

Abstract
Introduction
Accumulating evidence suggests that genomic structural variations, particularly copy number variations (CNV), are a common occurrence in humans that may bear phenotypic consequences for living individuals possessing the variant. While precise estimates vary, large-scale karyotypic abnormalities are present in 6–12% of stillbirths (SB). However, due to inherent limitations of conventional cytogenetics, the contribution of genomic aberrations to stillbirth is likely underrepresented. High-resolution copy number variant (CNV) analysis by genomic array-based profiling may overcome such limitations.
Methods
Prospectively acquired SB cases >22 weeks underwent classification of "unexplained" stillbirth by Wigglesworth and Aberdeen criteria after extensive testing and rigorous multidisciplinary audit. Genome-wide analysis was conducted using high resolution Illumina SNP arrays (HumanCNV370 Duo) on placental and fetal samples. Potential alternate detection methods were completed by one or more of three independent means (quantitative PCR, Illumina1M or Agilent105K CGH arrays).
Results
In our cohort of 54 stillbirths, 29 met strict unexplained criteria. Among these, we identified 24 putative novel CNVs. Subsequent interrogation detected18 of 24 CNVs (75%) in placental samples, 8 of which were also confirmed in available fetal samples; none were present in maternal blood.
Conclusion
We describe the potential of whole-genome placental profiling to identify small genomic imbalances which might contribute to a small proportion of well-characterized, unexplained stillbirths.
Keywords: Stillbirth, copy number variants, deletion syndromes, duplication syndromes, fetal death
INTRODUCTION
It is increasingly evident that genomic structural variations commonly occur in humans (1–2). These structural variations are comprised of inversions, translocations, deletions, duplications, or more complex combinations of such events. Deletions or duplications result in a change in the number of copies of a genomic segment and are referred to as copy number variations (CNVs). It has been suggested that structural variations such as CNVs contribute more to the genomic difference between individuals than single nucleotide polymorphisms (SNPs) (3). At least one potentially beneficial reproductive phenotype, increased fecundity, has been shown to be associated with a structural variation (4). However, a much larger number of deleterious effects (genomic disorders) have been shown to be associated with imbalances in array-based copy number analyses (5–8).
Recent estimates from a survey of 25,655 stillbirths after the 20th week of pregnancy supports an incidence of 6.2 per 1000 livebirths, or approximately 1/160 (9, 10). Identifiable causes of stillbirth are generally ascribed to one of three categories: fetal anomalies, placentation or cord morbidities, or maternal comorbidities. Accurate determination of the relative contribution of these and other factors, such as environmental exposures, to stillbirth is hampered by inaccurate fetal death records (11, 12) and incomplete evaluation of such potential comorbidities. As such, the cause of as great as 60% of stillbirths remains classified as “unknown” (13–16). The poorly understood epidemiology of stillbirths recently prompted the National Institute of Health to identify it as a high priority research focus, resulting in the Stillbirth Collaborative Research Network (13, 14). Because the empiric recurrence risk of stillbirth approximates 3–5% (10), identification of the etiology in index cases is essential to better stratify recurrence estimates, potentially reduce parental anxiety, and diminish future unnecessary and expensive antenatal testing in subsequent pregnancies (10, 4).
Previous studies to identify genomic abnormalities in stillbirth samples have primarily employed G-banded karyotyping and fluorescence in situ hybridization (FISH). G-banded chromosomal karyotyping at standard 450–600 band resolutions detects aneuploidy and chromosomal segmental abnormalities larger than 3 to 5 megabases (Mb). FISH can detect smaller-size abnormalities in the range of 50–150 Kb, but it is limited by the small number of probes (maximum ~5) that can be tested simultaneously requiring an informed selection of the specific probes to be used. Using these methods, cytogenetic abnormalities have been described in a small percentage (5–13%) of all stillbirths (13, 17, 18). Thus, array based approaches to identifying genomic abnormalities in stillbirths might be expected to have two main advantages over cytogenetic methods. First, depending on genomic coverage and density of the probes on the array, they can offer higher resolution analysis resulting in detection of smaller abnormalities to increase the test resolution by orders of magnitude. Secondly, array based approaches require preparation of DNA from tissue samples, avoiding the need for successful cell culturing. Other investigators have employed array comparative genomic hybridization (aCGH) in the genetic evaluation of anomalous stillbirths (19) as well as instances of fetal loss from 10 to 20 weeks of gestation (20). To date, employment of aCGH in the majority (60%) of instances of stillbirth (i.e., unexplained, non-anomalous stillbirths) has not been undertaken.
Given that CNVs are a common occurrence in humans and that stillbirths are generally only examined using low resolution cytogenetic methods if at all, it seems likely that some proportion of stillbirths ascribed to unknown causes may be due to CNVs. We hypothesized that in a well-described population-based cohort, a small but definable number of cases of unexplained stillbirth would be associated with small, cytogenetically undetected copy number imbalances identifiable by array-based platforms. Our overall objective was to estimate the genome-wide prevalence of copy number changes in truly unexplained stillbirths.
METHODS
IRB approval, subject enrollment, and sample acquisition
Following IRB approval, placental samples were prospectively acquired from stillbirths (n=33) and control livebirths (n=10) between January 2005 and November 2007 collected at the Policlinico Hospital of Modena, Italy. The diagnosis of stillbirth was based on the World Health Organization criteria defined as fetal death >22 weeks of gestation, or birth weight ≥500g if the gestational age was unknown (7, 29). The IRB protocol allowing the index study with diagnostic evaluation and sample repository with capacity for future use on stillborn infants was approved and granted by the Ministry of Education, University and Research. Subsequent IRB approval was obtained thereafter for genetics and DNA-based research.
Classification as stillbirth of unknown etiology
All stillbirths underwent evaluation according to a standardized protocol. Following initial evaluation, a panel of 2 obstetricians, 1 neonatologist and 1 pathologist reviewed the cases and classified them based on the Wigglesworth and Aberdeen stillbirth classification systems (21). The cause of stillbirth was assigned only when the majority (>2 of 4) of the clinician panel agreed. Complete parameters for evaluation entailed maternal serologies, expansive microbiology, and fetal karyotype. For maternal serologic testing, TORCH (HIV, toxoplasmosis, rubella, and CMV serologies), maternal serum parvovirus titers (IgG and IgM), maternal measures of thyroid function (TSH, free T3 and T4 in reflex), indirect Coombs, and testing for inherited and acquired thrombophilias (factor V Leiden, PT20210, APLS, anti-cardiolipin antibody, and protein C and S) were systematically obtained following enrolment. Gram staining, acid-fast bacilli (AFB), and aerobic and anaerobic cultures on vaginal, placental, fetal swabs (pre and pos-auricular, auricular, and nasal-pharyngeal cavity), and on fetal blood drawn by intra-cardiac fetal puncture prior to full autopsy. Fetal karyotype to a 600 G-banding resolution was performed on amniocentesis samples or fetal blood, cell cultures from placental biopsies, skin samples, or fascia lata. Finally, complete fetal autopsy was performed by a perinatal pathologist and a detailed placental gross and histological exam was undertaken. All data was recorded, and reviewed in a systematic and unbiased manner by the panel described as above.
DNA isolation
DNA isolation was attempted from frozen tissues and/or blood samples employing Gentra Puregene DNA purification kit (Qiagen, Valencia, CA) in accordance with the manufacturer’s protocol and was completed as needed until high purity was reached (Quant-iT PicoGreen dsDNA Genomic, Perkin Elmer, Waltham, MA).
Illumina Beadchips
The Illumina CNV370-Duo BeadChip (Illumina Corporation, San Diego, CA) was initially used to perform genome wide copy number profiling on DNA from placental samples. A subset of placental samples in which CNVs were identified was also interrogated using the Illumina 1M-Duo Beadchip (Illumina Corporation, San Diego, CA) following the same methodology. For these assays, a single-base pair extension reaction was performed, followed by staining with fluorescent dyes. The resulting products were imaged with the BeadArray Reader. Allele detection and genotype calling were performed using the BeadStudio software (Illumina Corporation, San Diego, CA) based on human genome build NCBI 36/UCSC hg18. CNVs were identified using the cnvPartition BeadStudio plugin. CNVs identified by cnvPartition that were smaller the 1000bp were removed from consideration. A total of 10 live born controls and 33 stillbirths were interrogated using the array. Of the 33 stillbirth arrays, 3 had a call rate less than 95% and were removed from subsequent analysis.
Agilent Comparative Genomic Hybridization Array
Three micrograms of genomic DNA from placentas and three micrograms of reference human female or male DNA prepared from single human placentas were labeled and hybridized to the Human Genome CGH Microarray Kit 105A (Agilent Technologies, Inc, Santa Clara, CA) in a rotating oven at 65ºC for 40 hours, according to the manufacturer’s recommended protocol with the following modifications. Briefly, DNA was digested with RsaI and AluI for 2 hours at 37ºC, followed by heat inactivation at 65ºC for 20 minutes. DNA was then labeled with the Agilent Genomic DNA Labeling Kit PLUS using random primers. Experimental samples were labeled with Cyanine-5 dUTP; reference samples were labeled with Cyanine-3 dUTP for two hours at 37ºC. After heat inactivation and sample clean-up, equal amounts of experimental and reference DNA were mixed, heat-denatured and blocked for 30 minutes at 37ºC with Cot-1 DNA in blocking buffer. After blocking, the mixture was applied to the arrays and hybridized in a rotating oven (20 rpm) at 65ºC for 40 hours. Slides were then washed and dried using Agilent’s oligo-array wash buffer 1 and 2, followed by one acetonitrile wash, drying and stabilization wash. Slides were scanned on an Agilent DNA microarray scanner model G2565BA and data extraction was performed with Agilent G2567AA Feature Extraction 9.1 software using the CGH014693 design file as template for automated gridding and the CGH-v4_91 protocol to assign spot-intensity values and ratios to each extraction set. Data were visualized and analyzed based on human genome build NCBI 36/UCSC hg18 with Agilent’s CGHanalytics and displayed as log2 ratios using following settings: 1-fold cut-off, ADM-2 aberration algorithm with threshold 10.0 and a 2 Mb moving-average window.
Exclusion of CNVs present in normal individuals
CNVs identified in stillbirths were compared to CNVs identified in controls and/or present in The Centre for Applied Genomics (TCAG) Database of Genomic Variants (http://projects.tcag.ca/variation/) based on human genome build NCBI 36/UCSC hg18. The TCAG contains CNVs identified in normal (live) individuals from a large number of studies. Stillbirth CNVs completely overlapped by CNVs in live born controls or in the TCAG database were removed from further consideration.
qPCR confirmation and detection of CNVs
Placental DNA sample qPCR analysis was performed for all 24 identified CNVs in Wigglesworth classification 2 cases. Fetal DNA sample qPCR detection was performed for 19 of 24 CNVs, because fetal samples were not available for cases 46 and 48. For cases not identified as Wiggleworths 2, qPCR analysis was performed on 5 of 6 identified CNVs in both placental and fetal samples. Maternal qPCR was performed on 13 of 24 CNVs for which samples were available. For validation, twenty ng of genomic DNA was amplified using PCR primers designed with Primer Express software (Applied Biosystems, Foster City, CA; Supplementary table). All reactions were done in triplicate. Each target region was amplified using the Biorad iQ5 optical system (Biorad Laboratories, Inc., Hercules, CA) for quantitative real-time PCR (SYBR-Green). Data were normalized against an endogenous reference gene (GAPDH) and a melting curve analysis was performed to verify PCR product specificity. Relative gene copy number was determined by the comparative threshold cycle method (Δ ΔCt) after standard curves with serial dilutions were obtained for each amplification reaction. To obtain ratios and generate graphs of copy numbers, we calculated the ΔCT of threshold cycles of normal female DNA [ΔCT(HF)] minus the ΔCT of threshold cycles of G17 DNA [ΔCT(G17)] and the ΔCT of threshold cycles of normal male DNA [ΔCT(HM)] minus the ΔCT of threshold cycle of G17 DNA [ΔCT(G17)]. As a control, we also calculated the ΔCT of threshold cycles of normal female DNA [ΔCT(HF)] minus the ΔCT of threshold cycles of normal male DNA [ΔCT(HM)]. Ratios for each amplification product and comparison were then calculated as 2ΔΔCT. qPCR 2-ΔΔCT ratios in the following ranges were considered to confirm the array CNV call: homozygous deletion 0–0.25; heterozygous deletion 0.25–0.75; duplication >1.25.
RESULTS
Classification as Stillbirth of Unknown Etiology
In order to identify stillbirth cases with unknown etiology, cases were examined and classified as described in Methods. The examination included fetal or placental G-banded karyotyping in order to identify cases with microscopically visible chromosomal abnormalities. 33 cases classified as Wigglesworth (21) 2 (unexplained antepartum fetal death) were chosen for this study, and 29 cases were ultimately successfully interrogated by array (Table I). These cases were further subdivided into stillborn fetuses with normal growth (n=17, ≥10th% adjusted for gestational age by birth weight and gender; 33, 34) and intrauterine growth restriction (n=12; <10th% adjusted for gestational age; 33, 34) cases. In addition to examining stillbirths classified as unexplained antepartum fetal death, we also interrogated 4 placental samples from stillbirths with a Wigglesworth classification of 3, 5, or 6 (Table II). As a control, 10 placental samples from liveborn infants were also interrogated by array and will be the subject of future publications. Suffice it to say, none of the large (>1 Mb) noted deletions nor duplications (Table III and IV) were present in the normal liveborn population (data not shown).
Table I.
Characteristics of Unexplained Stillbirth Cases by Wigglesworth and Aberdeen Criteria.
| Wigglesworth (W) and Aberdeen (A) Classification& | Case Number | Gestational Age, weeks | Fetal Weight, grams | Placental Pathology | Maternal Comorbidities or Incidental Conditions |
|---|---|---|---|---|---|
| #Appropriate for Gestational Age Growth (>10 | |||||
|
| |||||
| W2A5 | 36 | 35 and 1/7 | 2160 | Unremarkable | Preeclampsia, Prior stillbirth |
|
| |||||
| W2A8 | 49 | 40 and 1/7 | 3400 | Abruptio placenta, Umbilical vein thrombosis | None |
|
| |||||
| W2A9 | 4 | 31 and 5/7 | 1700 | Retroplacental hematoma | None |
|
| |||||
| W2A15 | 8 | 38 and 5/7 | 3700 | Chorioamnionitis, Multiple infarcts | Type I IDDM |
| 19 | 36 and 3/7 | 2910 | Fibrosis, Multiple infarcts | Type II IDDM | |
|
| |||||
| W2A16 | 37 | 33 and 5/7 | 2035 | Unremarkable | Group B strep positive |
|
| |||||
| W2A20 | 9 | 37 and 2/7 | 3550 | Chorioamnionitis | None |
| 23 | 40 and 6/7 | 3700 | Umbilical vein calcium deposits | MTHFR (heterozygote) | |
| 30 | 38 and 0/7 | 3780 | Fibrosis, Multiple infarcts | Gestational diabetes (A1) | |
| 33 | 40 and 0/7 | 3460 | Unremarkable | None | |
| 43 | 37 and 2/7 | 2600 | Unremarkable | None | |
| 50 | 40 and 4/7 | 3900 | Unremarkable | None | |
| 51 | 38 and 2/7 | 2610 | Unremarkable | None | |
|
| |||||
| W2A21 | 1 | 32 and 3/7 | 1800 | Vascular maldevelopment | None |
| 3 | 36 and 5/7 | 2110 | Chorioamnionitis | None | |
| 46 | 34 and 0/7 | 1800 | Chorioamnionitis, Multiple infarcts | None | |
| 48 | 34 and 3/7 | 2000 | Chorioamnionitis, Multiple infarcts | None | |
|
| |||||
| #Small for Gestational Age (Growth %) | |||||
|
| |||||
| W2A5 | 6 | 34 and 0/7 | 1210 (<3rd%) | Multiple infarcts | Preeclampsia |
| 16 | 26 and 0/7 | 580 (<10th%) | Retroplacental hematoma, Mulitple infarcts | Preeclampsia, factor V Leiden (heterozygote) | |
| 25 | 24 and 6/7 | 325 (<3rd%) | Chorioamnionits | Preeclampsia | |
|
| |||||
| W2A8 | 15 | 35 and 4/7 | 1635 (<5th%) | Abruptio placenta, Multiple infarcts | Gestational hypertension |
|
| |||||
| W2A16 | 21 | 32 and 0/7 | 950 (<3rd%) | Chorioamnionitis | IVF pregnancy, Group B strep positive |
| 44 | 38 and 4/7 | 2450 (<10th%) | Unremarkable | Group B strep positive | |
|
| |||||
| W2A20 | 42 | 41 and 1/7 | 2670 (<3rd%) | Multiple infarcts | None |
|
| |||||
| W2A21 | 11 | 28 and 3/7 | 665 (<5th%) | Multiple infarcts | Prior stillbirth |
| 18 | 40 and 0/7 | 2300 (<3rd%) | Chorioamnionitis | None | |
| 31 | 24 and 0/7 | 485 (<3rd%) | Unremarkable | None | |
| 40 | 32 and 0/7 | 910 (<3rd%) | Chorioamnionitis | None | |
| 45 | 28 and 3/7 | 680 (<10th%) | Unremarkable | None | |
Wigglesworth (W) and Aberdeen (A) classifications were employed as previously described and recently reassessed (Refs 10, 21). W2 in the extended Wigglesworth classification refers to unexplained antepartum fetal death (by contrast a live born infant which demises due to issues which arise during the antepartum interval would be coded W6). Subsequent Aberdeen classifications are: A5: Preeclampsia; A8: Abruptio placenta; A9: Antepartum hemorrhage of uncertain origins; A15: Other maternal disorder; A16: Maternal infection (asymptomatic); A20: Unexplained, ≥2500 grams; A21: Unexplained, <2500 grams.
Cases were further divided by virtue of fetal growth, into appropriate for gestational age (AGA) and small for gestational age (SGA) by previously defined criteria (29). For all SGA (<10th%), the centiles at <10th, <5th, and <3rd are as noted in column 4.
Table II.
Characteristics of Expanded Wigglesworth Classifications W3, W5 and W6 stillbirths.s
| Wigglesworth (W) Classification& | Case Number | Gestational Age, weeks | Fetal Weight, grams | Placental Pathology | Maternal Comorbidities or Incidental Conditions | Fetal Malformations |
|---|---|---|---|---|---|---|
| W3 | 7 | 29 and 0/7 | 990 | Areas of hypertrophy and fibrotic villi with chorioangiosis | *IVF multiple gestation pregnancy with intrapartum stillbirth of one multiple | None |
|
| ||||||
| W5 | 22 | 27 and 0/7 | 840 | Fibrotic and hydropic villia with thrombosis of 1 umbilical artery; Inflammation of decidua and parenchyma between villi | Parvovirus B19 infection | None |
| 47 | 27 and 0/7 | 825 | Fibrosis, Multiple infarcts | CMV and listeria infection | None | |
|
| ||||||
| W6 | 2 | 38 and 0/7 | 3750 | Subocclusive thrombosis of umbilical vein | None | Cranial asymmetry, hypotelorism, absent eyebrows, prominent tongue, short neck, bilateral hydrocoele, hydrops, hydrothorax |
Wigglesworth (W) was employed as previously described and recently reassessed (Refs 3–5). W3 in the extended Wigglesworth classification refers to unexplained intrapartum fetal death (death from “asphyxia”, “anoxia”, or “trauma”). W5: Infection, where there is clear microbiologic evidence of infection, and not incidental maternal screening of carrier status. W6: Other specific causes, such as recognizable fetal malformations.
Table III.
Novel CNV Deletions of Expanded Wigglesworth W2 Classification Stillbirths.
| Stillbirth Characterization | Illumina CNV370 Duo CNV/SNP Array | Deletion Detected on Higher (Illumina 1M) or Lower (Agilent 105K) Density Arrays& | qPCR Detection of CNV# | |||||
|---|---|---|---|---|---|---|---|---|
| Wigglesworth (W) Aberdeen (A) Classification | Case Number | Human Genome Coordinates | Length, Kb | Illumina 1M CNV/SNP Array (length, Kb) | Agilent 105K HD-CGH CNV Array (length, Kb) | Placental (Δ ΔCt ratio) | Fetal (Δ ΔCt ratio) | Maternal (Δ ΔCt ratio) |
| W2A21 | 3 | chr5:25507338–25597265 | 89.927 | Yes (71.161) | NA: No probes in region | Del (0.50) | Del (0.55) | Maternal DNA not available |
| chr6:33134224–33143208 | 8.984 | No | NA: No probes in region | Del (0.57) | Del (0.33) | |||
|
| ||||||||
| W2A10 | 4 | chr9:92249743–95136064 | 2886.321 | Yes (2917.954) | Yes (3918.72) | Del (0.55) | Del (0.52) | Maternal DNA not available |
| chr11:21899343–22024112 | 124.769 | Yes (158.539) | No | Nl (0.99) | Nl (0.83) | |||
|
| ||||||||
| W2A20 | 33 | chr3:85646536–85740299 | 93.763 | Yes (164.507) | NA: No probes in region | Del (0.33) | Del (0.43) | Del (0.31) |
|
| ||||||||
| W2A5 | 36 | chr3:106781069–106825705 | 44.636 | Not attempted | NA: No probes in region | Del (0.45) | Del (0.79) | Maternal DNA not available |
|
| ||||||||
| W2A20 | 43 | chr2:51728450–51781546 | 53.096 | Not attempted | NA: No probes in region | Nl (1.08) | Del (0.34) | Dup (2.15) |
|
| ||||||||
| W2A21 | 45 | chr11:41755318–41788391 | 33.073 | No | NA: No probes in region | Nl (0.87) | Nl (0.85) | Nl (1.05) |
|
| ||||||||
| W2A21 | 48 | chr11:4764803–4865576 | 100.773 | Not attempted | Yes ( 106.42) | Del (0.60) | Fetal DNA not available | Del (0.27) |
| chr12:18693321–18703198 | 9.877 | Not attempted | No | Del (0.74) | Del (0.20) | |||
|
| ||||||||
| W2A8 | 49 | chr18:61831188–62009085 | 177.897 | Not attempted | NA: No probes in region | Del (0.46) | Del (0.34) | Maternal DNA not available |
NA: Not attempted due to either (1) absence of probes available on array in region of identified interest from initial hybridization on CNV370 Duo, or (2) absence of ability to validate with qPCR initial finding on CNV370 Duo.
For qPCR validation, ΔΔCt ratio (approximating fold change) were calculated as described in Methods. A ratio approximating 1.0 may be interpreted as no variation in copy number (Nl), while one approximating 0.50 or less is indicative of a probable homozygous deletion (Del), approximating 0.75 a possible heterozygous deletion (Del) and >1.25 of a duplication (Dup). In the majority of cases, a maternal specimen was not available due to lack of willingness or consent for maternal DNA testing. In rare cases fetal DNA was not available due to quality of specimen. In no case was paternal consent for DNA testing obtained.
Table IV.
Novel CNV Duplications in Expanded Wigglesworth Classifications W2 Unexplained Stillbirths.
| Stillbirth Characterization | Illumina CNV370 Duo CNV/SNP Array | Duplication Detected on Higher (Illumina1M) or Lower (Agilent 105K) Density Arrays& | qPCR Detection of CNV# | |||||
|---|---|---|---|---|---|---|---|---|
| Wigglesworth (W) Aberdeen (A) Classification | Case Number | Human Genome Coordinates | Length, Kb | Illumina 1M CNV/SNP Array (length, Kb) | Agilent 105K HD-CGH CNV Array (length, Kb) | Placental (ΔΔCt ratio) | Fetal (ΔΔCt ratio) | Maternal (ΔΔCt ratio |
| W2A21 | 3 | chr1:72874351–72987563 | 113.212 | No | No | Del (0.69) | Del (0.48) | Maternal DNA not available |
|
| ||||||||
| W2A10 | 8 | chr2:118594504–118971963 | 377.459 | Not attempted | No | Dup (1.65) | Nl (1.14) | Dup (2.72) |
|
| ||||||||
| W2A20 | 9 | chr7:98338258–98427987 | 89.729 | Not attempted | NA: No probes in region | Dup (2.18) | Nl (1.25) | Dup (1.76) |
| chr8:141506008–142107091 | 601.083 | Not attempted | NA: No probes in region | Del (0.65) | Nl (1.07) | Nl (0.83) | ||
|
| ||||||||
| W2A5 | 16 | chr11:48090102–48999442 | 909.340 | Yes (806.674) | No | Dup (1.34) | Nl (0.99) | Maternal DNA not available |
|
| ||||||||
| W2A15 | 19 | chr19:60221847–60251552 | 29.705 | Not attempted | NA: No probes in region | Dup (1.28) | Dup (1.28) | Maternal DNA not available |
|
| ||||||||
| W2A20 | 30 | chr5:44813635–44975050 | 161.415 | Not attempted | NA: No probes in region | Nl (1.19) | Del (0.28) | Maternal DNA not available |
|
| ||||||||
| W2A16 | 37 | chr9:117927770–118062623 | 134.853 | Not attempted | Yes (120.09) | Dup (1.68) | Dup (1.70) | Maternal DNA not available |
|
| ||||||||
| W2A8 | 42 | chr2:70618196–70803486 | 185.29 | Yes chr2:70616413–70686526; chr2:70716420–70807493 | NA: No probes in region | Dup (2.38) | Nl (0.95) | Dup (3.05) |
|
| ||||||||
| W2A21 | 46 | chr10:96436622–96555640 | 119.018 | Not attempted | NA: No probes in region | Dup (1.95) | Fetal DNA not available | Dup (1.82) |
|
| ||||||||
| W2A21 | 48 | chr5:28272386–28317006 | 44.620 | Not attempted | No | Dup (1.50) | Fetal DNA not available | Nl (0.86) |
| 48 | chr12:91281594–91399738 | 118.144 | Not attempted | No | Dup (2.04) | Nl (1.08) | ||
|
| ||||||||
| W2A21 | 50 | chr2:153496689–153607849 | 111.160 | Not attempted | NA: No probes in region | Dup (2.47) | Dup (1.43) | Dup (2.00) |
NA: Not attempted as outlined in Table III.
For qPCR validation, ΔΔCt ratio (approximating fold change) were calculated as described in Methods. A ratio approximating 1.0 may be interpreted as no variation in copy number (Nl), while one approximating 0.50 or less is indicative of a probable homozygous deletion (Del), approximating 0.75 a possible heterozygous deletion (Del) and >1.25 of a duplication (Dup). In the majority of cases, a maternal specimen was not available due to lack of willingness or consent for maternal DNA testing. In rare cases fetal DNA was not available due to quality of specimen. In no case was paternal consent for DNA testing obtained.
Identified Novel CNVs on the Illumina CNV370-Duo
CNVs were initially identified in the 29 placental samples using the Illumina CNV370-Duo Beadchip arrays. CNVs with genomic coordinates that completely overlapped with those CNVs identified in liveborn controls (n=10) or contained in The Centre for Applied Genomics Database of Genomic Variants (http://projects.tcag.ca/variation/) were removed from further consideration. This resulted in the identification of 24 novel CNVs in 17 samples (Tables III and IV) representing 59% of samples interrogated. The CNVs were comprised of 11 deletions (Table III) and 13 duplications (Table IV) and ranged in size from 8,984 bp to 2,886,321 bp.
We further measured our ability to detect CNVs in non-W2 expanded Wigglesworth classification stillbirths (W5 and W6), by examining stillborns affected by either non-syndromic anomalies (case 2) or common perinatal infections (cases 7, 22, and 47). For example and as detailed in Table II, we interrogated case 2 (W6 classification) because although cranial asymmetry, hypotelorism, hydrops, and a hydrothorax was noted, these findings did not associate with a microscopically detected cytogenetic abnormality (karyotypically normal). Cases 22 and 47 were interrogated because although CMV, listeria, and parvovirus B19 serologies were positive, there were no associated abnormalities such as hydrops, microcephaly, nor fetal calcifications (Table II). Thus in each of these instances we were curious whether we could detect copy number imbalances with our ascribed methodologies. Our detection of novel deletions and duplications was akin to the W2 schema (Table V).
Table V.
Novel CNV Deletions and Duplications in Expanded Wigglesworth Classifications Stillbirths.
| Stillbirth Characterization | Illumina CNV370 Duo CNV/SNP Array | Deletion Detected on Higher (Illumina1M) or Lower (Agilent 105K) Density Arrays& | qPCR Detection of CNV# | |||||
|---|---|---|---|---|---|---|---|---|
| Wigglesworth (W) Aberdeen (A) Classification | Case Number | Human Genome Coordinates | Length, Kb | Illumina 1M CNV/SNP Array (length, Kb) | Agilent 105K HD-CGH CNV Array (length, Kb) | Placental (ΔΔCt ratio) | Fetal Tissue (ΔΔCt ratio) | Maternal Blood (ΔΔCt ratio) |
|
Deletions
| ||||||||
| W6 | 2 | chr6:31893432–31903349 | 9.917 | No | NA: No probes in region | Del (0.83) | Del (0.50) | Maternal DNA not available |
|
| ||||||||
| W5 | 22 | chr6:30504152–30504673 | 0.521 | Yes (0.489) | NA: No probes in region | Del (0.79) | Del (0.66) | Maternal DNA not available |
| chr11:40663613–40851253 | 187.640 | Yes (229.468) | Yes (187.640) | Del (0.47) | Del (0.27) | |||
| chr20:14312927–14343593 | 30.666 | Yes (52.429) | No | Del (0.42) | Del (0.39) | |||
|
| ||||||||
|
Duplications
| ||||||||
| W6 | 2 | chr7:125250953–125300258 | 49.305 | Yes (47.210) | NA: No probes in region | Dup (1.42) | Dup (1.70) | Maternal DNA not available |
|
| ||||||||
| W5 | 22 | chr8:87256311–87382591 | 126.280 | Yes (156.332) | No | Dup (1.44) | Dup (1.53) | Maternal DNA not available |
NA: Not attempted as outlined in Table III.
For qPCR validation, ΔΔCt ratio (approximating fold change) were calculated as described in Methods. A ratio approximating 1.0 may be interpreted as no variation in copy number (Nl), while one approximating 0.50 or less is indicative of a probable homozygous deletion (Del), approximating 0.75 a possible heterozygous deletion (Del) and >1.25 of a duplication (Dup). In the majority of cases, a maternal specimen was not available due to lack of willingness or consent for maternal DNA testing. In rare cases fetal DNA was not available due to quality of specimen. In no case was paternal consent for DNA testing obtained.
Detection of CNVs by Alternate Methodologies and Platforms
At present, it is unclear what platforms represent “optimal” methods to detect CNVs and, what methods, if any, actually serve to “validate” detected CNVs. In order to examine reproducibility of our findings quantitative PCR and two other platforms were employed. Specifically, the subset of placental samples with identified CNVs on the CNV370-Duo were also interrogated using the more expansive Illumina 1M-Duo Beadchip and the lower density Agilent 105K Comparative Genomic Hybridization Array.
QPCR Detection
Identified CNVs were examined by qPCR in both the interrogated placental samples and the associated fetal samples with the exception of cases 46 and 48 for which no fetal samples were available (Tables III and IV). Results of qPCR assays on the placental samples detected 18 of the 24 total identified CNVs (75%). When this was limited to only larger CNVs (>200 Kb) this was noted to be 96% (deletions and duplications; Tables III and IV) with all placental deletions (8/8 or 100%; Table III) detected on the CNV370-Duo also detected by qPCR.
Of the CNV deletions detected by qPCR on placental DNA, all (6/6, or 100%; Table III) were also confirmed by qPCR assays on fetal DNA samples in the 7 of 8 cases where fetal samples were available. In contrast, among the larger duplications detected by qPCR on placental DNA, only 2/7 (29%) were also detected in fetal tissue. This is likely due to the presence of probable maternal contamination, as validated by our agreement between available maternal samples and placental samples with discordance of fetal tissue (Table IV). These are therefore most likely to represent so-called benign duplications.
With respect to determination of CNV zygosity, three of the deletions were identified by cnvPartition as being homozygous. QPCR identified two of these putative homozygous deletions (sample 3, chr6:33134224–33143208 and sample 48, chr12:18693321–18703198) but the Ct values were consistent with heterozygous deletions rather than homozygous (ΔΔCt>0.25) and the remaining putative homozygous deletion (sample 45, chr11:41755318–41788391) was not detected by qPCR.
Alternate Array Detection
Placental samples with identified CNVs were also interrogated employing the Illumina 1M-Duo Beadchip in order to assess reproducibility of results using similar platforms, with the latter being of markedly higher density. The Illumina 1M-Duo Beadchip (1.2M) differs from the CNV370-Duo Beadchip (370K) only in the number of SNPs interrogated by the array. Of the 17 samples in which CNVs were identified using the CNV370-Duo, 6 samples (representing 9 identified CNVs) were also interrogated using the 1M-Duo Beadchip. The CNV370-Duo and 1M-Duo Beadchips agreed on 6 of the 9 (67%) CNV calls. In a comparison of only those CNVs also confirmed by qPCR, the CNV370-Duo and 1M-Duo Beadchips agreed on 5 the 6 (83%) confirmed CNVs. When we limited this analysis only to larger CNVs >50 Kb, 6 of 7 (87%) were detected.
Finally, placental samples were also interrogated using the less dense Agilent 105K Comparative Genomic Hybridization (CGH) Array. The Agilent 105K CGH array differs from the CNV370-Duo Beadchip in both the number and genomic location of probes, as well as basic analysis principles. The Agilent aCGH platforms rely on a comparative hybridization of a differentially labeled interrogated DNA sample and a reference DNA sample, with copy number aberrations derived from the comparative strength of the hybridization signal from each call sample. The Illumina platforms, however, rely on a single sample hybridization, whereby copy number information is derived from combined analysis of absence of SNP heterozygosity and signal strength data at various oligonucleotides in aggregation. Of the 17 samples in which CNVs were identified using the CNV370-Duo Beadchip, 6 samples, representing 12 identified CNVs, were also interrogated using the Agilent 105K CGH Array. A total of 3 samples, representing 4 identified CNVs, were interrogated using all three array platforms. All 4 CNVs interrogated by all three platforms were confirmed by qPCR. As reported in Tables III and IV, the vast majority of CNVs detected on either Illumina platform but not the Agilent platform were due to lack of probe coverage on the latter, less dense array. When we again limit our analysis to CNV deletions 100% of the cases would demonstrate a deletion regardless of the platform employed (Table III). However, this was not true for duplications (Table IV).
Detection of CNVs in Expanded Wigglesworth Classification Stillbirths
Interestingly and as shown in Table V, in cases 2 and 22 all duplications and deletions detected by the Illumina CNV370Duo were detected by qPCR of both placental and fetal tissue (6/6; 100%) but only the deletions were detected by the Agilient 105K array (Table V).
Genes Affected By Confirmed Deletions
Deletions affecting genes are most likely to play a causative role in stillbirth. A total of 4 deletions confirmed by qPCR on the placental and fetal samples, with the exception of case 48 for which there was no fetal sample, affected genes (Table VI).
Table VI.
Genes Affected by Confirmed Novel CNV Deletions and Duplications in Stillbirths of Expanded Wigglesworth Classifications.
| Stillbirth Characterization | Genomic Region | Affected Gene(s) in Region Contained in Structural Variant | |||
|---|---|---|---|---|---|
| Wigglesworth (W) Aberdeen (A) Classification | Case Number | Human Genome Coordinates | Length, Kb | Exonic Loci | Intronic Loci |
|
Deletions
| |||||
| W2A21 | 3 | chr6:33134224–33143208 | 8.984 | None | HLA-DPA1 3′ UTR and part of preceding intron |
|
| |||||
| W2A9 | 4 | chr9:92249743–95136064 | 2886.321 | ANKRD19, ASPN, AUH, BICD2, C9orf44, C9orf89, CENPP, DIRAS2, ECM2, FGD3, IARS, IPPK, NFIL3, NINJ1, NOL8, OGN, OMD, ROR2, SNORA84, SPTLC1, SUSD3, SYK, WNK2, ZNF484 (completely deleted), C9orf129 (partially deleted) | None |
|
| |||||
| W2A20 | 33 | chr3:85653649–85740299 | 93.763 | None | CADM2 intron |
|
| |||||
| W2A21 | 48 | chr11:4764803–4861778 | 100.773 | OR52R1, OR51F2, OR51S1, OR51T1(completely deleted) | None |
|
| |||||
| W5 | 22 | chr11:40663613–40851253 | 187.640 | None | LRRC4C intron |
| chr20:14312927–14343593 | 30.666 | None | MACROD2 intron | ||
|
| |||||
|
Duplications
| |||||
| W2A16 | 37 | chr9:117927770–118062623 | 185.299 | PAPP-A | None |
|
| |||||
| W2A21 | 48 | chr12:91281594–91399738 | 118.144 | CLLU1 OS, CLLU1 | None |
By far the most compelling evidence for an identifiable deletion with a potential causative role for the stillbirth was observed for case 4 in which 25 contiguous genes were deleted (Table VI).
Other less notable and less expansive deletion occurred. For example in case 3 this involved deletion of the HLA-DPA1 3′ UTR. The potential mechanism from this deletion resulting in dysregulation of gene expression might arise from the concomitant observations that conserved miRNAs have been identified that target HLA-DPA1 (MicroCosm Targets http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/), and miRNA regulation occurs primarily through binding to the 3′ UTR. The genes deleted in case 48 are all olfactory receptor encoding genes and as such seem unlikely to play a causative role even though deletions of olfactory receptors have also been identified in spontaneous abortions between 10–20 weeks (15). As a third example, a deletion occurring in an intron of CADM2 in case 33 could play a causative role as such a deletion can result in the gene being incorrectly spliced. An additionally likely possibility for cases 33 and 48 are that these variants are in fact not de novo but may arise from the maternal germline. However, the most compelling case for a likely de novo and contributing variant to stillbirth was observed in case 4.
Case 4 Demonstrates Potential Clinical Utility of Array Based Copy Number Analysis
The available clinical data on case 4 indicated that there were no fetal abnormalities or maternal comorbidities, and prior karyotype was reported as normal. Array based copy number analysis resulted in the identification of a 2.89 Mbp heterozygous deletion on chromosome 9 in this case (Figure 1), which was detected by all array platforms (Tables III, IV, VI) and confirmed by qPCR assays on the placenta and fetal tissue, with the high qPCR ΔΔCt in the fetal blood sample most likely resulting from sample contamination by maternal blood (Figure 1B). The deletion resulted in the loss of 25 RefSeq Genes (Table 3 and Figure 1C). The large size and high gene content of this deletion strongly suggests that it likely played a causative role in the stillbirth.
Figure 1.
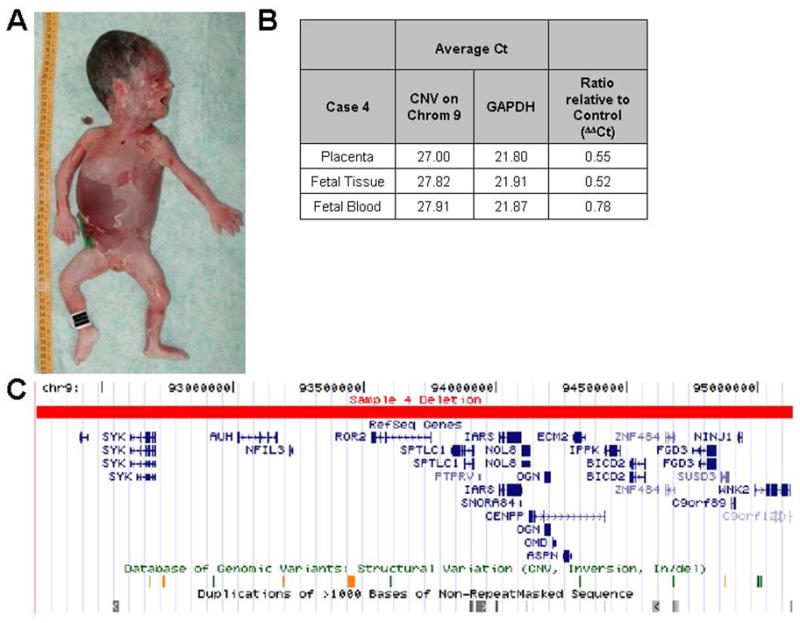
A 2.89 Mb deletion identified in case 4. A) Autopsy photograph of fetus, with complete anatomic pathology revealing no evidence of gross nor histologic abnormalities. B) SYBR Green qPCR results showing a ratio relative to control (ΔΔCt) approximating 0.5 confirming the deletion. A ΔΔCt in fetal blood was 0.78, possibly due to maternal blood contamination. C) UCSC Genome Browser view of the genomic region deleted. The deletion affected 25 known (RefSeq) genes (with complete listing as noted in Table VI). Known structural variations and segmental duplications in this region are also shown.
DISCUSSION
We have demonstrated for the first time that genome-wide array-based profiling of placentas from well-characterized, systematically classified unexplained stillbirths enables the detection of CNVs that were not observed by cytogenetic methods. These findings suggest that small genomic imbalances might contribute to a small proportion of non-anamalous, unexplained stillbirths. While in this study the likelihood of truly significant imbalances was low, it was not negligible (Table IV) and occurred in a carefully controlled research setting where karyotypes had been already performed.
Previous studies to identify genomic abnormalities in stillbirth samples have primarily employed G-banded karyotyping and fluorescence in situ hybridization (FISH). Using these methods, cytogenetic abnormalities have been described in a small percentage (5–13%) of all stillbirths (13, 17, 18). While in two studies this number does not demonstrably increase when fetal anatomic anomalies are present (17, 18), in a 3rd study chromosomal abnormalities were significantly higher (p<0.001) and observed in up to 38% of dysmorphic stillbirths (13). Karyotyping and FISH are generally accompanied by a high failure rate due to the inability to culture cells from fetal demise tissue samples (16%; 17). Presently, two types of arrays are available for genome-wide analysis of copy number imbalances: single nucleotide polymorphism (SNP) based arrays and array comparative genomic hybridization without SNP analysis.
CNVs detectable by arrays can be quite large as demonstrated by the 2.89 Mbp deletion in case 4, and yet would still be under the threshold of detection by broadly utilized traditional cytogenetic methods. Thus, the higher resolution provided by arrays suggests that they can be used as an alternative or at least a complementary method to standard karyotype analysis and FISH for examining genomic abnormalities in unexplained stillbirths. While the cost of clinical array-based copy number analysis is still higher than that of standard karyotyping, it is expected to continue to decrease as development will improve automation to facilitate high-throughput sample processing, data analysis, and reliable result reporting (22). We propose that the nominal difference in cost between the two approaches is well balanced by the higher resolution of genomic abnormalities in stillbirths provided by an array. In addition, besides its capacity to detect clinically significant copy number changes, array based analysis also avoids the low rate of detection of cytogenetic abnormalities resulting from failure of fetal or placental cell growth in vitro (17). A third advantage is that results can be more rapidly available because there is no need for precedent cell culture (23).
There are some inherent limitations to our study. First, we did not have uniformly available maternal DNA for the establishment of the de novo status of described CNVs. Moreover, in no instance was paternal DNA available. Optimal characterization of de novo status and likely contributory role of detected CNVs would require comparison to both parental genomes. Second, the samples were not initially collected with the purpose of array-based copy number analysis in mind, and thus in not all cases were we able to obtain stringent quality fetal DNA needed for confirmation. We believe that protocols optimized for maintenance of DNA integrity (including immediate DNA extraction) could have potentially allowed for a higher detection rate as well as greater ability for confirmation. Third, there are a number of inherent limitations to the array based technology itself. For example, CGH arrays cannot detect balanced rearrangements and are unreliable in detection of triploidy (24, 25). That said, de novo truly balanced rearrangements are unlikely to be a cause of stillbirth.
Despite these limitations, the strengths of our study are several-fold. First, we employed a number of practical applications in our protocol design. For example, we chose to use arrays to interrogate placental samples in order to examine the applicability of using placental DNA as a substitute for assaying DNA from fetal samples for a number of clinically practical reasons. In some cases direct fetal DNA assay is possible because fascia lata (or other fetal tissue) can be acceptably obtained. However, in other instances the parents may not be comfortable with consenting for direct fetal tissue sampling because they have concerns about tissue extraction on the fetus (14, 26). In other instances, extreme maceration in cases of longer interval fetal demise samples may necessitate utilization of placental tissue as the only viable alternative (27).
A second strength to our study was the strong concordance between results from placental and fetal DNA with respect to detection of deletions. While this was not true for detection of CNV duplications, there are likely several reasons. First, relatively small (<1 Mb) duplications are of unlikely significance in our sample set since the overwhelming majority detected in placenta were also detected in maternal blood but not in the fetus (Table IV). Second, of the four cases where CNV deletions (cases 4 and 48) and duplications (cases 37 and 48) were detected by multiple modalities (Illumina and/or Agilent platforms and/or qPCR) we observed concordance between placenta and fetus. The exception to this was case 37, which could not be tested secondary to the unavailability of fetal DNA.
Alternatively, CNVs detected solely in placental DNA and not in fetal tissue DNA may also be of interest if they represent genomic abnormities that are deleterious to the placenta, thereby contributing to the stillbirth. Chromosomal aneuploidy differences between placental and fetal genomes, known as confined placental mosaicism, have been observed by others to be associated with stillbirths (28). Thus, it seems possible that smaller scale CNV differences between the placenta and fetus may play a similar role and it would be of great value to interrogate both placenta and fetal tissue in future analyses of normal pregnancies and those complicated by stillbirth or other prenatal complications. A potential pertinent example of this is with case 37 which demonstrated a duplication of chr9 at the PAPP-A region. Lower circulating levels in PAPP-A on maternal serum screening have been associated with miscarriage (30); CNV duplications may result in either increased or decreased expression depending upon the genomic disruption (1).
A third strength of our study is our utilization of multiple measures for detection of structural variants. qPCR detection of putative CNVs can be time consuming and costly and does not necessarily imply “validation”. Alternately, confirmation using a second array platform of similar or higher resolution (i.e., the Illumina 1M but not Agilent 105K) might offer an alternative to qPCR especially in cases where multiple CNVs must be validated in a single sample because those CNVs can be validated by a single array platform rather than multiple qPCR assays. Warren et al. (20) initially identified CNVs in spontaneous abortions between 10–20 weeks using a low resolution BAC based array and then validated identified CNVs using a higher resolution CGH array. We initially identified CNVs using an array with 370K SNP probes and attempted to additionally detect those found using a lower resolution 105K CGH array. The low validation rate by the second array of 30% for CNVs also confirmed by qPCR is largely due to the lower resolution of the second array compared to the first. Validation by a second array platform would be an alternative to qPCR if the resolution is similar or greater on the second array. This will become more important as higher resolution arrays are used, with an inherently greater chance of detecting more than one CNV. Furthermore, it would allow for detection of additional CNVs that may be found by one type of array technology, but not by another (i.e., SNP based versus aCGH). This would be rare for large deletions and duplications (as illustrated by Case 4) but could be important for small yet clinically significant copy number deletions.
A fourth strength of this study is its robust characterization of stillbirths as “unexplained”, with further sub-classification by virtue of gestational age and gender-adjusted fetal growth. While others (19) interrogated 15 anomalous and non-anomalous stillbirths with aCGH, this is the first such report of determination of structural genetic variation in systematically acquired and well-characterized unexplained fetal deaths. By benefitting from the robust experience of a panel of experts in the characterization of these stillbirths, we avoided potential bias by not including anomalous stillbirths in the W2 criterion.
A fifth strength of our study is its potential for broader clinical implications. Unexplained stillbirths are clinically problematic, as they comprise the vast majority of stillbirths and occur most notably in the third trimester. The ability to utilize newer genome-wide technologies in a relatively inexpensive manner in this population would constitute an efficacious use of available resources to obtain a definitive diagnosis and thereby minimize parental stress and grief. For example, consider a hypothetical couple who present with a 39 week non-anomalous, unexplained stillbirth. In the course of evaluation, an array was completed which demonstrated a novel or known clinically significant 2.9 Mbp deletion which encompassed 34 genes. Both parents are then interrogated, found not to contain the structural variant, thus confirming an association between the fetal demise and a spontaneous de novo event. In any future pregnancies, the couple could choose to employ CVS or amniocentesis for targeted detection of the same structural variant. If the structural variant is not present,, then expensive other antenatal testing (i.e., once or twice weekly non-stress tests through the third trimester) might be further avoided. Larger multicenter studies will likely be needed to confirm that such an approach will be cost-effective and clinically beneficial.
Finally, such larger studies on stillbirth samples may reveal new copy number imbalances, associated with an important proportion of cases that have not previously been ascertained because we lacked the technologies to do so. Such discoveries have clearly resulted from postnatal application of array-based copy number analysis in children with congenital anomalies, dysmorphic features, and developmental delays. This will not only result in better understanding of causes of stillbirth, but also in improved diagnosis (ACOG Committee Opinion 440, 2009). Array based copy number analysis has now replaced karyotyping as the first-line diagnostic test in pediatric genetic analyses. Similar developments are predicted for prenatal diagnosis and the evaluation of fetal demise.
In sum, we have employed multiple modalities in detecting and analyzing copy number variants in unexplained stillbirths. We have observed that in at least one instance, this yielded a likely deletion potentially causative of stillbirth which was not detected with cytogenetic analysis. While the overall yield may be conceived of as low and is most likely to be of significance when CNV deletions are detected, it bears mention that under conventional protocols such detection would avoid the emotionally and clinically troublesome classification of this fetal death as having been “unknown”. Further studies with protocols optimized for such advancing genetic technologies are essential in this common perinatal disorder.
Supplementary Material
Acknowledgments
This work was funded by the NIH New Innovator Award to K.A.T. (DP2OD001500-01).
References
- 1.Estivill X, Armengol L. Copy number variants and common disorders: filling the gaps and exploring complexity in genome-wide association studies. PLoS Genet. 2007;3:1787–99. doi: 10.1371/journal.pgen.0030190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eichler EE, Nickerson DA, Altshuler D, Bowcock AM, Brooks LD, Carter NP, et al. Completing the map of human genetic variation. Nature. 2007;447:161–5. doi: 10.1038/447161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–37. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 5.Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–7. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 6.Vissers LELM, de Vries BBA, Osoegawa K, Janssen IM, Feuth T, Choy CO, et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet. 2003;73:1261–70. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rickman L, Fiegler H, Carter NP, Bobrow M. Prenatal diagnosis by array-CGH. Eur J Med Genet. 2005;48:232–40. doi: 10.1016/j.ejmg.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacDorman MF, Munson ML, Kirmeyer S. Fetal and perinatal mortality, United States, 2004. Natl Vital Stat Rep. 2007;56:1–19. [PubMed] [Google Scholar]
- 10.Facchinetti F, Reddy U, Stray-Pedersen B, Baronciani D, Requejo JH. International issues in stillbirth. J Matern Fetal Neonatal Med. 2008;21:425–8. doi: 10.1080/14767050802040849. [DOI] [PubMed] [Google Scholar]
- 11.Greb AE, Pauli RM, Kirby RS. Accuracy of fetal death reports: comparison with data from an independent stillbirth assessment program. Am J Public Health. 1987;77:1202–6. doi: 10.2105/ajph.77.9.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korteweg FJ, Bouman K, Erwich JJHM, Timmer A, Veeger NJGM, Ravise JM, et al. Cytogenetic analysis after evaluation of 750 fetal deaths: proposal for diagnostic workup. Obstetrics Gynecology. 2008;111:865–74. doi: 10.1097/AOG.0b013e31816a4ee3. [DOI] [PubMed] [Google Scholar]
- 13.Dudley DJ, Goldenberg R, Conway D, Silver RM, Saade GR, Varner MW, et al. A new system for determining the causes of stillbirth. Obstet Gynecol. 2010;116:254–60. doi: 10.1097/AOG.0b013e3181e7d975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silver RM, Heuser CC. Stillbirth workup and delivery management. Clin Obstet Gynecol. 2010;53:681–90. doi: 10.1097/GRF.0b013e3181eb3297. [DOI] [PubMed] [Google Scholar]
- 15.Menten B, Swerts K, Chiaie BD, Janssens S, Buysse K, Philippe J, Speleman F. Array comparative genomic hybridization and flow cytometry analysis of spontaneous abortions and mor in utero samples. BMC Med Genet. 2009;10:89–94. doi: 10.1186/1471-2350-10-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith GCS, et al. Maternal and biochemical predictors of antepartum stillbirth among nulliparous women in relation to gestational age of fetal death. BJOG. 2007;114:705–714. doi: 10.1111/j.1471-0528.2007.01343.x. [DOI] [PubMed]
- 17.Pinar H, Carpenter M, Martin BJ, Tantravahi U. Utility of fetal karyotype in the evaluation of phenotypically abnormal stillbirths. Pediatr Dev Pathol. 2009;12:217–21. doi: 10.2350/07-07-0307.1. [DOI] [PubMed] [Google Scholar]
- 18.Smith A, Bannatyne P, Russell P, Ellwood D, den Dulk G. Cytogenetic studies in perinatal death. Aust N Z J Obstet Gynaecol. 1990;30:206–10. doi: 10.1111/j.1479-828x.1990.tb03214.x. [DOI] [PubMed] [Google Scholar]
- 19.Raca G, Artzer A, Thorson L, Huber S, Modaff P, Laffin J, et al. Array-based comparative genomic hybridization (aCGH) in the genetic evaluation of stillbirth. Am J Med Genet A. 2009;149A:2437–43. doi: 10.1002/ajmg.a.33083. [DOI] [PubMed] [Google Scholar]
- 20.Warren JE, Turok DK, Maxwell TM, Brothman AR, Silver RM. Array comparative genomic hybridization for genetic evaluation of fetal loss between 10 and 20 weeks of gestation. Obstet Gynecol. 2009;114:1093–102. doi: 10.1097/AOG.0b013e3181bc6ab0. [DOI] [PubMed] [Google Scholar]
- 21.Wigglesworth JS. Classification of perinatal deaths. Soz Praventivmed. 1994;39:11–4. doi: 10.1007/BF01369938. [DOI] [PubMed] [Google Scholar]
- 22.Van den Veyver IB, Beaudet AL. Comparative genomic hybridization and prenatal diagnosis. Curr Opin Obstet Gynecol. 2006;18:185–91. doi: 10.1097/01.gco.0000192986.22718.cc. [DOI] [PubMed] [Google Scholar]
- 23.Bi W, Breman AM, Venable SF, Eng PA, Sahoo T, Lu X, et al. Rapid prenatal diagnosis using uncultured amniocytes and oligonucleotide array CGH. Prenat Diagn. 2008;28:943–9. doi: 10.1002/pd.2087. [DOI] [PubMed] [Google Scholar]
- 24.Van den Veyver IB, Patel A, Shaw CA, Pursley AN, Kang SL, Simovich MJ, et al. Clinical use of array comparative genomic hybridization (aCGH) for prenatal diagnosis in 300 cases. Prenat Diagn. 2009;29:29–39. doi: 10.1002/pd.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaeffer AJ, Chung J, Heretis K, Wong A, Ledbetter DH, Lese Martin C. Comparative genomic hybridization-array analysis enhances the detection of aneuploidies and submicroscopic imbalances in spontaneous miscarriages. Am J Hum Genet. 2004;74:1168–74. doi: 10.1086/421250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith GCS, Fretts RC. Stillbirth. Lancet. 2007;370:1715–25. doi: 10.1016/S0140-6736(07)61723-1. [DOI] [PubMed] [Google Scholar]
- 27.Bonetti LR, Ferrari P, Trani N, Maccio L, Laura S, Giuliana S, et al. The role of fetal autopsy and placental examination in the causes of fetal death: a retrospective study of 132 cases of stillbirths. Arch Gynecol Obstet. 2010 doi: 10.1007/s00404-009-1317-4. [DOI] [PubMed] [Google Scholar]
- 28.Kalousek DK, Barrett I. Confined placental mosaicism and stillbirth. Pediatr Pathol. 1994;14:151–9. doi: 10.3109/15513819409022034. [DOI] [PubMed] [Google Scholar]
- 29.Gardosi J, Kady SM, McGeown P, Francis A, Tonks A. Classification of stillbirth by relevant condition at death (ReCoDe): population based cohort study. BMJ. 2005;331:1113–7. doi: 10.1136/bmj.38629.587639.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Ravenswaaij R, Tesselaar-van der Goot M, de Wolf S, van Leeuwen-Spruijt M, Visser GH, Schielen PC. First-trimester serum PAPP-A and fβ-hCG concentrations and other maternal characteristics to establish logistic regression-based predictive rules for adverse pregnancy outcome. Prenat Diagn. 2011;31:50–57. doi: 10.1002/pd.2610. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.