

Abstract
Biphalin is a dimeric opioid peptide that exhibits affinity for three types of opioid receptors (MOP, DOP and KOP). Biphalin is undergoing intensive preclinical study. It was recognized that activation of δ-opioid receptor elicits neuroprotection against brain hypoxia and ischemia. We compare the effect of biphalin and morphine and the inhibition of opioid receptors by naltrexone on survival of neurons in rat organotypic hippocampal cultures challenged with NMDA. Findings: (1) 0.025–0.1 μM biphalin reduces NMDA-induced neuronal damage; (2) biphalin neuroprotection is abolished by naltrexone; (3) reduced number of dead cells is shown even if biphalin is applied with delay after NMDA challenge.
Keywords: Neuroprotection, Opioid, Opioid receptor, Excitotoxicity, Biphalin, Morphine
Introduction
Excitotoxicity is a leading cause of neurodegeneration observed in progressive and acute brain diseases [1–3]. Despite many years of research on the mechanisms of neuronal death and search for effective neuroprotectants there is still no effective therapy [4–6]. Among agents tested so far, those designed to combine multiple neuroprotective mechanisms such as the AM-36, seem to have the greatest neuroprotective effect [7].
Excitotoxicity is associated with pathological changes (such as excess release of excitatory amino acids, disruption of ionic homeostasis due to Na+ and Ca2+ influx and generation of toxic free radicals) as well as with generation and transmission of pain signal. Pain is a signal of acute (e.g. wound) or chronic (e.g. inflammation) pathological changes within the body. Therefore, involvement of opioids, which are currently used in severe pain treatments, in neurodegeneration/neuroprotection mechanisms is important field of studies [8, 9]. Most of available study indicated that all opioid receptors are involved in neuroprotection. Therefore activation of all opioid receptors could result in more effective neuroprotection than selective ligands interacting with one type of opioid receptor.
Biphalin (BIPH) is a peptide [10] that hybridizes two opioid pharmacophores in one. The compound is promoting as a new analgesics following idea that multitarget receptor ligands could be more effective than specific to one type of opioid receptor. Indeed, biphalin exhibits a high affinity for opioid receptors types MOP and DOP and lower but significant to receptors KOP [11–13]. When administrated directly to central nervous system (intracerebroventricularly or intrathecally) it has been shown to be more potent than morphine and ethorphine at eliciting antinociception [11]. Moreover, BIPH induces less physical dependence than morphine [14] and express several positive additional effects that further rationalize of its development as a new analgesic [11, 15–17]. Our present study refers to neuroprotective potential of biphalin and compares it to the known morphine protection [18, 19] in the organotypic hippocampal cultures (OHC) challenged with NMDA.
Materials and Methods
Organotypic Hippocampal Culture
The Local Committee for Ethics in Animal Experiments approved all the experimental procedures on rat organotypic hippocampal culture (OHC). Hippocampal slices were prepared from 6 to 7 days old Wistar rats according to the method of Stoppini [20] with slight modifications [21]. After brief anesthesia with Vetbutal (pentobarbital; Sigma) ice-cooled pups were plunged into 70% alcohol solution, decapitated with scissors, and then brains were quickly removed to ice-cold HBSS (Gibco). Hippopcampi were separated and cut into 400 μm slices using McIlwain tissue chopper. Slices were transposed to Millicell-CM (Millipore) membranes for further growth. Millicell-CM membranes in 6-well plates were pre-equilibrated with 1 ml of culture medium (HEPES pH 7.2, DMEM 50%, HBSS 25%, Horse Serum 25% (Gibco), 2 mmol/l l-glutamine, 5 mg/ml glucose, 1% amphotericine B and 0.4% penicillin–streptomycin). Cultures were started in a regular, 25% horse serum-containing medium which was gradually replaced (from DIV 4th until 7th) by a serum-free, defined-solution-based medium. This medium contained DMEM/F12 50% and additionally N2A (1:10; Gibco) and B27 supplement (1:100; Gibco) without serum (the rest of compounds remained the same). Cultures were maintained in a moist atmosphere of air and 5% CO2, at 36°C for 14–16 days.
Induction of Glutamatergic Stress
After 10 days in culture, excitotoxic stress was induced by adding 100 μM N-methyl-D-aspartate (NMDA, Sigma), for 3 h. Then the slices were transferred to the fresh culture medium. Biphalin (0.025–0.1 μM) or morphine (0.1 or 3 μM) were applied together with NMDA or with the delay of 0.5, 1 or 1.5 h and were present throughout the experiment (up to 24 h). To block opioid receptors 10 nM naltrexone (Sigma) was added together with NMDA and biphalin or morphine and was present throughout the experiment according to the paradigm shown in Fig. 1.
Fig. 1.

Experimental protocol used to study the effect of single dose of biphalin (BIPH) or morphine (MPH) on neurons survival in vitro in the model of organotypic hippocampal cultures (OHC) after 100 μM NMDA injury. DIV days in vitro, PI propidium iodide, HS horse serum, Ntx naltrexone
Analysis of Cell Death
Cell death was quantified in the manner described previously [21]. The fluorescent cell-death marker propidium iodide (PI) was present in the medium from 24 h prior to the experiments and throughout the recovery period. The relative extent of cell death was calculated from each standardized CA1 region as follows: % of dead cells = (experimental fluorescent intensity [FI] − background FI)/(maximal FI − background FI) × 100, where maximal FI was obtained by killing all cells with exposure to 1 mM NMDA.
Results
Neuroprotection Exerted by Biphalin After Glutamatergic Stress In Vitro
We have found that biphalin, in all tested concentrations, revealed significant cell protection in vitro, in stable temperature conditions (36°C), reducing the number of PI labeled cells after injury by more than half. A gradual increase in cell death was observed from 0 to 24 h after the insult (Fig. 2a). At 24 h after NMDA stress 61.9 ± 0.18% (n = 24) of CA1 layer neurons were PI positive. Application of 0.025, 0.05 or 0.1 μM biphalin decreased the amount of dead cells to 21.3 ± 0.17% (n = 16), 29.3 ± 0.3% (n = 16), and 22.5 ± 0.24% (n = 24), respectively in NMDA challenged slices (Fig. 2b). Biphalin alone did not change the viability of the slices. In such a same experimental setup, similar protection was given by 3 μM morphine decreasing the number of PI positive cells in CA1 region up 30.9 ± 0.19% (n = 8), as well as application of morphine in 0.1 μM concentration was resulted in 29.9 ± 0.46% (n = 8) PI positive cells after NMDA injury (Fig. 2b).
Fig. 2.
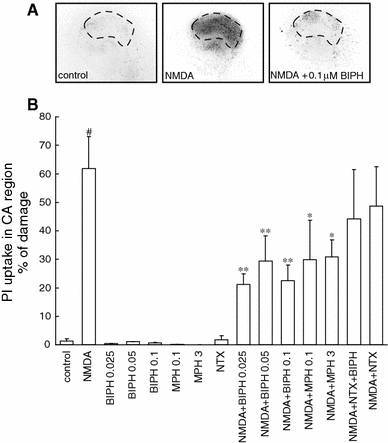
Neuroprotective effect evoked by biphalin (BIPH) in vitro in organotypic hippocampal cultures (OHC). a Inverted fluorescent images of propidium iodide-stained hippocampal slices 24 h after transient glutamatergic (100 μM NMDA, 3 h) stress. Damage was detected mostly in the CA1 area (defined by dotted lines). BIPH in the different concentrations (0.025–0.1 μM), morphine (MPH) (0.1, 3 μM) or naltrexone (Ntx) (10 nM) were added to the medium together with NMDA and were present till the end of the experiment. b Quantitative analysis of cell death of OHC, 24 h after glutamatergic stress and single dose of BIPH, MPH, naltrexone (Ntx) or combination of the drugs. The results are expressed as the mean ± SD (n = 9–24) of propidium iodide (PI) positive cells from at least three independent experiments. Values are considered significant where *P < 0.05 or **P < 0.01 versus NMDA treated cultures or #P < 0.01 versus control
Involvement of Opioid Receptors in Neuroprotection Exerted by Biphalin In Vitro
To explore the involvement of opioid receptors in biphalin-evoked protection in OHC, together with 0.1 μM biphalin and excitotoxic stress naltrexone, known multi-opioid receptor blocker was added. The optimal concentrations of naltrexone was set based on the data from studies testing the 0.5, 1, 10, 50 nM naltrexone on PI staining of neurons in control, unchallenged OHC (data not shown). To further experiments 10 nM naltrexone was applied; it was the highest tested concentration that did not impair the neurons in the control slices. Here we show that after NMDA injury and naltrexone application, the neuroprotective potential of 0.1 μM biphalin was abolished and resulted in 44.2 ± 0.39% (n = 8) of PI stained cells versus 22.5 ± 0.24% (n = 24) being observed in naltrexone free samples (Fig. 2b). While 10 nM naltrexone was applied with NMDA the number of PI positive cells was 48.7 ± 0.28% (n = 8) and did not significantly differ from NMDA alone challenged OHC.
Therapeutic Window of Biphalin Neuroprotection In Vitro
Next we have shown that biphalin was a potent neuroprotectant even it was applied 1.5 h after NMDA application (Fig. 3). Application of 0.1 μM biphalin 0.5, 1 or 1.5 h after NMDA challenge decreased the amount of dead cells to 23.1 ± 0.43% (n = 16), 33 ± 0.3% (n = 16), and 29.7 ± 0.3% (n = 24), what resulted in 63, 47 and 52% of protection, respectively.
Fig. 3.
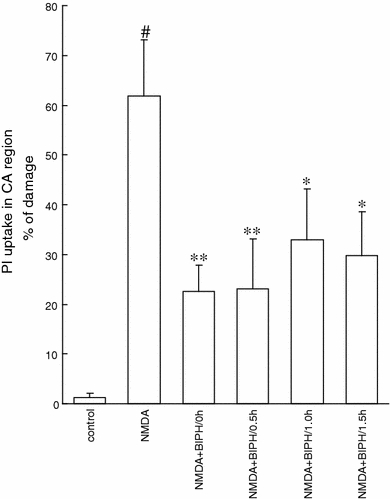
Neuroprotection evoked by delayed application of the single dose of biphalin (BIPH) in vitro in organotypic hippocampal culture (OHC) challenged with NMDA (100 μM) for 3 h. Quantitative analysis of cell death of OHC, 24 h after glutamatergic stress and 0.1 μM BIPH application at 0.5, 1 or 1.5 h after NMDA. The results are expressed as the mean ± SD (n = 16–24) of propidium iodide (PI) positive cells. Values are considered significant where *P < 0.05 or **P < 0.01 versus NMDA treated cultures or #P < 0.05 versus control
Discussion
In the reported experiments, the organotypic hippocampal cultures challenged with NMDA to assess the neuroprotective potential of biphalin and to compare it to the known protection effects of opioid analgesic “gold standard”, morphine [18, 19] have been used. In primary experiments, as was reported previously [22], relatively high dose of morphine (3 μM) has been used. In our studies the administrated dose induced survival of about 50% of hippocampal cells. Furthermore, we were observed a similar protective effect at a dose of morphine reduced even 30 times. The obtained ceiling protective effect may be directly related to proportion of cells containing MOP receptor types on cell membranes.
The application of biphalin, multitarget opioid ligand caused also a protective effect even at the lowest dose as 0.025 μM. Similar to morphine the application of three different doses of biphalin resulted in ceiling effect. Although this effect has been observed in protection of larger proportion, almost 65% of hippocampal cells. This results may indicates of the synergic neuroprotective interaction(s) of all types of opioid receptors.
The strong neuroprotective effect of biphalin was abolished by naltrexone, opioid multireceptor antagonist. This suggests exclusive involvement of opioid receptors in the mechanisms of biphalin neuroprotection. The small dose dependent effect of both ligands possibly depends on the unequal expression of opioid receptors in neurons populations.
Although, most of available studies indicate that all opioid receptors are involved in neuroprotection, some authors report contradictory results. Ammon-Treiber et al. [23] demonstrated that morphine exposure increases the neurotoxic effect of hippocampal hypoxia/hypoglycemia in a concentration dependent manner. They showed that 1 h morphine perfusion, immediately followed by a short hypoxic/hypoglycemic episode, resulted in an impaired restoration of evoked field potentials in the CA1 region as compared to untreated control brain slices undergoing hypoxia/hypoglycemia without drug pretreatment. In contrary, Zhao et al. [8] reported that exposure to morphine immediately or at 24 h before oxygen–glucose deprivation, reduced the oxygen–glucose deprivation-induced neuronal death in the CA1 region of the rat hippocampal slice cultures [8]. Morphine has preferential affinity to MOR receptors however, the studies have suggested that its protection from myocardial or neuronal injury occurs by activation of DOP-opioid receptor [24, 25]. DOPs may be topically involved in neuroprotection through a Gi-dependent manner. Extensive studies with DOR selective ligands in vivo and in vitro confirm neuroprotective effects of DOP activation [26]. DOP activation attenuates oxidative injury in the brain exposed to ischemia/reperfusion by enhancing antioxidant ability and inhibiting caspase activity [27]. It has been suggested that KOP activation is also involved in morphine protection mechanisms as well [28, 29]. Reported neurodegenerative properties of dynorphins, endogenous KOR ligands are caused by their metabolites, des-Tyr-dynorphins [30].
Our results excellently harmonize with just published results of Yang et al. [31]. They described biphalin ability for reducing brain edema formation using both in vitro and in vivo models of stroke. For the in vitro model of ischemia, hippocampal slices were exposed to oxygen glucose deprivation (OGD) conditions, what resulted in increased hippocampal water content. Interestingly, biphalin exhibited a greater effect in decreasing water content in OGD-exposed hippocampal slices, compared with MOR, DOR, and KOR selective opioid agonists. Furthermore, biphalin decreased edema and infarct ratios, and neuronal recovery from stroke in a permanent middle cerebral artery occlusion (MCAO) model of focal ischemia.
In conclusion, our data confirm that opioid ligands, in addition to their primary antinociceptive activity, may play neuroprotective role in neuropathological conditions resulted from brain ischemia. Biphalin expressed similar neuroprotective effect to that caused by morphine. However biphalin can be administrated in much smaller doses, which probably is possible due to the simultaneous interaction with DOP, MOP, KOP, three types of opioid receptors.
Acknowledgments
Support from Polish Mitochondrial Network MitoNet.pl is gratefully acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Maria Kawalec and Joanna E. Kowalczyk have equally contributed to this work.
References
- 1.Di Filippo M, Tozzi A, Costa C, et al. Plasticity and repair in the post-ischemic brain. Neuropharmacology. 2008;55:353–362. doi: 10.1016/j.neuropharm.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 2.Martin LJ. Neuronal cell death in nervous system development, disease, and injury (review) Int J Mol Med. 2001;7:455–478. [PubMed] [Google Scholar]
- 3.Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 4.Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Gunn AJ, Thoresen M. Hypothermic neuroprotection. NeuroRx. 2006;3:154–169. doi: 10.1016/j.nurx.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callaway JK. Investigation of AM-36: a novel neuroprotective agent. Clin Exp Pharmacol Physiol. 2001;28:913–918. doi: 10.1046/j.1440-1681.2001.03547.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhao P, Huang YM, Zuo ZY. Opioid preconditioning induces opioid receptor-dependent delayed neuroprotection against ischemia in rats. J Neuropathol Exp Neurol. 2006;65:945–952. doi: 10.1097/01.jnen.0000235123.05677.4b. [DOI] [PubMed] [Google Scholar]
- 9.Barry U, Zuo Z. Opioids: old drugs for potential new applications. Curr Pharm Des. 2005;11:1343–1350. doi: 10.2174/1381612053507459. [DOI] [PubMed] [Google Scholar]
- 10.Lipkowski AW, Konecka AM, Sroczynska I. Double-enkephalins-synthesis, activity on guinea-pig ileum, and analgesic effect. Peptides. 1982;3:697–700. doi: 10.1016/0196-9781(82)90173-5. [DOI] [PubMed] [Google Scholar]
- 11.Kosson D, Klinowiecka A, Kosson P, et al. Intrathecal antinociceptive interaction between the NMDA antagonist ketamine and the opioids, morphine and biphalin. Eur J Pain. 2008;12:611–616. doi: 10.1016/j.ejpain.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 12.Lipkowski AW, Carr DB, Bonney I, et al. Biphalin: a multireceptor opioid ligand. In: Chang K-J, Porreca F, Woods J, et al., editors. The delta receptor. New York: Informa Healthcare; 2003. pp. 245–259. [Google Scholar]
- 13.Abbruscato TJ, Williams SA, Misicka A, et al. Blood-to-central nervous system entry and stability of biphalin, a unique double-enkephalin analog, and its halogenated derivatives. J Pharmacol Exp Ther. 1996;276:1049–1057. [PubMed] [Google Scholar]
- 14.Yamazaki M, Suzuki T, Narita M, et al. The opioid peptide analogue biphalin induces less physical dependence than morphine. Life Sci. 2001;69:1023–1028. doi: 10.1016/S0024-3205(01)01194-8. [DOI] [PubMed] [Google Scholar]
- 15.Lazarczyk M, Matyja E, Lipkowski AW. A comparative study of morphine stimulation and biphalin inhibition of human glioblastoma T98G cell proliferation in vitro. Peptides. 2010;31:1606–1612. doi: 10.1016/j.peptides.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Wojciechowski P, Szereda-Przestaszewska M, Lipkowski AW. Respiratory and cardiovascular effects of biphalin in anaesthetized rats. Eur J Pharmacol. 2009;602:50–53. doi: 10.1016/j.ejphar.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 17.Tang JL, Lipkowski AW, Specter S. Molecular assessment of the potential combination therapy of cytokines with biphalin and AZT for Friend leukemia virus infection in vitro. Pharmacol Rep. 2008;60:190–198. [PubMed] [Google Scholar]
- 18.Fanjun M, Junfa L, Bingxi Z, et al. nPKCepsilon and NMDA receptors participate in neuroprotection induced by morphine pretreatment. J Neurosurg Anesthesiol. 2006;18:119–124. doi: 10.1097/00008506-200604000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Yang TT, Hung CF, Lee YJ, et al. Morphine inhibits glutamate exocytosis from rat cerebral cortex nerve terminals (synaptosomes) by reducing Ca2+ influx. Synapse. 2004;51:83–90. doi: 10.1002/syn.10290. [DOI] [PubMed] [Google Scholar]
- 20.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-M. [DOI] [PubMed] [Google Scholar]
- 21.Sarnowska A, Beresewicz M, Zablocka B, et al. Diazepam neuroprotection in excitotoxic and oxidative stress involves a mitochondrial mechanism additional to the GABAAR and hypothermic effects. Neurochem Int. 2009;55:164–173. doi: 10.1016/j.neuint.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Li J, Yang J, et al. Inhibition of PKCgamma membrane translocation mediated morphine preconditioning-induced neuroprotection against oxygen-glucose deprivation in the hippocampus slices of mice. Neurosci Lett. 2008;444:87–91. doi: 10.1016/j.neulet.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 23.Ammon-Treiber S, Stolze D, Schroder H, et al. Effects of opioid antagonists and morphine in a hippocampal hypoxia/hypoglycemia model. Neuropharmacology. 2005;49:1160–1169. doi: 10.1016/j.neuropharm.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 24.Lim YJ, Zheng S, Zuo Z. Morphine preconditions Purkinje cells against cell death under in vitro simulated ischemia-reperfusion conditions. Anesthesiology. 2004;100:562–568. doi: 10.1097/00000542-200403000-00015. [DOI] [PubMed] [Google Scholar]
- 25.Schultz JJ, Hsu AK, Gross GJ. Ischemic preconditioning and morphine-induced cardioprotection involve the delta (delta)-opioid receptor in the intact rat heart. J Mol Cell Cardiol. 1997;29:2187–2195. doi: 10.1006/jmcc.1997.0454. [DOI] [PubMed] [Google Scholar]
- 26.Iwata M, Inoue S, Kawaguchi M, et al. Effects of delta-opioid receptor stimulation and inhibition on hippocampal survival in a rat model of forebrain ischaemia. Br J Anaesth. 2007;99:538–546. doi: 10.1093/bja/aem220. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Xia X, Zhang Y, et al. Delta-opioid receptor activation attenuates oxidative injury in the ischemic rat brain. BMC Biol. 2009;7:55–64. doi: 10.1186/1741-7007-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schunk E, Aigner C, Stefanova N et al (2010) Kappa opioid receptor activation blocks progressive neurodegeneration after kainic acid injection. Hippocampus. doi:10.1002/hipo.20813 [DOI] [PubMed]
- 29.Ela C, Barg J, Vogel Z, et al. Distinct components of morphine effects on cardiac myocytes are mediated by the kappa and delta opioid receptors. J Mol Cell Cardiol. 1997;29:711–720. doi: 10.1006/jmcc.1996.0313. [DOI] [PubMed] [Google Scholar]
- 30.Sherwood TW, Askwith CC. Dynorphin opioid peptides enhance acid-sensing ion channel 1a activity and acidosis-induced neuronal death. J Neurosci. 2009;29:14371–14380. doi: 10.1523/JNEUROSCI.2186-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Wang H, Shah K, et al. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011;1383:307–316. doi: 10.1016/j.brainres.2011.01.083. [DOI] [PubMed] [Google Scholar]