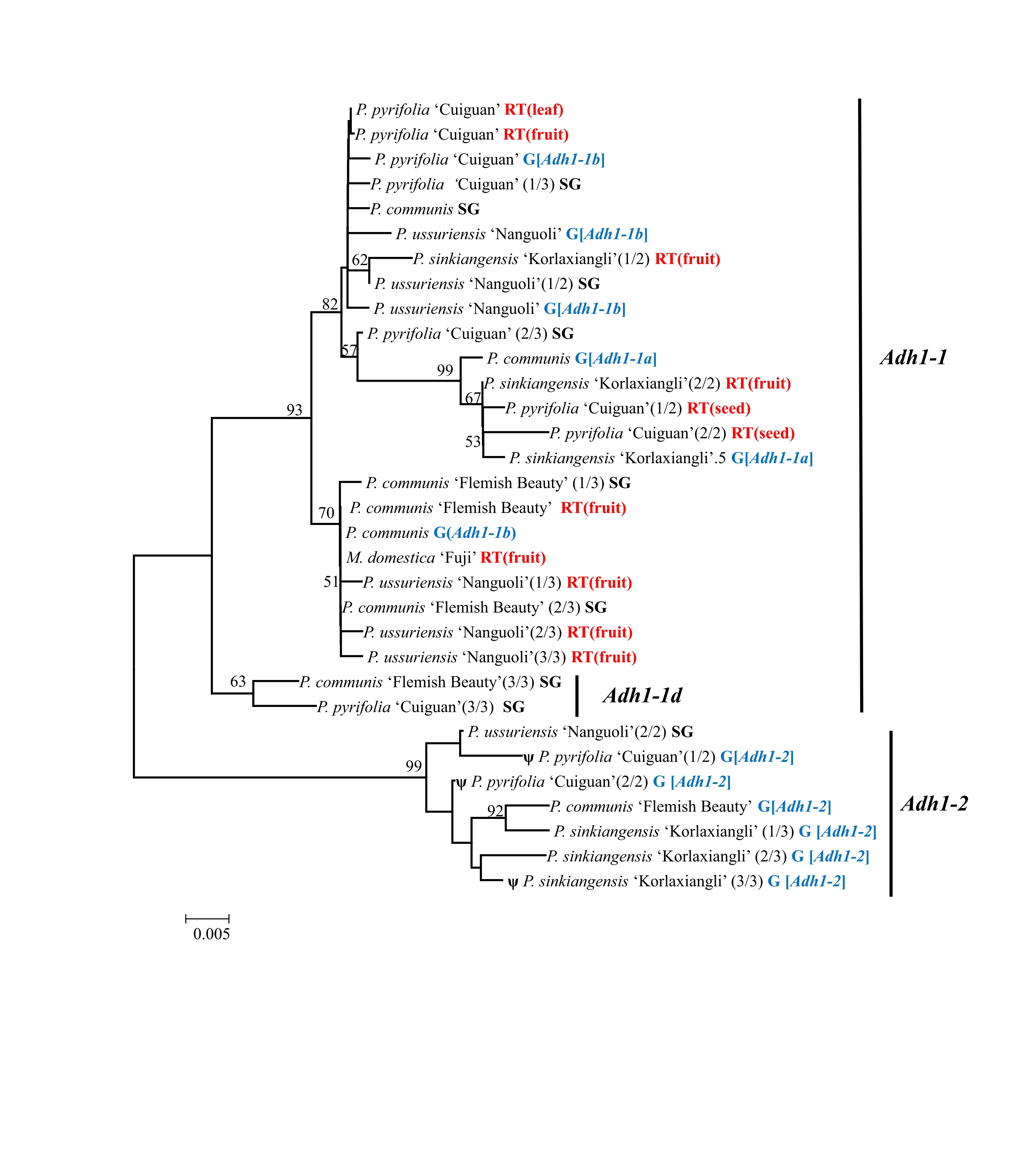
Only a short region containing intron 2 and intron 3 of Adh1 (reduced Adh1) were sequenced from more Pyrus species to investigate the phylogenetic utility of this region. Adh1-1 was preferentially amplified and only two Adh1-2 sequences were obtained in P. amygaliformis and 'Dangshansuli' (P. pyrifolia, CWP). Three subparalogs, Adh1-1a, Adh1-1b, Adh1-1c, were supported by the tree topologies (Figure 3). These were outparalogs that occurred before Malus and Pyrus diversification. Only Adh1-1c was obtained in all Pyrus accessions, while all of the three Adh1-1 outparalogs were derived in the same nine accessions. Multiple intra-individual polymorphic sequences representing one Adh1-1 outparalog could be recovered in some individuals. For example, five Adh1-1c sequences of P. × hopeiensis displaying autapomorphic mutations were monophyletic in the tree, but only one sequence was retained in the final dataset. Based on our previous experience, such small mutations were more probably caused by Taq polymerase errors during cloning and PCR. Among these subparalogs, the length of intron 2 varied from 228 to 262 bp while that of intron 3 varied from 114 to 117 bp, and subparalog associated indels were observed (data not shown). A string of thymine residues from 8 bp to 22 bp among Adh1-1c copies made alignment difficult, thus this region was removed from phylogenetic analyses. Two sequences possessing characteristics (indels and substitutions) of different Adh1-1 outparalogs were identified as recombinants by Recombination Detection Program (RDP) [54], and 13 more similar putative recombinants were identified manually by observing their conflicting positions within the alignment. These putative recombinants were probably artificial products created during PCR and were excluded from all analyses.
Figure 3.

Bayesian majority-rule consensus tree for reduced Adh1. Posterior probabilities and bootstrap values greater than 50 are provided above and below the branches, respectively. Outgroup accessions are highlighted in blue, while occidental species are in green. Multiple intra-individual clones for each of the three major clades (Adh1-1a, Adh1-1b and Adh1-1c) are differentiated by the fraction in the parenthesis following the taxon names, and those within the Adh1-1a clade are highlighted by different shapes and colors. The 'G' in the end of the taxa name indicates sequences obtained by G-PCR.
The final dataset contained 101 Adh1-1 and six Adh1-2 copies with an aligned length of 677 bp. The NSD within each Adh1-1 subparalog was very low, ranging from 0.011 to 0.013 (excluding Malus accessions) (Table 4). Adh1-2 from 'Ralls' (M. domestica) was selected as the outgroup for phylogenetic analyses. MP (data not shown) and Bayesian analyses (Figure 3) of this dataset resulted in similar phylogenetic trees with little difference in support values (the Bayesian posterior probabilities are generally higher than bootstrap percentage). Adh1-1 and Adh1-2 formed two separate clades. Within the Adh1-1 clade, Adh1-1a and Adh1-1b were closely related and formed a sister clade to Adh1-1c. Adh1-1c was obtained in all accessions, but the relationships were poorly resolved with extensive polytomies. The occidental species, P. communis, P. amygdaliformis, P. elaeagrifolia and P. cossonii, were not monophyletic. Most intra-individual polymorphic sequences were polyphyletic, e.g. P. calleryana, P. xerophila, 'Nijisseiki' (P. pyrifolia) and 'Korlaxiangli' (P. sinkiangenesis). Adh1-a and Adh1-b were only obtained in some accessions, thus the phylogenetic relationships in these two clades were incomplete, but the occidental species were monophyletic in both clades.
Table 4.
Sequence variations of Adh1-1 and LEAFY subparalogs in Pyrus (excluding the Malus accessions)
| Reduced Adh1 | The second intron of LEAFY | ||||
|---|---|---|---|---|---|
| Adh1-1a | Adh1-1b | Adh1-1c | LFY1int2 | LFY2int2 | |
| N a | 37 | 30 | 17 | 53 | 43 |
| NSD b | 0.012 | 0.013 | 0.011 | 0.019 (0.016)d | 0.029 (0.028)e |
| PI c | 25/617 | 21/633 | 11/645 | 61/653 (42/653)d | 46/562 (40/553)e |
a The number (N) of sequences used for analyses.
b Mean sequence divergence of nucleotide sequence (NSD) calculated by the K-2P method.
c The number of parsimony-informative (PI) sites after alignment.
d PI sites and NSD in brackets were calculated after excluding LFY1int2-c.
e PI sites and NSD in brackets were calculated after excluding LFY2int2-Ins8 and LFY2int2-Del2.
Transcription of Adh homologs
Specific genomic PCR (SG-PCR) and RT-PCR using locus specific primers produced expected bands in the genomic DNA and cDNA samples. Both Adh1 and Adh2 were transcribed in all investigated tissues and cultivars (Figure 4). To investigate the transcription of different Adh homologs, phylogenetic analyses including sequences derived from SG-PCR, G-PCR and RT-PCR were conducted for Adh1 and Adh2 separately, and the putative pseudogenes identified above were also included to enhance the findings on functionality of Adh homologs.
Figure 4.

Genomic (SG)-PCR and RT-PCR of Adh1 and Adh2. Bands of the molecular marker from the bottom up denote size standards of 0.1 kb, 0.2 kb, 0.5 kb, 0.75 kb, 1 kb and 2 kb. Row a is SG-PCR of Adh1 and Adh2 in 'Cuiguan'(C1, C2), 'Korlaxiangli' (K1, K2), 'Nanguoli' (N1, N2) and 'Flemish Beauty'(F1, F2); Rows b, c and d are RT-PCR of Adh1, Adh2 and control Actin gene in investigated tissues and cultivars. Cs, Cl and Cf denote seed, leaf and fruit of 'Cuiguan', respectively; Kf, Nf, Ff and Mf-fruit denote fruit of 'Korlaxiangli', 'Nanguoli', 'Flemish beauty' and 'Fuji', respectively.
As indicated in Additional file 2 the transcription of an Adh1-2 copy was not observed. We speculate that Adh1-2 is a nonfunctional outparalog, since putative pseudogenes have been identified and its 3'UTR region is divergent from Adh1-1 as described above. Among the Adh1-1 outparalogs the most frequently cloned Adh1-1c was not recovered by RT-PCR, indicating that it was a degenerate outparalog. This also explains its preferential amplification. Transcription of the other Adh1-1 outparalogs was detected among different tissues or cultivars. Transcription of Adh2-1 could be detected in all investigated tissues and cultivars. An Adh2-1 sequence was not obtained in the three Malus accessions by G-PCR, but its transcription was found in 'Fuji' (M. domestica) (Additional file 3). Adh2-2 was only observed in Malus taxa, and transcription of this paralog was not detected. We could not deduce distinct tissue-specific or cultivar-specific expression for either Adh1-1 or Adh2-1. Several anomalous Adh2-1 copies with only intron 6 were recovered by cloning of RT-PCR products in 'Flemish Beauty' (P. communis) and 'Nanguoli' (P. ussuriensis) (HQ912054, HQ912055 and HQ912056). Since intron-containing cDNA has not been reported, those copies may be amplified due to genomic DNA contamination and indicate existence of an additional Adh2 paralog with more intron loss in Pyrus.
Sequence variation and paralogs of LEAFY
Twenty-six LFY1 and 27 LFY2 sequences including a partial exon 2 were obtained. Four groups with different length variations in the exon region were observed: LFY1-Malus (407 bp), LFY1-Pyrus (395 bp), LFY2-Malus (410 bp) and LFY2-Pyrus (401 bp), which were congruent with those from RT-PCR in P. pyrifolia 'Housui' (LFY1-AB162029, LFY2-AB162035) and M. domestica 'Fuji' (LFY1-AB162028 LFY2-AB162034), respectively. The length of these indels only had an effect on the length of the deduced amino acid sequences. Among Pyrus accessions, NSD of this partial exon 2 between LFY1 and LFY2 was relatively high (0.076), while that within LFY1 and LFY2 was low at 0.013 and 0.015, respectively (data not shown).
The entire intron 2 of LFY1 among Pyrus species ranged from 774-783 bp, while that of LFY2 ranged from 670 to 700 bp. LFY1int2 and LFY2int2 were amplified in all accessions using specific primers, but LFY2int2 of M. rockii and M. domestica subsp. chinensis were amplified by another forward primer MLFY2-F (Table 2) due to a large deletion (approximately 220 bp, GU991522 vs DQ535886) in these two accessions. LFY2int2 of all Malus accessions contained a 211-bp insertion, which made alignment difficult and was removed from the analyses. NSD within LFY1int2 and LFY2int2 among Pyrus accessions was 0.019 and 0.029, respectively (Table 4), while that between Malus and Pyrus was much higher at 0.057 for LFY1int2 and 0.066 for LFY2int2 (data not shown).
Sequence variation and phylogenetic analyses of LFY1int2 suggest two subparalogs, LFY1int2-a and LFY1int2-b, among Pyrus species. Compared with LFY1int2-a, LFY1int2-b contains a 6-bp insertion. Recovery of LFY1int2-a and LFY1int2-b in each accession is shown in Table 1. Among oriental species, NSD within LFY1int2-a was lower than that within LFY1int2-b for both ORF regions (0.002, 0.009) and intron regions (0.005, 0.013). Between LFY1int2-a and LFY1int2-b, NSD was 0.007 for the ORF region and 0.017 in for the intron region. For LFY2int2, subparalogs with an 8-bp insertion (LFY2int2-Ins8) and 2-bp deletion (LFY2int2-Del2) were only recovered in a few accessions. The common LFY2int2-N, with no indel, was recovered in all accessions but 'Yali' (P. pyrifolia, CWP) (Table 1). Coexistence of these subparalogs in one individual could be detected by direct sequencing due to the fixed position of indels. A minimum of three clones were sequenced. It was found that PCR or direct sequencing sometimes did not reflect the subparalogs existing in one genome, probably due to amplification preference of different nuclear alleles. For example, direct sequencing of LFY2int2 in P. calleryana identified LFY2int2-N, but a LFY2int2-Del2 sequence was obtained by cloning.
Several anomalous LFY2int2 copies were exclusively found in particular accessions. LFY2int2 from P. communis had a 21-bp deletion. LFY2int2 from P. fauriei had a 525-bp insertion that was partially homologous (reverse and complement) to the noncoding region of the S-RNase gene (AB308360), and was named LFY2int2-S. To eliminate the possibility of genetic recombination during PCR, all accessions were tested with an insert-specific upstream forward primer 'LFY2S-F' (Table 1) and the reverse primer LFY2-R. As a result, the LFY2int2-S was detected in 'Yali' (P. pyrifolia, CWP), 'Jianbali' (P. pyrifolia CWP), and 'Yaguang' (P. ussuriensis), but the sequences were not included in the phylogenetic analyses. LFY2int2-S in P. fauriei was still included in the dataset after the exclusion of its large insertion.
Phylogenetic analyses of LFY1int2 and LFY2int2
Malus sieboldii and M. domestica (DQ535885-ALF1, DQ535886-ALF2) without the 220-bp deletion in LFY2int2 were used as outgroups in phylogenetic analyses of LFY1int2 and LFY2int2. Putative recombinants were identified by RDP or by investigating abnormal substitution patterns and ambiguous alignment positions. The putative recombinants displayed unique substitutions of sequences from two distinct subclades and always formed well-separated clades in the tree, thus they were excluded from the final analyses (data not shown). A total of 57 LFY1int2 (four from Malus) and 46 LFY2int2 (three from Malus) sequences were included in two separate datasets. Excluding sequences from Malus, the LFY1int2 dataset had an aligned length of 653 sites, of which 61 (9.3%) were parsimony informative, while LFY2int2 had an aligned length of 562 sites and 46 (8.2%) were parsimony informative (Table 4). Similarly, only Bayesian trees were shown for both LFY1int2 and LFY2int2 datasets.
As shown in Figure 5, LFY1int2-a formed a monophyletic clade. The relationships within this clade were largely unresolved; however, close relationships among P. × serrulata, P. calleryana and 'Yali' (P. pyrifolia, CWP) were resolved with high bootstrap support. Most of the subclades of LFY1int2-b were unresolved polytomies sister to LFY1int2-a. The occidental species were not monophyletic, since three sequences from P. amygdaliformis and P. elaeagrifolia formed a highly supported independent clade. These three sequences, representing a paralog termed LFY1int2-c, displayed many unique variations and shared several substitutions with outgroup accessions. LFY1int2-c was reamplified in these four occidental species, and could be frequently cloned in P. amygdaliformis and P. elaeagrifolia. The origin of LFY1int2-c is unclear.
Figure 5.

Bayesian majority-rule consensus trees for LFY1int2. Posterior probabilities and bootstrap values greater than 50 are provided above and below the branches, respectively. Outgroup accessions are highlighted in blue, while occidental species are in green. Multiple intra-individual clones are differentiated by the fraction in the parenthesis following the taxon names. Accessions possessing both LFY1int2-a and LFY1int2-b are marked by different shapes and colors.
Relationships inferred by LFY2int2 were much better resolved, with higher support values and fewer polytomies (Figure 6), than those of LFY1int2., but the relationships within most subclades were still unresolved. Two major clades were resolved. In clade I, the four occidental species and a clone of 'Korlaxiangli' (P. sinkiangensis) formed a subclade sister to that of oriental pear including 'Yaguang' (P. ussuriensis), P. betulaefolia, P. × phaeocarpa and P. × hopeiensis. In clade II LFY2int2-Ins8 and LFY2int2-Del2 were monophyletic and mixed with LFY2int2-N sequences, thus they were inparalogs of recent origin. It is notable that the four LFY2int2-N sequences from P. xerophila were putative recombinants. They shared mutations with sequence from multiple subclades and also had unique mutations. Phylogenetic positions of these P. xerophila sequences were unresolved: P. xerophila (2/4) and P. xerophila (3/4) were sisters and formed a separate clade, P. xerophila (4/4) also formed a separate clade, and P. xerophila (1/4) shared mutations with occidental species and formed a separate subclade within clade I. It seems impossible that divergent copies in one genome are all recombinants, thus these sequences were included in the phylogenetic analyses and are highlighted in bold in Figure 6. Similarly, the LFY2int2-S of P. fauriei formed an unresolved separate clade.
Figure 6.

Bayesian majority-rule consensus trees for LFY2int2. Posterior probabilities and bootstrap values greater than 50 are provided above and below the branches, respectively. Outgroup accessions are highlighted in blue, and occidental species in green. Multiple intra-individual sequences are differentiated by the fraction in the parenthesis following the taxon names. Accessions possessing both LFY2int2-N and LFY1int2-Del2 or LFY1int2-Ins8 are marked by different shapes and colors. Excluding the two inparalogs of LFY1int2-Del2 or LFY1int2-Ins8, intra-individual sequences representing LFY2int2-N are highlighted in red.
Discussion
Frequency of Adh and LEAFY duplication
Gene duplication plays an important role in increasing the diversity of gene function and expression, which can enable plants to colonize diverse habitats. Most monocots and dicots have at least two Adh genes, indicating that an initial Adh gene duplication occurred before the divergence of these plant taxa, and separate duplications have subsequently taken place [46]. Two major loci in Malus and Pyrus, Adh1 and Adh2, are outparalogs derived from an ancient gene split. Based on an estimated 0.66% rate of nucleotide substitution per million year of Adh in Drosophila [55] and 0.2-0.3% in mammalian nuclear genes [56], the split occurred approximately 50 million years ago. In maize Adh1 and Adh2 share 87% identity at the amino acid sequence level but are located on different chromosomes and differ in the level of tissue-specific expression [57]. The expression of three Adh genes with 85% and 87% shared amino acid identity in Vitis vinifera varied in developmental stage of grape berries and affinity to either ethanol or acetaldehyde as a substrate [58]. ADH from apple had optimal acetaldehyde activity at pH 5.5-6.0 and ethanol activity at pH 7.0-10.0 [59]. Therefore, Adh1-1 and Adh2-1 in Pyrus (ASD = 0.170, Table 3) with less shared identity, likely also have diversified their expression patterns and substrate affinity. ADH plays an essential role in the biosynthetic pathway of aroma volatiles in apple and pear fruits by reducing aldehydes to alcohols [60-62]. It will be interesting to determine functional divergence of Adh genes and kinetic properties of the corresponding ADH enzymes in Pyrus and Malus.
Two paralogs representing Adh1 (Adh1-1 and Adh1-2) and Adh2 (Adh2-1 and Adh2-2) were observed by G-PCR. Among these the Adh2-1 may be the most ancestral since it has the classical nine-intron Adh structure, which is widely conserved among angiosperms and gymnosperms [53]. Intron losses have been found in Adh genes of diverse taxa like Arabidopsis thaliana [63,64] and Mangifera indica [65]. In some species of Leavenwortia, an expressed intronless Adh3 locus occurs and is thought to have arisen by an mRNA intermediate [66]. The single intron loss found in our study lends support to the 'intron exclusion hypothesis', which suggests that a single intron could be precisely removed by double strand breaks from a multiple-intron gene [67]. Adh1-2, with the same gene structure as Adh1-1, was a putative nonfunctional outparalog derived by gene duplication (loss of intron 7, Figure 1). Gene duplication leading to Adh1-2, as inferred by the tree topology (Figure 2), occurred before Malus and Pyrus diverged, but probably after diversification of Pyreae taxa, since the orthologous Adh gene in Fragaria (Rosoideae) displayed the classical nine-intron structure. Additionally, the NSD between Adh1-1 and Adh1-2 in Pyrus was 0.06 (Table 3), which was much lower than that between Adh1-1 in Pyrus and Adh in Fragaria (0.10) (data not shown). However, the NSD in the coding region between these outparalogs was too low to confirm the non-functionality of Adh1-2. Adh2-1 was not obtained in Malus accessions by G-PCR, probably due to an amplification preference for Adh2-2, but its transcription was detected in 'Fuji' (Additional file 3). Adh2-2 was recovered neither in Pyrus by G-PCR and SG-PCR nor in Malus or Pyrus by RT-PCR. Compared with Adh1-1 and Adh1-2, the ASD between Adh2-1 and Adh2-2 was much higher (Table 3) and exon/intron structure also varied (Figure 1). Due to the lack of highly homologous Adh2 sequences from other Rosaceae taxa, is the origin of Adh2-2 is uncertain. It may be a duplicated inparalog derived from Adh2-1 and restricted to Malus taxa or a functional outparalog that appeared before Malus and Pyrus diverged and was subsequently lost during diversification of Pyrus species. The latter theory is similar to the paralog sorting of RPB1 and RPB2 in different core eudicots taxa [68]. Specific RT-PCR for additional tissues and specific G-PCR for more Rosaceae taxa will be needed to examine the origin and transcription of Adh2-2. Three subparalogs representing Adh1-1 (Adh1-1a, Adh1-1b, Adh1-1c) were identified by accession sequencing (Figure 3), although Adh1-1c may not be transcribed. It is unknown whether similar subparalogs have evolved for Adh1-2 and Adh2-2.
Our study revealed that ancient and recent duplications led to the complex structure of Adh outparalogs in Pyrus and Malus resulting in neofunctionalization, nonfunctionalization and possible subfunctionalization, the three common fates of gene duplications. Adh homologs in Malus and Pyrus were more complex than those in other angiosperms like Paeonia [69], grasses [44] or legumes [70], but similar to those in Gossypium [48]. Gossypium has at least seven Adh loci of two primary lineages in diploid species and the Adh gene family is dynamic with pseudogenization and gene elimination. Genomic data suggest that almost all angiosperms, perhaps even all plant groups, have experienced one to several rounds of polyploidy [71,72]. Though Malus and Pyrus accessions used in this study were all diploid (x = 17), Pyreae taxa with x = 17 are derived from autopolyploidization of the formerly Spiraeoid ancestors with x = 9 [30]. This apparently accounts for such complex paralogs, and similar Adh gene structures could be imputed for other Pyreae taxa with x = 17.
LEAFY was first found to be a homeotic gene encoding a transcription regulator for differentiation of the floral meristem and flowering time in Arabidopsis and was expected to be a single-copy gene in diploid angiosperms [73]. In our study, two major lineages, LFY1 and LFY2, were recovered in both Pyrus and Malus, as in many other Pyreae taxa including the formerly Spiradeae taxa with x = 9 [39,41], suggesting gene duplication of these two paralogs before diversification of the Rosaceae. LFY1 and LFY2 in apple are located on distinct chromosomes and thus are not alleles [74]. In a study including the pear cultivars 'Housui' (P. pyrifolia) and 'Barlett' (P. communis), the transcriptional patterns of two LEAFY homologs differed in developmental stages and tissues, and each homolog varied among plant taxa [41]. In our study, genus-specific and locus-specific indels were discovered in coding regions, which would alter the length of the corresponding amino acid sequences. These might be responsible for the diversification of LEAFY gene functions.
Multiple inparalogs of LFY1int2 and LFY2int2 were observed, and their recovery varied among our accessions (Table 1). LFY1int2-a and LFY1int2-b were unequally observed among Pyrus species (Table 1). LFY1int2-a is monophyletic with shorter branch lengths than LFY1int2-b (Figure 5), suggesting it might be an inparalog derived from LFY1int2-b by a recent duplication. This would have occurred after the divergence of occidental and oriental pears, because only LFY1int2-a is not found in occidental pears. Consequently, LFY1int2-b was lost during diversification of some oriental species, which explains paralog sorting during diversification of Pyrus. Three LFY1int2-c sequences in two west Asian species (P. amygdaliformis and P. elaeagrifolia) formed a separate clade (Figure 5). We suggest two possible explanations for their origin: 1) they are pseudogenes derived from LFY1int2-b, but have evolved more rapidly, and thus are highly divergent from LFY2int2-b; 2) they represent another outparalog of LFY1int2, derived from gene duplication that occurred before diversification of the occidental species, that was subsequently lost in some occidental species. However, only the second intron of LFY1int2-c was sequenced. To help differentiate between these two possibilities the entire exonic region must be obtained and its presence in more Pyrus species investigated. For LFY2int2, the common LFY2int2-N was recovered in all accessions but 'Yali' (P. pyrifolia, CWP). LFY2int2-Ins8 and LFY2int2-Del2 are inparalogs that originated recently after Pyrus diversification and were only recovered in a few accessions (Figure 6). LFY2int2-S, with a long insertion homologous noncoding region of S-RNase gene, was similar to functional AFL1a copies found in some apple cultivars [74]. Genomic Southern analysis also showed that apple had other homologues in addition to AFL1 and AFL2 [75]. However, the relationships among these homologs have not been published. Only a few accessions contained LFY2int2-S (Table 1), and only the one from P. fauriei was included in our analyses. It is unknown how the intron of the RNase gene was inserted in the second intron of LFY2 and whether LFY2int2-S is functional. Additional research found both LFY2int2-S and LFY2int2-N in multiple species, and showed the LFY2int2-S sequences were all highly divergent from LFY2int2-N even after exclusion of the large insertion (unpublished data).
Incongruence and poor resolution
Paralogs and lineage sorting are two major challenges when conducting phylogenetic analyses based on LCNGs, because they can lead to incongruent patterns similar to those resulting from hybridization and polyploidization [31]. Paralogs reflect a horizontal event, the gene duplication in one species, while orthologs reflect a vertical event, the speciation in a lineage [36,76]. Thus, it is crucial to differentiate paralogs from orthologs by investigating their origins and monophyletic positions in the tree. We have clearly identified the outparalogs and inparalogs for both Adh and LEAFY genes. If inparalogs representing LFY1int2 and LFY2int2 were not identified, P. calleryana possessing both LFY1int2-a and LFY1int2-b as well as LFY2int2-N and LFY2int2-Del2 would be polyphyletic in both gene trees (Figure 5 and 6) and presumed to be hybrids involving other Pyrus species. However, P. calleryana is one of the most ancestral species in Pyrus and should not be a hybrid of other Pyrus species. Additionally, Pyrus ussuriensis, P. × hopeiensis, P. × phaeocarpa and P. hondoensis containing only LFY1int2-a fell in the same clade. This shows a close relationship (Figure 5). However, P. betuleafolia was not in the clade and only contained LFY1int2-b. These findings were inconsistent with the hypothesis that P. betuleafolia was involved in the origin of P. × hopeiensis and P. × phaeocarpa and to the phylogeny based on LFY2int2-N.
Interspecific hybridization has been considered the major mode of evolution for Pyrus [52], and LCNG has been useful for testing the hypothesis of hybridization, since homologs of a nuclear locus from both parents could be detected in putative hybrids through cloning [31]. Excluding the possibility of paralogs, incongruence caused by hybridization and lineage sorting could be differentiated by comparing phylogenies of multiple unlinked nuclear loci. Only Adh1-1c and LFY2int2-N are shown to be two independent orthologs, and they were recovered in all accessions (except LFY2int2-N in 'Yali'). As described above, relationships revealed by Adh1-1c were poorly resolved, and most intra-species and intra-individual sequences were polymorphic. As shown in Figure 3, intra-individual sequences of P. calleryana, P. hondoensis and P. dimorphophylla were polyphyletic, and the occidental species were not monophyletic. However, the putative interspecific hybrids, P. × hopeinensis and P. × phaeocarpa, were monophyletic, which was incongruent with other gene trees and our previous understanding of these species. In contrast, only four accessions were polymorphic in the LFY2int2-N tree, including P. × phaeocarpa, P. × hopeiensis, and 'Korlaxiangli' (P. × sinkiangensis) (marked in red in Figure 6), all of which were putative interspecific hybrids. Therefore, lineage sorting of ancestral polymorphic Adh1-1c alleles may have occurred during diversification of Pyrus.
The phylogenetic relationships revealed by LFY2int2-N were mostly congruent to other orthologous gene trees and previous studies based on other data. Close relationships among P. calleryana, P. dimorphophylla, P. pashia, P. pyrifolia, and/or P. × serrulata were supported by all gene trees, suggesting a close relationship among these species. Two distinct LFY2int2-N sequences of 'Korlaxiangli' (P. sinkiangensis) were grouped with occidental species and P. pyrifolia, respectively (Figure 6). Similar relationships were found in Adh1-1a, Adh1-1c and Adh1-2 clades (Figure 3), which supports the hypothesis that P. sinkiangensis is an interspecific hybrid involving at least P. communis and P. pyrfolia [11]. Intra-individual copies of P. × hopeiensis and P. × phaeocarpa were grouped with P. ussuriensis, P. hondoensis or P. betulaefolia in both the Adh1-1a clade (Figure 3) and LFY2int2-N (Figure 6) clade. Pyrus × phaeocarpa was a putative hybrid involving P. betulaefolia and P. ussuriensis, and P. × hopeiensis was a hybrid involving P. × phaeocarpa and P. ussuriensis. Pyrus hondoensis, which was once classified as a variety of P. ussuriensis by morphological data [3], and P. ussuriensis were found to be closely related [12]. Phylogenetic relationships among these species were supported by multiple orthologous gene data, suggesting ancient hybridization rather than lineage sorting. More wild individuals of these species are needed to test such complex evolutionary histories.
The relationships based on all separate orthologs were mostly poorly resolved. In our study, different Adh and LEAFY paralogs showed a relative low sequence divergence (< 0.03). LFY2int2-N showed the highest proportion of informative sites (38/562, 6.8%, Table 4) which was similar to results in Neillia and Stephanandra (7.4%) [39]. Low sequence divergence of multiple DNA regions suggests rapid radiation during divergence [77] and this has been hypothesized for many Pyreae taxa (the former Maloideae taxa) [26,40]. This may also explain the poor resolution of the gene trees. Another contribution to the poor resolution in this study is the conflicting signals caused by recombinants. Recombinants are derived from two homologous chromosomes in one genome during meiosis (genic recombinants) or PCR (artifacts), leading to incorrect phylogenetic inferences [78-80]. As predicted by statistical principles, we found that putative recombinants formed separated clades. Recombinants represent substitutions of two distinct lineages, and thus receive no bootstrap support from either of the lineages in a cladistic phylogeny (data not shown). In this study, most putative recombinants represent one of the intra-individual polymorphic copies and were excluded from analyses. The four LFY2int2-N copies of P. xerophila all displayed the characteristics of recombinants, and formed separate clades in the tree (Figure 6). Polymorphic LFY2int2-N copies in P. xerophila may all be ancient genetic recombinants that arose by interspecific hybridization involving both oriental and occidental species. More individuals of P. xerophila and occidental species are necessary to confirm this hypothesis and investigate the origin of this species.
Phylogenetic utility of the introns
Among three Adh1-1 subparalogs, Adh1-1c was orthologous and recovered in all accessions, but it resulted in a poorly resolved phylogeny due to lack of informative sites and possible lineage sorting (Table 3). This makes it inadequate for the phylogenic reconstruction of Pyrus. The two introns of Adh1-2, Adh2-1 and Adh2-2, were not sequenced and analyzed in the current study, and it is unknown whether multiple subparalogs also exist for these paralogs. Adh2-2 might not exist in Pyrus species as discussed above. The phylogenetic utility of the introns of Adh2-1 and Adh1-2 needs to be estimated, which will require primers designed specifically to the paralogs.
LFY1int2 was not suitable for studying interspecific relationships due to sorting of LFY1int2-a and LFY1int2-b paralogs and the unclear origin of LFY1int2-c. In contrast, LFY2int2-N showed the highest sequence divergence, resulting in the best-resolved tree. Inparalogs of LFY2int2-Ins8 and LFY2int2-Del2, as well as the LFY2int2-S of unclear origin, could be easily identified and removed from phylogenetic inferences. Most importantly, relationships based on LFY2int2-N were congruent to previous studies based on morphological and molecular marker data. Conflicting placement of species may be resolved by using LFY2int2-N. It provides reliable evidence of ancient hybridization, since incomplete lineage sorting was not imputed for LFY2int2-N. Phylogenetic studies of Pyrus based on nuclear gene regions have been rare. Only the ITS region has been applied to a wide range of East Asian Pyrus species, but it resulted in a poorly resolved tree [28]. One study based on the 18S gene focused only on two species, P. pyrifolia and P. communis [81]. We conclude that LFY2int2-N is currently the most useful nuclear gene region for phylogenetic inference in Pyrus. It is as yet unknown whether additional inparalogs representing LFY2int2 will be found by analyzing more occidental species and individuals of oriental species.
Conclusion
This is the first study that explores LCNGs for phylogenetic analyses in Pyrus. It is also the first to document the gene structures and transcription of Adh homologs in the Rosaceae taxa. We demonstrated that frequent gene duplications contributed to complex outparalogs and inparalogs of Adh genes with functional diversification or nonfunctionalization. Paralogs, lineage sorting of alleles, and recombinants are three major problems when applying LCNGs in plant phylogenetic analyses. One ortholog of LEAFY, LFY2int2-N, is currently the best nuclear marker for studying interspecific relationships of Pyrus. Complex reticulate histories likely complicate the phylogenetic reconstruction of some Pyrus species. To better resolve interspecific relationships and examine the evolutionary processes of Pyrus, we are extending our phylogenetic studies with plastid DNA and nuclear DNA, including LFYint2-N, and by sampling a wider assortment of species and individuals.
Methods
Taxon sampling, DNA extraction, primer design and amplification
Twenty-five accessions from 13 oriental species and four occidental species of genus Pyrus were included. Six accessions of four Malus species were used as outgroups (Table 1). Total genomic DNA was isolated from fresh leaf tissue using a modified sodium dodecyl sulfate (SDS) method [12,82].
To date, a complete cDNA sequence representing Adh1 from 'Granny Smith' (M. domestica, Z48234) and two 5' partial (beginning at the 3' end of exon 2) cDNA sequences representing two distinct Adh loci (AF031900-Adh1, AF031899-Adh2) from 'Packham's Triumph' (P. communis) are available. The Adh series (Adh1 and Adh2) were named randomly and do not correspond to previously named alleles. A forward primer (Adh-F1) based on sequence of Z48243 and three downstream primers (Adh1-R1, Adh1-R2 and Adh2-R) based on AF031900-Adh1 and AF031899-Adh2 were designed to obtain the entire gene region in several accessions including P. communis, 'Flemish Beauty' (P. communis), 'Nanguoli' (P. ussuriensis), 'Cuiguan' (P. pyrifolia), 'Korlaxiangli' (P. sinkiangensis), 'Ralls' (M. domestica), M. rockii and M. domestica subsp. chinensis. However, these primer pairs only succeeded in amplifying 12 Adh1 sequences in select accessions. Therefore, two additional forward primers, Adh1-F2 and Adh2-F, were designed based on AF031900-Adh1 and AF031899-Adh2, respectively, targeting a partial Adh region lacking exon 1 and intron 1. These primers amplified Adh2 sequences and additional Adh1 sequences. All of the above PCR products were designated as long partial genomic Adh sequences (G-PCR). Considering labor costs and difficulties in amplifying and sequencing fragments greater than 2 kb, a smaller region covering only introns 2 and 3 (about 650 bp) of Adh1 (reduced Adh1) was used in all accessions to construct a phylogeny.
For LEAFY, a long partial region of LFY1 and LFY2 spanning exon 2 and intron 2 was first amplified in some accessions using the primer pair 'LFY-F+LFY-R' developed in an exonic region of M. domestica 'Pinova' (DQ535885, DQ535886). After initial sequence analyses, specific primer pairs of 'LFY1-F + LFY1-R' and 'LFY2-F + LFY2-R' were developed to amplify partial intron 2 of LFY1 and LFY2 (LFY1int2, LFY2int2), respectively. This was done independently in all accessions except the three commercial cultivars ('Cuiguan', 'Ralls', 'Flemish Beauty' and 'Fuji'). Sequence information for primers used in this study is listed in Table 2 and their locations illustrated in Figure 1.
PCR was carried out in a final reaction volume of 50 μL, containing 10-20 ng total DNA, 2.5 mM MgCl2, 0.4 μM of each primer, 5% DMSO (v/v), 0.2 mM dNTP, 2 U Taq DNA polymerase (Takara Biotechnology Company Co., Ltd, Kyoto, Japan) and 1 × PCR buffer supplied by the manufacturer. Amplification of the long partial Adh region was performed for 4 min at 94°C, followed by 35 cycles of 40 s at 94°C, 40 s at 58°C, 2 min of 20 s at 72°C, and a final extension for 7 min at 72°C. For other shorter regions like locus-specific RT-PCR as described below, the PCR procedure was identical, but only 1 min was needed for the extension step.
Cloning and sequencing
PCR products were verified by 2% agarose gel electrophoresis, and the target bands were separated and purified using 3S spin DNA Agarose Gel Purification (Shenergy Biocolor, Shanghai, China). For long partial genomic Adh amplified by G-PCR, the purified PCR products were cloned using TA cloning kit Pmd19 (Takara) and more than three clones per sample were sequenced using M13+, M13- primers and internal primers located at exonic regions (Adh1-F5 for Adh1, and Adh2-F5 for Adh2, Table 2). For the reduced Adh1 region, five to ten clones per sample were sequenced using the M13+ primer. For the long partial LEAFY region, more than three clones were sequenced to obtain reads representing putative LFY1 and LFY2. For LFY1int2 and LFY2int2, the purified PCR products were directly sequenced by amplification primers. Additionally, more than three clones were sequenced to capture all the copies indicated by direct sequencing results.
Sequence analyses
Intronic and exonic boundaries were determined by comparison with available cDNA sequences and preservations of the 'GT' and 'AG' at two ends of introns. Sequences were aligned with Clustal X [83]. Sequence divergence within and between different homologs was calculated using MEGA4 [84] with gaps treated as pairwise deletions. Putative recombinants were detected using RDP3 software package [54], and some putative recombinants were identified manually.
Transcription of Adh homologs
The expression patterns of LFY1 and LFY2 have been well documented in Malus and Pyrus [41], but little was known about expression patterns of Adh1 and Adh2 in Pyrus. Therefore, 'Cuiguan' (P. pyrifolia), 'Nanguoli' (P. ussuriensis), 'Korlaxiangli' (P. sinkiangensis) and 'Flemish Beauty' (P. communis), representing four major pear cultivar groups together with 'Fuji' (M. domestica) were selected to examine transcription of Adh1 and Adh2. For 'Cuiguan' (P. pyrifolia), fresh young leaves, ripe fruits and seeds were collected in our campus yard for Adh expression analyses, while for the other accessions, only ripe fruits were used. The plant tissues were frozen in liquid nitrogen and stored at -80°C. Total RNA was isolated using a modified CTAB method. First strand cDNA was synthesized from 1.0 μg of total RNA using a poly (T)18 as primer and AMV reverse transcriptase (Bio Basic Inc, New York, USA) according to the manufacturer's instructions. To efficiently detect transcription of Adh1 and Adh2 independently, two specific primer pairs 'spAdh1-F+spAdh1-R' and 'spAdh2-F+spAdh2-R' (Table 2) targeting a shorter region were used for RT-PCR and specific genomic PCR (SG-PCR). PCR products were directly sequenced followed by cloning to identify copies involved in transcription. Actin was analyzed as a reference gene. The primers Pact-F and Pact-R were designed based on the Actin gene sequences from P. communis and 'Yali' (P. pyrifolia, CWP) (AB190176, GU830958) (Table 2).
Phylogenetic analyses
The predicted amino acid sequences of long partial Adh genes in Pyrus and Malus from G-PCR were compared with those from other well-studied plant taxa by conducting NJ and MP analyses using PAUP 4.0b10 [85]. For reduced Adh1, nucleotide sequences including the exonic and intronic regions were both included. LFY1int2 and LFY2int2 were analyzed separately, since their sequence homology was too low to be aligned. MP analyses were conducted using PAUP 4.0b10 with gaps treated as missing data. MP analyses were performed using a heuristic search with the TBR and Multree options. To estimate support for the clades, non-parametric bootstraps were estimated with 1000 replicates. Bayesian analyses were performed with MrBayes 3.1 [86]. The best fitting substitution models for each dataset were determined with the Akaike Information Criterion (AIC) using ModelTest 3.06 [87]. The AIC favored the HKY+G for the reduced Adh1 and the K81uf+Gfor both LFY1int2 and LFY2int2. Markov chains were run for 10,000,000 generations with sample frequency of 100. The average standard deviation of split frequency was 0.003 for reduced Adh1, 0.005 for LFY1int2 and 0.002 for LFY2int2, indicating the runs have reached convergence for each dataset. The first 25% of the trees were discarded as burn-in. Clade posterior probabilities were calculated from the combined sets of trees. Both MP and Bayesian analyses resulted in largely congruent tree topologies. Sequences included in the final phylogenetic analyses were deposited in GenBank (Accessions GU991401-991522, HM003976-004066, HQ912028-HQ912076). Alignments of these datasets are deposited as additional files 4, 5, 6 and 7.
Authors' contributions
XZ carried out primer design, molecular phylogenetic analyses, and drafted the manuscript. CH carried out genomic DNA and RNA isolation, participated in the amplifications and cloning. DS helped in writing the manuscript. JL participated in the amplification and collection of fresh leaves. JC participated in the revision. YT conceived of the study and participated in the revision. All authors read and approved the final manuscript.
Supplementary Material
50% majority-rule consensus tree based on amino acid sequences of Adh loci from diverse plant taxa. ADH sequences in Rosaceae are highlighted in blue. Numbers above the branches or near the branch nodes indicate bootstrap values (1000 replicates). Accession number was given for sequences from GenBank. Multiple intraindividual sequences for Adh1 (Adh1-1 and Adh1-2) and Adh2 (Adh2-1 and Adh2-2) are differentiated by the number in the brackets following the taxa name*: Though Adh2-1 was not obtained by G-PCR in Malus, its transcription was detected by RT-PCR in 'Fuji' (M. domestica), which was described in the text.
{kind=link}
Transcription of Adh1 homologs revealed by neighbor joining (NJ) analyses. Sequences obtained from genomic PCR are marked by G followed by the corresponding Adh1-1 subparalogs name in the square brackets. Sequences obtained from RT-PCR are marked by RT followed by the plant tissues used in parenthesis. Sequences obtained from specific genomic PCR are marked by SG. Multiple intraindividual sequences obtained from different PCR or plant tissues are differentiated by the fraction in the brackets following the taxa name. Putative pseudogenes obtained by G-PCR are marked by 'ψ'.
{kind=link}
Transcription of Adh2 homologs revealed by neighbor joining (NJ) analyses. Sequences obtained from genomic PCR are marked by G followed by the paralogs name in the square brackets. Sequences obtained from RT-PCR are marked by RT followed by the plant tissues used in parenthesis. Sequences obtained from specific genomic PCR are marked by SG. Multiple intraindividual sequences obtained from different PCR or plant tissues are differentiated by the fraction in the brackets following the taxa name. Putative pseudogenes obtained by G-PCR are marked by 'ψ.
{kind=link}
Alignment of ADH amino acid sequences from different plant taxa.
Alignment of the reduced Adh1 nucleotide sequences from Malus and Pyrus.
Alignment of LFY1int2 nucleotide sequences from Malus and Pyrus.
Alignment of LFY2int2 sequences from different plant taxa.
Contributor Information
Xiaoyan Zheng, Email: zxy419@zju.edu.cn.
Chunyun Hu, Email: huchunyun2@163.com.
David Spooner, Email: David.Spooner@ars.usda.gov.
Jing Liu, Email: liujing0521@zju.edu.cn.
Jiashu Cao, Email: jshcao@zju.edu.cn.
Yuanwen Teng, Email: ywteng@zju.edu.cn.
Acknowledgements
This work has been financed by the project (No. 30871690) from the National Natural Science Foundation of China, Project (R307605) from Zhejiang Provincial Natural Science Foundation of China, and Project (No. 20090451480) of the China Postdoctoral Science Foundation.
References
- Potter D, Eriksson T, Evans RC, Oh S, Smedmark JEE, Morgan DR, Kerr M, Robertson KR, Arsenault M, Dickinson TA, Campbell CS. Phylogeny and classification of Rosaceae. Pl Syst Evol. 2007;266:5–43. doi: 10.1007/s00606-007-0539-9. [DOI] [Google Scholar]
- Bailey LH. Standard cyclopedia of horticulture. Vol. 5. New York: Macmillan; 1917. Pyrus; pp. 2865–2878. [Google Scholar]
- Kikuchi A. Horticulture of fruit trees. Tokyo: Yokendo; 1948. (in Japanese) [Google Scholar]
- Kikuchi A. Assessment of Chinese pear species and cultivars. Collec Rec Hort Res Fac Agr Kyoto Univ. 1946;3:1–11. (in Japanese) [Google Scholar]
- Yu T. Taxonomy of the fruit tree in China. Beijing: China Agriculture Press; 1979. (in Chinese) [Google Scholar]
- Yu T, Kuan K. Taxa nava Rosacearum (1) Acta Phytotaxon Sinica. 1963;8:202–236. (in Chinese with English summary) [Google Scholar]
- Aldasoro JJ, Aedo C, Garmendia FM. The genus Pyrus L. (Rosaceae) in south-west Europe and North Africa. Bot J Linn Soc. 1996;121:143–158. [Google Scholar]
- Challice JS, Westwood MN. Numerical taxonomic studies of the genus Pyrus using both chemical and botanical characters. Bot J Linn Soc. 1973;67:121–148. doi: 10.1111/j.1095-8339.1973.tb01734.x. [DOI] [Google Scholar]
- Iketani H, Manabe T, Matsuta N, Akihama T, Hayashi T. Incongruence between RFLPs of chloroplast DNA and morphological classification in east Asian pear (Pyrus spp.) Genet Resour Crop Evol. 1998;45:533–539. doi: 10.1023/A:1008646016181. [DOI] [Google Scholar]
- Monte-Corve L, Cabrita L, Oliveira C, Leitao J. Assessment of genetic relationships among Pyrus species and cultivars using AFLP and RAPD markers. Genet Resour Crop Evol. 2000;47:257–265. doi: 10.1023/A:1008794809807. [DOI] [Google Scholar]
- Teng Y, Tanabe K, Tamura F, Itai A. Genetic relationships of pear cultivars in Xinjiang, China, as measured by RAPD markers. J Hort Sci Biotech. 2001;76:771–779. [Google Scholar]
- Teng Y, Tanabe K, Tamura F, Itai A. Genetic relationships of Pyrus species and cultivars native to East Asia revealed by randomly amplified polymorphic DNA markers. J Amer Soc Hort Sci. 2002;127:262–270. [Google Scholar]
- Yamamoto T, Kimura T, Sawamura Y, Manabe T, Kotobuki K, Hayashi T, Ban Y, Matsuta N. Simple sequence repeats for genetic analysis in pear. Euphytica. 2002;124:129–137. doi: 10.1023/A:1015677505602. [DOI] [Google Scholar]
- Bao L, Chen K, Zhang D, Cao Y, Yamamoto T, Teng Y. Genetic diversity and similarity of pear (Pyrus L.) cultivars native to East Asia revealed by SSR (simple sequence repeat) markers. Genet Resour Crop Evol. 2007;54:959–971. doi: 10.1007/s10722-006-9152-y. [DOI] [Google Scholar]
- Bassil N, Postman JD. Identification of European and Asian pears using EST-SSRs from Pyrus. Genet Resour Crop Evol. 2010;57:357–370. doi: 10.1007/s10722-009-9474-7. [DOI] [Google Scholar]
- Yao L, Zheng X, Cai D, Gao Y, Wang K, Cao Y, Teng Y. Exploitation of Malus EST-SSRs and the utility in evaluation of genetic diversity in Malus and Pyrus. Genet Resour Crop Evol. 2010;57:841–851. doi: 10.1007/s10722-009-9524-1. [DOI] [Google Scholar]
- Bao L, Chen K, Zhang D, Li X, Teng Y. An assessment of genetic variability and relationships within Asian pears based on AFLP (amplified fragment length polymorphism) markers. Sci Hort. 2008;116:374–380. doi: 10.1016/j.scienta.2008.02.008. [DOI] [Google Scholar]
- Morgan DR, Solits DE, Robertson KR. Systematics and evolutionary implications of rbcL sequence variation in Rosaceae. Amer J Bot. 1994;81:890–903. doi: 10.2307/2445770. [DOI] [Google Scholar]
- Campbell CS, Donoghue MJ, Baldwin BG, Wojciechowski MF. Phylogenetic relationships in Maloideae (Rosaceae), evidence from sequences of the internal transcribed spacer of nuclear ribosomal DNA and ITS congruence with morphology. Amer J Bot. 1995;82:903–918. doi: 10.2307/2445977. [DOI] [Google Scholar]
- Alice LA, Campbell CS. Phylogeny of Rubus based on nuclear ribosomal DNA internal transcribed spacer region sequences. Amer J Bot. 1999;86:81–97. doi: 10.2307/2656957. [DOI] [PubMed] [Google Scholar]
- Robinson JP, Harris SA, Juniper BE. Taxonomy of the genus Malus Mill. (Rosaceae) with emphasis on the cultivated apple, Malus domestica Borkh. Plant Syst Evol. 2001;226:35–58. doi: 10.1007/s006060170072. [DOI] [Google Scholar]
- Potter D, Gao F, Bortiri E, Oh S, Baggett S. Phylogenetic relationships in Rosaceae inferred from chloroplast matK and trnL-trnF nucleotide sequence data. Plant Syst Evol. 2002;231:77–89. doi: 10.1007/s006060200012. [DOI] [Google Scholar]
- Eriksson T, Hibbs M, Yoder AD, Delwiche CF, Donoghue MJ. The phylogeny of Rosoideae (Rosaceae) based on sequences of the internal transcribed spacer (ITS) of nuclear ribosomal DNA and the trnL-F region of chloroplast DNA. Int J Plant Sci. 2003;164:197–211. doi: 10.1086/346163. [DOI] [Google Scholar]
- Wissemann V, Ritz CM. The genus Rosa (Rosoideae, Rosaceae) revisited: molecular analysis of nrITS-1 and atpB-rbc L intergenic spacer (IGS) versus conventional taxonomy. Bot J Linn Soc. 2005;147:275–290. doi: 10.1111/j.1095-8339.2005.00368.x. [DOI] [Google Scholar]
- Ohta S, Yamamoto T, Nishitani C, Katsuki T, Iketani H, Omura M. Phylogenetic relationships among Japanese flowering cherries (Prunus subgenus Cerasus) based on nucleotide sequences of chloroplast DNA. Plant Syst Evol. 2007;263:209–225. doi: 10.1007/s00606-006-0474-1. [DOI] [Google Scholar]
- Campbell CS, Evans RC, Morgan DR, Dickinson TA, Arsenault MP. Phylogeny of subtribe Pyrinae (formerly the Maloideae, Rosaceae): Limited resolution of a complex evolutionary history. Plant Syst Evol. 2007;266:119–145. doi: 10.1007/s00606-007-0545-y. [DOI] [Google Scholar]
- Iwata H, Kato T, Ohno S. Triparental origin of Damask roses. Gene. 2000;259:53–59. doi: 10.1016/S0378-1119(00)00487-X. [DOI] [PubMed] [Google Scholar]
- Zheng X, Cai D, Yao L, Teng Y. Non-concerted ITS evolution, early origin and phylogenetic utility of ITS pseudogenes in Pyrus. Mol Phylogenet Evol. 2008;48:892–903. doi: 10.1016/j.ympev.2008.05.039. [DOI] [PubMed] [Google Scholar]
- Kimura T, Iketani H, Kotobuki K, Matsuta N, Ban Y, Hayashi T, Yamamoto T. Genetic characterization of pear varieties revealed by chloroplast DNA sequences. J Hort Sci Biotech. 2003;78:241–247. [Google Scholar]
- Bailey CD, Doyle JJ. Potential phylogenetic utility of the low-copy nuclear gene pistillata in Dicotyledonous Plants: Comparison to nrDNA ITS and trnL intron in Sphaerocardamum and other Brassicaceae. Mol Phylogenet Evol. 1999;13:20–30. doi: 10.1006/mpev.1999.0627. [DOI] [PubMed] [Google Scholar]
- Sang T. Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit Rev Biochem Mol Biol. 2002;37:121–147. doi: 10.1080/10409230290771474. [DOI] [PubMed] [Google Scholar]
- Evans RC, Campbell CS. The origin of the apple subfamily (Maloideae; Rosaceae) is clarified by DNA sequence data from duplicated GBSSI genes. Amer J Bot. 2002;89:1478–1484. doi: 10.3732/ajb.89.9.1478. [DOI] [PubMed] [Google Scholar]
- Mort ME, Crawford DJ. The continuing search: low-copy nuclear sequences for lower-level plant molecular phylogenetic studies. Taxon. 2004;53:257–261. doi: 10.2307/4135604. [DOI] [Google Scholar]
- Ané C, Larget B, Baum DA, Smith SD, Rokas A. A Bayesian estimation of concordance among gene trees. Mol Biol Evol. 2007;24:412–426. doi: 10.1093/molbev/msl170. [DOI] [PubMed] [Google Scholar]
- Lawton-Rauh A. Evolutionary dynamics of duplicated genes in plants. Mol Phylogenet Evol. 2003;29:396–409. doi: 10.1016/j.ympev.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Sonnhammer ELL, Koonin EV. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002;18:619–620. doi: 10.1016/S0168-9525(02)02793-2. [DOI] [PubMed] [Google Scholar]
- Meng C, Kubatko LS. Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: A model. Theor Popul Biol. 2009;75:35–45. doi: 10.1016/j.tpb.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Evans RC, Alice LA, Campbell CS, Kellogg EA, Dickinson TA. The granule-bound starch synthase (GBSSI) gene in the Rosaceae: multiple loci and phylogenetic utility. Mol Phylogenet Evol. 2000;17:388–400. doi: 10.1006/mpev.2000.0828. [DOI] [PubMed] [Google Scholar]
- Oh SH, Potter D. Phylogenetic utility of the second intron of LEAFY in Neillia and Stephanandra (Rosaceae) and implications for the origin of Stephanandra. Mol Phylogenet Evol. 2003;29:203–215. doi: 10.1016/S1055-7903(03)00093-9. [DOI] [PubMed] [Google Scholar]
- Lo EYY, Stefanović S, Christensen KI, Dickinson TA. Evidence for genetic association between East Asian and Western North American Crataegus L. (Rosaceae) and rapid divergence of the Eastern North American lineages based on multiple DNA sequences. Mol Phylogenet Evol. 2009;51:157–168. doi: 10.1016/j.ympev.2009.01.018. [DOI] [PubMed] [Google Scholar]
- Esumi T, Tao R, Yonemori K. Isolation of LEAFY and TERMINAL FLOWER 1 homologs from six fruit tree species in the subfamily Maloideae of the Rosaceae. Sex Plant Reprod. 2005;17:277–287. doi: 10.1007/s00497-004-0239-3. [DOI] [Google Scholar]
- Shafqat J, El-Ahmad M, Danielsson O, Martínez MC, Persson B, Parés X, Jornvall H. Pea formaldehyde-active class III alcohol dehydrogenase: common derivation of the plant and animal forms but not of the corresponding ethanol-active forms (classes I and P) Proc Natl Acad Sci USA. 1996;93:5595–5599. doi: 10.1073/pnas.93.11.5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MC, Achkor H, Persson B, Shafqat J, Farrés J, Jörnvall H, Parés X. Arabidopsis formaldehyde dehydrogenase. Molecular properties of plant class III alcohol dehydrogenase provide further insights into the origins, structure and function of plant class p and liver class I alcohol dehydrogenases. Eur J Biochem. 1996;241:849–857. doi: 10.1111/j.1432-1033.1996.00849.x. [DOI] [PubMed] [Google Scholar]
- Gaut BS, Peek AS, Morton BR, Clegg MT. Patterns of genetic diversification within the Adh gene family in the grasses (Poaceae) Mol Biol Evol. 1999;16:1086–1097. doi: 10.1093/oxfordjournals.molbev.a026198. [DOI] [PubMed] [Google Scholar]
- Petersen G, Seberg O, Aagesen L, Frederiksen S. An empirical test of the treatment of indels during optimization alignment based on the phylogeny of the genus Secale (Poaceae) Mol Phylogenet Evol. 2004;30:733–742. doi: 10.1016/S1055-7903(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Thompson CE, Salzano FM, de-Souza ON, Freitas LB. Sequence and structural aspects of the functional diversification of plant alcohol dehydrogenases. Gene. 2007;396:108–115. doi: 10.1016/j.gene.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Small RL, Wendel JF. Copy number lability and evolutionary dynamics of the Adh gene family in diploid and tetraploid cotton. Genetics. 2000;155:1913–1926. doi: 10.1093/genetics/155.4.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roalson EH, Friar EA. Phylogenetic analysis of the nuclear alcohol dehydrogenase (Adh) gene family in Carex section Acrocystis (Cyperaceae) and combined analyses of Adh and nuclear ribosomal ITS and ETS sequences for inferring species relationships. Mol Phylogenet Evol. 2004;33:671–686. doi: 10.1016/j.ympev.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Chervin C, Truett J, Speirs J. Alcohol dehydrogenase expression and the production of alcohols during pear fruit ripening. J Amer Soc Hort Sci. 1999;124:71–75. [Google Scholar]
- Wolyn DJ, Jelenkovic G. Nucleotide sequence of an alcohol dehydrogenase gene in octoploid strawberry (Fragaria × ananassa Duch.) Plant Mol Biol. 1990;14:855–857. doi: 10.1007/BF00016518. [DOI] [PubMed] [Google Scholar]
- Korban SS, Skirvin RM. Nonmenclature of the cultivated apple. HortScience. 1984;19:177–180. [Google Scholar]
- Rubstov GA. Geographical distribution of the genus Pyrus and trends and factors in its evolution. Amer Nat. 1944;78:358–366. doi: 10.1086/281206. [DOI] [Google Scholar]
- Perry DJ, Furnier GR. Pinus banksiana has at least seven expressed alcohol dehydrogenase genes in two linked groups. Proc Natl Acad Sci USA. 1996;93:13020–13023. doi: 10.1073/pnas.93.23.13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn VH, Thompson MA, Moore GP. Nucleotide sequence comparison of the Adh gene in three drosophilids. J Mol Evol. 1984;20:31–37. doi: 10.1007/BF02101983. [DOI] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge, Cambridge University Press; 1983. [Google Scholar]
- Dennis ES, Sach MM, Gerlach WL, Finnegan EJ, Peacock WJ. Molecular analysis of the alcohol dehydrogenase 2 (Adh2) gene in maize. Nucleic Acids Res. 1985;13:727–743. doi: 10.1093/nar/13.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesnié C, Verriès C. Molecular cloning and expression of cDNAs encoding alcohol dehydrogenase from Vitis vinifera L. during berry development. Plant Science. 2000;157:77–88. doi: 10.1016/S0168-9452(00)00274-0. [DOI] [PubMed] [Google Scholar]
- Bartley IM, Hindley SJ. Alcohol dehydrogenases of apple. J Exper Bot. 1980;31:449–459. doi: 10.1093/jxb/31.2.449. [DOI] [Google Scholar]
- Bartley IM, Stoker PG, Martin ADE, Hatfield SGS, Knee M. Synthesis of aroma compounds by apples supplied with alcohols and methyl esters of fatty acids. J Sci Food Agric. 1985;36:567–574. doi: 10.1002/jsfa.2740360708. [DOI] [Google Scholar]
- Dixon J, Heweit EW. Factors affecting apple aroma/flavour volatile concentration: a review. New Zealand J Crop Hort Sci. 2000;28:155–173. doi: 10.1080/01140671.2000.9514136. [DOI] [Google Scholar]
- Chervin C, Speirs J, Loveys B, Patterson BD. Influence of low oxygen storage on aroma compounds of whole pears and crushed pear flesh. Postharvest Biol Technol. 2000;19:279–285. doi: 10.1016/S0925-5214(00)00096-X. [DOI] [Google Scholar]
- Chang C, Meyerowitz EM. Molecular cloning and DNA sequence of the Arabidopsis thaliana alcohol dehydrogenase gene. Proc Natl Acad Sci USA. 1986;83:1408–1412. doi: 10.1073/pnas.83.5.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita NT, Innan H, Terauchi R. Intra- and interspecific variation of the alcohol dehydrogenase locus region in wild plants Arabis gemmifera and Arabidopsis thaliana. Mol Biol Evol. 1996;13:433–436. doi: 10.1093/oxfordjournals.molbev.a025603. [DOI] [PubMed] [Google Scholar]
- Singh RK, Sane VA, Misra A, Ali SA, Nath P. Differential expression of the mango alcohol dehydrogenase gene family during ripening. Phytochemistry. 2010;71:1485–1494. doi: 10.1016/j.phytochem.2010.05.024. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Liu FL, Zhang L. The evolution of the alcohol dehydrogenase gene family by loss of introns in plants of the genus Leavenworthia (Brassicaceae) Mol Biol Evol. 1998;15:552–559. doi: 10.1093/oxfordjournals.molbev.a025955. [DOI] [PubMed] [Google Scholar]
- Hu K. Intron exclusion and the mystery of intron loss. FEBS Letters. 2006;580:6361–6365. doi: 10.1016/j.febslet.2006.10.048. [DOI] [PubMed] [Google Scholar]
- Luo J, Yoshikawa N, Hodoson MC, Hall BD. Duplication and paralog sorting of RPB2 and RPB1 genes in core eudicots. Mol Phylogenet Evol. 2007;44:850–862. doi: 10.1016/j.ympev.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Sang T, Donoghue MJ, Zhang D. Evolution of alcohol dehydrogenase genes in Peonies (Paeonia) phylogenetic relationships of putative non hybrid species. Mol Biol Evol. 1997;14:994–1007. doi: 10.1093/oxfordjournals.molbev.a025716. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Yokoyama J, Nakamura T, Song I, Ito T, Ochiai T, Kanno A, Kameya T, Maki M. Molecular phylogeny and evolution of alcohol dehydrogenase (Adh) genes in legumes. BMC Plant Biol. 2005;5:6. doi: 10.1186/1471-2229-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel JF. Genome evolution in polyploids. Plant Mol Biol. 2000;42:225–249. doi: 10.1023/A:1006392424384. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Patterson AH, Zheng C, Sankoff D, DePamphilis CW, Wall PK, Soltis PS. Polyploidy and angiosperm diversification. Amer J Bot. 2009;96:336–348. doi: 10.3732/ajb.0800079. [DOI] [PubMed] [Google Scholar]
- Blázquez MA, Soowal LN, Lee I, Weigel D. LEAFY expression and flower initiation in Arabidopsis. Development. 1997;124:3835–3844. doi: 10.1242/dev.124.19.3835. [DOI] [PubMed] [Google Scholar]
- Wada M, Ureshino A, Cao Q, Bessho H. Genomic varieties of apple AFL genes. Plant Sci. 2007;193:559–566. [Google Scholar]
- Wada M, Cao Q, Kotoda N, Soejima J, Masuda T. Apple has two orthologues of FLORICAULA/LEAFY involved in flowering. Plant Mol Biol. 2002;49:567–577. doi: 10.1023/A:1015544207121. [DOI] [PubMed] [Google Scholar]
- Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–113. doi: 10.2307/2412448. [DOI] [PubMed] [Google Scholar]
- Calviño CI, Martínez SG, Downie SR. The evolutionary history of Eryngium (Apiaceae, Saniculoideae): Rapid radiations, long distance dispersals, and hybridizations. Mol Phylogenet Evol. 2008;46:1129–1150. doi: 10.1016/j.ympev.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc Natl Acad Sci USA. 2001;98:13757–13762. doi: 10.1073/pnas.241370698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Hewitt GM. Nuclear DNA analyses in genetic studies of populations: practice, problems and prospects. Mol Ecol. 2003;12:563–584. doi: 10.1046/j.1365-294X.2003.01773.x. [DOI] [PubMed] [Google Scholar]
- Poke FS, Martin DP, Vaillancourt RE, Reid JB. The impact of intragenic recombination on phylogenetic reconstruction at the sectional level in Eucalyptus when using a single copy nuclear gene (cinnamoyl CoA reductase) Mol Phylogenet Evol. 2006;39:160–170. doi: 10.1016/j.ympev.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Kim CS, Lee CH, Park KW, Kang SJ, Shin IS, Lee GP. Phylogenetic relationships among Pyrus pyrifolia and P. communis detected randomly amplified polymorphic DNA (RAPD) and conserved rDNA sequences. Sci Hort. 2005;106:491–501. doi: 10.1016/j.scienta.2005.04.009. [DOI] [Google Scholar]
- Dellaporta SL, Wood J, Hicks JB. A plant DNA minipreparation, Version II. Plant Mol Biol Rpt. 1983;1:19–21. doi: 10.1007/BF02712670. [DOI] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*: Phylogenetic analysis using parsimony (and other methods) 4.0 beta. Sunderland, Massachusetts: Sinauer Associates; 2002. [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
50% majority-rule consensus tree based on amino acid sequences of Adh loci from diverse plant taxa. ADH sequences in Rosaceae are highlighted in blue. Numbers above the branches or near the branch nodes indicate bootstrap values (1000 replicates). Accession number was given for sequences from GenBank. Multiple intraindividual sequences for Adh1 (Adh1-1 and Adh1-2) and Adh2 (Adh2-1 and Adh2-2) are differentiated by the number in the brackets following the taxa name*: Though Adh2-1 was not obtained by G-PCR in Malus, its transcription was detected by RT-PCR in 'Fuji' (M. domestica), which was described in the text.
Transcription of Adh1 homologs revealed by neighbor joining (NJ) analyses. Sequences obtained from genomic PCR are marked by G followed by the corresponding Adh1-1 subparalogs name in the square brackets. Sequences obtained from RT-PCR are marked by RT followed by the plant tissues used in parenthesis. Sequences obtained from specific genomic PCR are marked by SG. Multiple intraindividual sequences obtained from different PCR or plant tissues are differentiated by the fraction in the brackets following the taxa name. Putative pseudogenes obtained by G-PCR are marked by 'ψ'.
Transcription of Adh2 homologs revealed by neighbor joining (NJ) analyses. Sequences obtained from genomic PCR are marked by G followed by the paralogs name in the square brackets. Sequences obtained from RT-PCR are marked by RT followed by the plant tissues used in parenthesis. Sequences obtained from specific genomic PCR are marked by SG. Multiple intraindividual sequences obtained from different PCR or plant tissues are differentiated by the fraction in the brackets following the taxa name. Putative pseudogenes obtained by G-PCR are marked by 'ψ.
Alignment of ADH amino acid sequences from different plant taxa.
Alignment of the reduced Adh1 nucleotide sequences from Malus and Pyrus.
Alignment of LFY1int2 nucleotide sequences from Malus and Pyrus.
Alignment of LFY2int2 sequences from different plant taxa.