

Abstract
Objective
Determine the probability of attaining complete remission in children with nonsyndromic epilepsy over the course of ≥10 years from initial diagnosis; identify early predictors of complete remission; and assess the risk of relapse after achieving complete remission.
Methods
In a prospective community-based cohort, complete remission was defined as 5 years seizure-and medication-free. Any subsequent seizure for any reason was a relapse. Uni- and bivariate analyses were conducted with standard methods including the Kaplan-Meier approach. Proportional hazards modeling was used for multivariable analysis.
Results
Of 613 cohort members, 347 had nonsyndromic epilepsies of whom 294 (85%) were followed ≥10 years (max=17.9). 170/294(58%) achieved complete remission of whom 10 (6%) relapsed. Seizure outcome at 2 years (remission, pharmacoresistant, unclear) (p<0.0001) and underlying cause (p<0.0001) distinguished groups with complete remission ranging from ~20% to ~75%. Older age at onset was independently associated with a poorer chance of complete remission. Relapses occurred up to 7.5 years after attaining complete remission and were marginally associated with underlying cause (p=0.06).
Interpretation
Complete remission occurs in over half of young people with nonsyndromic epilepsy and generally persists. Meaningful but imperfect predication is possible based on underlying cause and early seizure control. The finding of age effects may play a role in meaningful identification of phenotypes which could become fruitful targets for genetic and imaging investigations in these otherwise poorly differentiated epilepsies.
Complete cure or remission of seizures with no further need of treatment is the most desired seizure outcome and an ultimate goal in epilepsy care. How often this occurs and in whom is not fully answered. Numerous studies have examined remission and relapse in epilepsy 1–6 and relapse after stopping medication 7, 8; however, these approaches have yet to be integrated to provide an overall assessment of the chances for complete remission.
Another difficulty is definitional. Studies use the term “terminal remission 9. In fact, this outcome cannot be truly ascertained without following patients until death. Consequently a reasonable marker is needed. We examined one such marker, five years seizure and drug-free, to determine the probability of achieving such an outcome, whether it endures, and the clinical factors associated with it. A third consideration is that the literature contains studies of mixed ages including adults in whom electro clinical syndromes are relatively unhelpful given the current state of knowledge and children in whom 40–50% can be diagnosed with specific syndromes which have well defined natural histories. Finally most of the literature is based on cohorts in whom neuroimaging was not routinely available. In this analysis, we focused on cases whose epilepsy did not meet any of the criteria for well-defined electro-clinical syndromes and who had either focal seizures or generalized convulsive seizures (“nonsyndromic epilepsy,” NSE). These are the types of epilepsies most commonly seen in adults 10.
Methods
In a prospectively identified and followed community-based cohort of 613 children with onset of epilepsy between the ages of 1 month and 16 years, we studied those followed ≥10 years after initial diagnosis of epilepsy. Information was collected through initial parent interview, repeated follow-up calls every 3–4 months, review of all baseline medical records and continued review of new records as they accumulated 11. Electroclinical syndromes were classified by pediatric epileptologists at onset and then two years later based upon accumulated information 12. For these analyses, we focused on only those epilepsies that did not fit well-defined electroclinical syndromes based on information available at initial diagnosis and for up to two years after diagnosis. Subjects with developmental encephalopathies or whose epilepsy partially fit well defined syndromes but who could not be confidently diagnosed were also excluded. We included children with nonsyndromic epilepsy with focal onset seizures or convulsions of unclear onset.
Subjects were further separated into those with and without evidence of an underlying brain disorder to which the cause of the epilepsy might be attributed. Such evidence included structural abnormalities on imaging, abnormal neurological exam, a history of a neurological insult, or intellectual disability or autism spectrum disorder (ASD) preceding the onset of epilepsy. Most of this information was ascertained at initial onset or shortly thereafter. Approximately 2/3s of the study group had MRIs during the initial clinical evaluation 13. We performed research scans on over half of this study group (for some it was the only MRI performed). When research scans were not performed, we attempted to obtain original scans for central reinterpretation14. We used the best imaging information available with priority given in descending order to research MRIs (unless post-surgical), reinterpreted MRIs, non-reinterpreted MRIs, and CT scans. This sometimes meant that we included information from MRIs performed later (e.g.. research scans). When scans were done as part of the initial evaluation, abnormalities were generally found on those scans except in a few instances when CT was used. In fact recent recommendations are that children should have an MRI at onset 15, 16, consequently the abnormalities that were found later would have been found at initially. We adopted recent recommendations regarding terminology 17 and refer to these epilepsies as either having structural/ metabolic causes or as being of unknown cause. Potential prognostic factors examined included early seizure outcomes at 2 years post diagnosis: early remission (≥12 months seizures free at 2 years),early pharmacoresistance (failed trials of 2 drugs by 2 years),neither seizure free or had failed 2 drugs (“unclear”). A drug was considered to have failed if it was appropriate for the form of epilepsy, and was given an adequate trial with evidence that the physician had made attempts to optimize the dose. This definition was used in previous reports from this study 18, 19 and its main components were recently adopted in a consensus statement from the International League Against Epilepsy 20. We also considered initial seizure frequency, any status epilepticus up through the second year after diagnosis, and family history of epilepsy in a first degree relative.
The outcome (“complete remission”) required a period of at least five years drug-free in addition to being five years seizure-free. Thus, someone could be five or even ten years seizure-free but not be considered in “complete remission” if still on medication or if medications were stopped within the past five years. Any seizure that occurred for any reason after attaining complete remission was considered a relapse.
Analyses were performed using standard techniques for bivariate comparisons. Kaplan-Meier survival curves were used to analyze time to complete remission and for relapse after complete remission. Bivariate and multivariable analyses were performed with proportional hazards models (SAS, 9.2).
Initially, parents provided informed written consent and study subjects, when able, informed assent. As subjects reached their 18th birthday and unless they were unable to do so due to intellectual disability, they were invited to participate as adults and written informed consent was obtained from them directly. Ethics committees of all involved institutions approved all procedures used in this investigation.
Results
Of the original 613, 347 (57%) met our criteria for NSE of whom 294 (85%) were followed at least 10 years. Those followed <10 years were 1.5 years older at onset of epilepsy (p=0.02) and had higher mortality (p=0.04) than those followed ≥10 years. Other wise those followed ≥10 versus <10 years were similar with respect to other clinical factors including early seizure outcomes and underlying cause (Table 1).
Table 1.
Comparison of those followed <10 versus ≥10 years
| Followed < 10 years (N=53) | Followed>=10 years (N=294) | p-value | |
|---|---|---|---|
| Age at onset | 7.0 (4.2) | 5.5 (4.1) | 0.02 |
| Underlying brain disorder | |||
| Present | 21 (39.6%) | 91 (31.0%) | 0.21 |
| Died | 3 (5.7%) | 3 (1.0%) | 0.04 |
| Early seizure control* | |||
| In remission at 2 years | 24 (53.2%) | 156 (53.1%) | 0.84 |
| Unclear | 15 (32.6%) | 102 (34.7%) | |
| Pharmacoresistant | 7 (15.2%) | 36(12.2%) | |
| IQ <80** | 15 (28.3%) | 74 (25.2%) | 0.63 |
Based on cohort members followed at least 2 years
As previously described 29
In those followed ≥10 years, 262 (89%) participants had MRIs of whom 170 had study scans, 48 re-interpreted scans, and 44 non-reinterpreted scans. Another 17 (6%) had a CT scan and 15 (5%) had no imaging. The average age at onset was 5.5 years (SD 4.0), 146(49%) were female, and the average length of follow-up was 14.4 years (max 17.9).
Of the 294,170 (58%) achieved complete remission (both seizure and drug free for ≥5 years; Figure 1a). The probability of achieving complete remission by 5, 8, 10, and 15 years after diagnosis of epilepsy was 3%, 31%, 46%, and 60%. The average duration of follow-up after attaining complete remission was 6.4 years (range 0.2 to 11.3). Ten (6%) of subjects in complete remission experienced a relapse. Relapses occurred 0.4 to 7.5 years after the date of attaining complete remission; the probability of relapse at 1, 5, and 8 years after complete remission was 1%, 5%, and 8% (Figure 1b). Another 46 individuals had a five year seizure, but not drug-free remission. Sixteen (33%) relapsed, 8 once drugs were discontinued.
Figure 1.

Cumulative probability of (a) attaining complete remission and of (b) relapse after remission
Predictors of Complete Remission
Good early seizure outcomes (Figure 2) and epilepsy of unknown cause (Figure 3) were associated with a higher likelihood of complete remission whereas status epilepticus and older age at onset were associated with a poorer chance. Other factors (including family history of epilepsy, history of febrile seizures, and initial seizure frequency) were not associated with complete remission (Table 2). Early outcome and underlying cause together distinguished groups with a chance of complete remission ranging from about 19% to 77% (Table 3, Figure 4).
Figure 2.
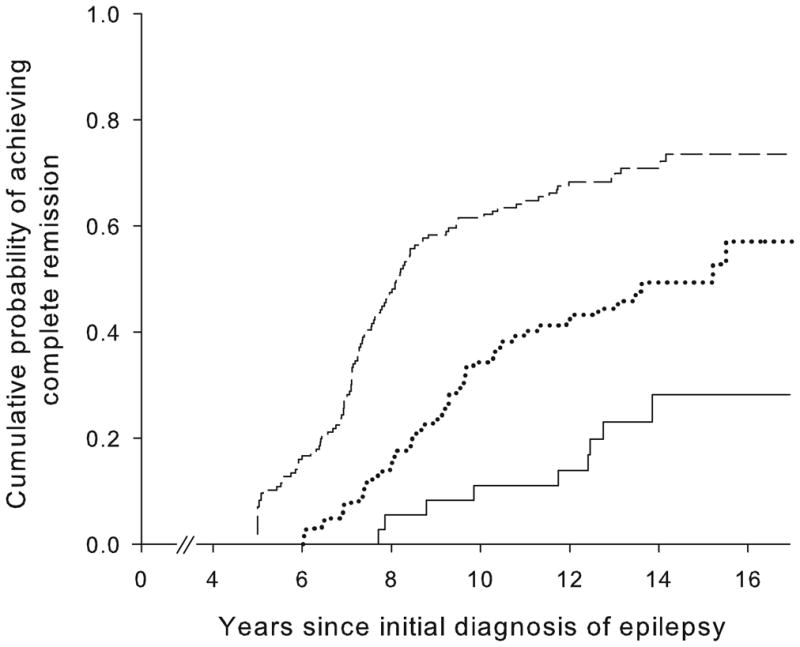
Complete remission as a function of early seizure outcome: remission at two years (---), unclear, (....) and pharmacoresistant (—)
Figure 3.
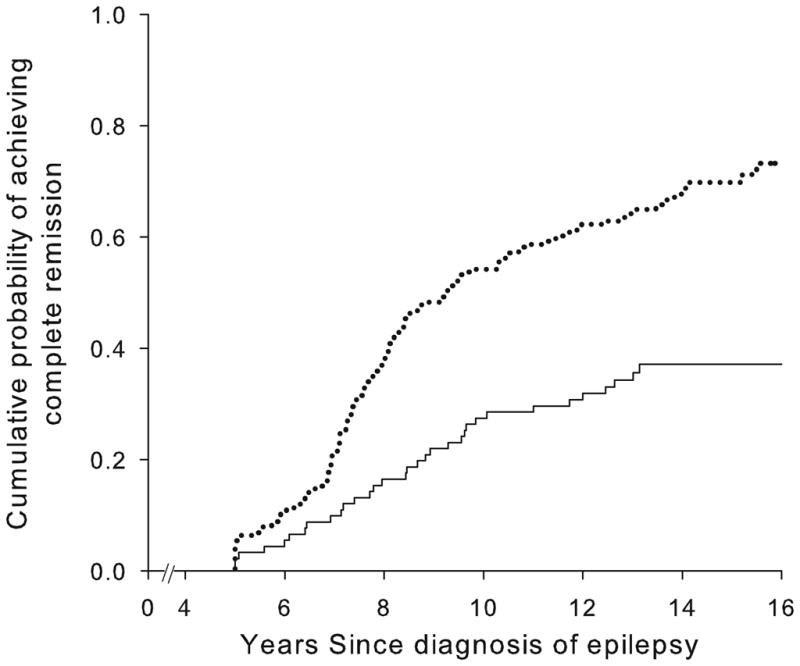
Complete remission as a function of the presence or absence of an underlying brain disorder: no identified underlying disorder (....) and underlying disorder identified (—).
Table 2.
Bivariate associations for complete remission and clinical factors of potential prognostic significance.
| Prognostic Factor | N | N (%) Complete remission | Hazards Ratio (95% CI)* | p-value |
|---|---|---|---|---|
| Early seizure outcome | ||||
| Remission | 156 | 111 (71.5%) | 1.00** + | |
| Unclear | 102 | 50 (49%) | 0.46 (0.33, 0.65) | <0.0001 |
| Pharmacoresistant | 36 | 9 (25%) | 0.20 (0.10, 0.39) | <0.0001 |
| Evidence of underlying brain disorder | ||||
| Absent | 203 | 137 (67.5%) | 1.00** | |
| Present | 91 | 33 (36.3%) | 0.39 (0.27, 0.58) | <0.0001 |
| Abnormal MRI | 55 | 18 (32.7%) | 0.35 (0.21, 0.57) | <0.0001 |
| Abnormal exam | 43 | 16 (37.2%) | 0.42 (0.25, 0.71) | 0.001 |
| Intellectual Disability only | 11 | 3 (37.3%) | 0.31 (0.10, 0.97) | 0.04 |
| Autism Spectrum Disorder only | 4 | 1 (25%) | 0.25 (0.03, 1.76) | 0.16 |
| Age at onset | ||||
| <2 years | 66 | 43 (65.2%) | 1.00** + | |
| 2− | 88 | 52(59.1%) | 0.72 (0.48, 1.11) | 0.11 |
| 5− | 85 | 51 (60.0%) | 0.78 (0.52, 1.18) | 0.24 |
| 10+ years | 55 | 24 (43.6%) | 0.50 (0.30, 0.82) | 0.006 |
| Status epilepticus | ||||
| None within 2 years | 254 | 154 (60.6%) | 1.00** | |
| 1+ Episodes | 40 | 16 (40.0%) | 0.48 (0.29, 0.81) | 0.006 |
| Convulsive seizures | ||||
| None | 112 | 61 (54.5%) | 1.00** | |
| Present | 182 | 109 (59.9%) | 1.16 (0.85, 1.58) | 0.36 |
| History of febrile seizures** | ||||
| None | 250 | 143 (57.2%) | 1.00** | |
| Present | 42 | 26 (61.9%) | 1.01 (0.66, 1.53) | 0.97 |
| Family history of epilepsy–first degree relative*** | ||||
| None | 267 | 156 (58.4%) | 1.00** | |
| 1+ relatives | 23 | 12 (52.2%) | 0.77 (0.43, 1.39) | 0.39 |
| Initial seizure frequency | ||||
| <1/month | 70 | 39 (55.7%) | 1.00**++ | |
| 1–9/month | 168 | 100 (59.2%) | 1.00 (0.99, 1.01) | 0.84 |
| 10–49/month | 33 | 19 (57.6%) | ||
| ≥50/month | 23 | 12 (52.1%) |
(95% CI):95% confidence interval
Referent group. For underlying brain disorders, each group defined by the presence of specific underlying disorder is tested relative to the referent group only. The underlying disorders, however, are not mutually exclusive.
Treated as an ordinal variable, the effect estimate for early outcome is 0.45 (95% CI 0.35, 0.58, p<0.0001) for a change from remission to unclear and unclear to pharmacoresistant. For age at onset grouping, the estimates effect when treated as an ordinal variable was 0.83 (95% CI, 0.72, 0.97, p=0.02).
Missing on 2 individuals who were adopted for febrile seizures and four individuals for family history.
Tested as an ordinal variable only. The hazards ratio estimate reflects the change in complete remission rates with each incremental change in seizure frequency category.
Table 3.
Combined influences of early seizure outcome and underlying brain disorder on the chance of attaining complete remission:
| Early Seizure outcome | Underlying structural/metabolic disorder (N=91) | Unknown Cause | p-value** | ||||
|---|---|---|---|---|---|---|---|
| N | CR+ | % (95% CI) | N | CR+ | % (95% CI) | ||
| Remission | 35 | 18 | 51% (35, 68) | 121 | 93 | 77% (69, 84) | 0.004 |
| Unclear | 35 | 11 | 31% (16, 47) | 67 | 39 | 58% (46, 70) | 0.01 |
| Pharmacoresistant | 21 | 4 | 19% (2, 36) | 15 | 5 | 33% (9, 57) | 0.32 |
| p-value* | 0.01 | 0.0001 | |||||
CR=Complete Remission
p-value is the association of early outcome with complete remission within each group defined by the presence of a brain disorder, tested as a trend.
p-value is for the association between presence of an underlying brain disorder and complete remission within each early outcome group.
Figure 4.

Cumulative probability of attaining complete remission in those (a) without and (b) with an underlying brain disorder as a function of early seizure outcomes: remissions at two years (---), unclear, (....) and pharmacoresistant (—)
In a multivariable proportional hazards model, epilepsy of unknown cause (rate ratio (RR)= 2.1; 95% CI= 1.4, 3.1, p=0.0001) and better early seizure outcome ( RR=2.1 for each increment from early pharmacoresistance to unclear, and unclear to early remission, 95% CI 1.6, 2.7; p<0.0001) were associated with a greater chance of achieving complete remission. Older age at onset was associated with a poorer chance of complete remission; however, almost all of the effect appeared to be due to the oldest age group. Those who were 10 years old at the time of their first seizure were less likely to achieve complete remission (RR=0.57, 95% CI 0.370.88, p=0.01). After adjustment for these three factors, status epilepticus was no longer statistically significant (RR=0.65, 95% CI 0.391.11, p=0.12). None of the other factors considered in table 2 was statistically significant after adjustment for these first three factors.
Of the ten individuals who relapsed, one had difficulty regaining seizure control, and eight were seizure-free for periods of greater than 1 year, one of whom became 5 years seizure-free but not medication-free. The last was not consistently taking medication. Early seizure outcome was not significantly associated with relapse after complete remission;, 5%, 8% and 0% relapsed in those who were seizure-free, with unclear outcome and pharmacoresistant at two years respectively (p=0.6). Six (4%) in the group with unknown cause relapsed versus 4(12%) in the group with structural brain lesions or related disorders (p=0.06, based upon product-limit estimate).
Epilepsy surgery was performed on 17 individuals, 14 five of whom achieved complete remission including 4 individuals with tumors and one with a hypothalamic hamartoma. Two had been pharmacoresistant prior to surgery (including one with a tumor).
Discussion
The prognosis for cure of seizures is of particular interest to people with epilepsy. There is no consensus definition of complete remission or cure of epilepsy short of one that would require following a patient to the end of life and demonstrating no further seizure activity. Our definition, 5 years seizure-free is frequently used in long-term studies of epilepsy 1, 2, 5, 9, 21. The added requirement that patients also be five years drug-free is not explicitly used in the other studies; however, several refer to five years seizure-free off drugs 1, 9, 21. We found complete remission to be a meaningful but not absolute marker for continued remission, at least over the additional time patients have so far been followed. A small but definite risk of relapse remained for several years with the last relapse observed thus far occurring almost eight years after achieving complete remission. Most patients who relapsed attained some degree of seizure control. Our finding that one went on to have difficulty with seizure control is consistent with that reported in a Canadian cohort 8. Earlier reports, some from this study, demonstrated that brief remission (≥1 year) are relatively nonrobust indicators of long-term seizure outcome as many epilepsies follow a relapsing-remitting course and pharmacoresistance can develop after a significant but still relatively brief period of remission 19, 22, 23. Complete remission as we have defined it is a meaningful target for long-term seizure outcome.
Our definition of complete remission is composed of two factors, the actual cessation of the seizure propensity and the decision to stop medications. The first is a function of the underlying biology. The second is a personal and clinical decision on the part of the patient or parent and the physician and is likely influenced by perceived risk (e.g. underlying cause, early seizure outcome, etc).
The same consideration arises in determining early treatment outcomes and control of seizures. Identification of early pharmacoresistance depends on the physician prescribing an appropriate medication, following a reasonable titration scheduled and then deliberately increasing the dose in the face of continued seizures until it is determined that a drug has not succeeded, and then moving on to another drug and doing the same. There is likely variation across practitioners in how aggressively drug failure is determined. Patients and care givers play a role as well in that good compliance is necessary. As we have defined these outcomes, early seizure control at 2 years can be temporally independent of complete remission.
There are several report son prediction of seizure outcome, variously defined and ranging from remission to pharmacoresistance. We have previously made the point that one is not the full complement of the other and that there is a range of outcomes between the two 24. Regardless, for the purposes of summarizing the literature, it may be reasonable to expect that factors associated with an increased likelihood of remission should generally be associated with a decreased likelihood of pharmacoresistance and vice versa.
Comparisons among studies are further complicated by differences in the study groups. The cohorts published in the literature tend to use one of two main approaches. Either they include patients of all ages (children and adults combined) or they focus only on the pediatric age range. In the pediatric cohorts and in addition to electro-clinical syndromes, the evidence of a brain abnormality (including abnormal motor exam) is usually associated with a poorer seizure outcome6, 21, although in one analysis from the Dutch study this was not the case after adjustment for of a set of electroclinical syndromes (previously termed “idiopathic”) with a known good seizure prognosis 9. More selective series of temporal lobe epilepsy also report that an MRI abnormality is predictive of poor seizure outcome 25, 26. In the first ever epidemiological study of remission, the presence of a secondary cause (e.g. acquired lesion) was associated with a poorer chance of attaining remission off medication but was not clearly associated with a lower chance of remission on medication 1. By contrast, patients with neurological deficits presumed to be present from birth (e.g. cerebral palsy) and who would mostly have had childhood onset epilepsy, had a much lower chance of remission regardless of treatment. A similar distinction was made in the British study in which cases were characterized as “idiopathic” (no identified underlying cause), remote symptomatic (acquired lesions), and congenital deficits (e.g. cerebral palsy) 5. Similar to the previous study, the group with congenital deficits but not with secondary acquired lesions had a poorer chance of remission relative to their “idiopathic” group. No distinctions were made regarding whether patients continued on medication. In two other studies of mixed ages underlying structural/metabolic conditions or indicators of a brain disorder (abnormal exam) were not associated with remission 4 or pharmacoresistance 3.
Our approach strips away the group of epilepsies with well-defined prognoses and whose inclusion, especially in mixed age cohorts, creates difficulties for interpretation of findings. In children with nonsyndromic epilepsies similar to those seen in adults, underlying structural/metabolic causes are an important determinant of seizure outcome. This is likely what the findings for “congenital” neurological abnormalities reflects in the US 1 and British cohorts 5.
The results of these various studies also suggest that the prognosis for nonsyndromic epilepsy secondary to underlying structural-metabolic conditions differs in children versus adults. Certainly the causes themselves vary with more acquired insults (stroke, trauma, tumor) seen in adults and more congenital insults (intrauterine insults, malformations of cortical development, genetic encephalopathies) seen in children. The lack of routine neuroimaging in most of these studies also raises questions about how thoroughly structural and other causes were identified.
Age at onset is an important feature of many of the electroclinical syndromes. The studies of mixed ages as well as some of the pediatric studies tend to find that younger age at onset (variously defined as <12 or <16, or <20) is associated with a better outcome than older age at onset 1, 4–6. In the mixed-ages studies, some of this may be due to the large proportion of pediatric patients with specific self-limited electroclinical syndromes such as childhood absence or benign rolandic epilepsy. In a combined analysis of two pediatric cohorts, older age at onset (>12 years) was associated with poorer outcome after adjustment for some but not all electroclinical syndromes and other factors 6. Our finding of poorer prognosis associated with onset in the second decade should be viewed cautiously as it is somewhat novel and not truly demonstrated in other cohorts free of the influences of specific age-related electroclinical syndromes. If it holds up, this could provide some initial clues for defining meaningful clinical phenotypes to guide not only counseling of patients but also targeted investigation into mechanisms underlying these poorly differentiated epilepsies.
In addition to underlying cause, and possibly age, early seizure control is also an important indicator of later outcome. This has now been seen in several studies 2, 3, 9, 21. For nonsyndromic epilepsy in our cohort, cause and early seizure control alone provided meaningful prognostic information, separating patients into those with only a ~20% to those with a~75% chance of complete remission.
Strictly speaking, this is not a population-based cohort. We have previously compared our community-based cohort to another population-based cohort from Nova Scotia, Canada with respect to a number of benchmark characteristics and demonstrated that our study is highly comparable to one that is generally accepted as population-based consequently we can expect our nonsyndromic group to be representative of nonsyndromic epilepsies in the target population as well 14. As those lost before 10 years are similar to those followed ≥10 years in terms of their early seizure outcomes and proportion with underlying conditions, both key predictors of complete remission, it is unlikely that substantial bias has been introduced by the exclusion of 15% of the study group who were lost early. We believe that our results provide a valid assessment of complete remission for nonsyndromic childhood-onset epilepsy diagnosed in the community.
The length of follow-up is over a decade although other, older studies, have much longer follow-up periods 27, 28 due to when the studies began. An advantage of our study is the close prospective follow-up with frequent (3–4 times per year) family contact combined with access to medical records. In addition, 95% of our study group had neuroimaging, 89% with MRI, which permitted detection of structural brain abnormalities that might otherwise have been found only if patients were extensively investigated for pharmacoresistance.
In conclusion, our results provide clinically relevant prognostic information about a large group of childhood-onset epilepsies for which such information was not previously available. They also underscore the importance of identifying underlying structural or metabolic causes early, including the use of MRI in most forms of epilepsy and the importance of rapidly determining drug responsiveness 20.
Acknowledgments
Funding: This work was supported by a grant from the National Institutes of Health, National Institute of Neurologic Disorders and Stroke, R37-NS-31146.
We are very grateful to all the physicians in Connecticut who have made it possible for us to recruit and follow their patients all these years. We also would like to thank Eugene Shapiro who provided essential administrative help throughout and Shlomo Shinnar and Francis DiMario who participated in other phases of this study. Drs. Richard Bronnen and Robert Fulbright interpreted MRI scans for this study. This study was made possible by the generous help of the many families who have participated over the course of the last many years.
References
- 1.Annegers JF, Hauser WA, Elveback LR. Remission of seizures and relapse in patients with epilepsy. Epilepsia. 1979;20:729–737. doi: 10.1111/j.1528-1157.1979.tb04857.x. [DOI] [PubMed] [Google Scholar]
- 2.Lindsten H, Stenlund H, Forsgren L. Remission of seizures in a population-based adult cohort with a newly diagnosed unprovoked epileptic seizure. Epilepsia. 2001;42:1025–1030. doi: 10.1046/j.1528-1157.2001.0420081025.x. [DOI] [PubMed] [Google Scholar]
- 3.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;3:314–319. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- 4.Collaborative Group for the Study of Epilepsy. Prognosis of epilepsy in newly referred patients: A multi center prospective study of the effects of monotherapy on the long-term course of epilepsy. Epilepsia. 1992;33:45–51. doi: 10.1111/j.1528-1157.1992.tb02281.x. [DOI] [PubMed] [Google Scholar]
- 5.Cockerell OC, Johnson AL, Sander JWAS, et al. Remission of epilepsy: results from the national general practice study of epilepsy. The Lancet. 1995;346:140–144. doi: 10.1016/s0140-6736(95)91208-8. [DOI] [PubMed] [Google Scholar]
- 6.Geelhoed M, Boerrigter AO, Camfield P, et al. The accuracy of outcome prediction models for childhood-onset epilepsy. Epilepsia. 2005;46:1526–1532. doi: 10.1111/j.1528-1167.2005.07405.x. [DOI] [PubMed] [Google Scholar]
- 7.Berg AT, Shinnar S. Relapse following discontinuation of antiepileptic drugs: a meta-analysis. Neurology. 1994;44:601–608. doi: 10.1212/wnl.44.4.601. [DOI] [PubMed] [Google Scholar]
- 8.Camfield P, Camfield C. The frequency of intractable seizures after stopping AEDs in seizure-free children with epilepsy. Neurology. 2005;64:973–975. doi: 10.1212/01.WNL.0000154517.82748.A7. [DOI] [PubMed] [Google Scholar]
- 9.Geerts AT, Arts WF, Stroink H, et al. Course and outcome oc childhood epilepsy: A 15 year follow-up of the Dutch study of epilepsy in childhood. Epilepsia. 2010;51:1189–1197. doi: 10.1111/j.1528-1167.2010.02546.x. [DOI] [PubMed] [Google Scholar]
- 10.Jallon P, Loiseau P, Loiseau J. Newly diagnosed unprovoked epileptic seizures: presentation at diagnosis in CAROLE study. Epilepsia. 2001;42:464–475. doi: 10.1046/j.1528-1157.2001.31400.x. [DOI] [PubMed] [Google Scholar]
- 11.Berg AT, Shinnar S, Levy SR, Testa FM. Newly diagnosed epilepsy in children: presentation at diagnosis. Epilepsia. 1999;40:445–452. doi: 10.1111/j.1528-1157.1999.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 12.Berg AT, Shinnar S, Levy SR, et al. How well can epilepsy syndromes be identified at diagnosis? A reassessment two years after initial diagnosis. Epilepsia. 2000;41:1269–1275. doi: 10.1111/j.1528-1157.2000.tb04604.x. [DOI] [PubMed] [Google Scholar]
- 13.Berg AT, Testa FM, Levy SR, Shinnar S. Neuroimaging in children with newly diagnosed epilepsy: A community-based study. Pediatrics. 2000;106:527–532. doi: 10.1542/peds.106.3.527. [DOI] [PubMed] [Google Scholar]
- 14.Berg AT, Mathern GW, Bronen RA, et al. Frequency, prognosis, and surgical treatment of MRI structural abnormalities in childhood epilepsy. Brain. 2009;132:2785–2797. doi: 10.1093/brain/awp187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirtz D, Ashwal S, Berg A, et al. Practice parameter: Evaluating a first nonfebrile seizure in children. Report of the quality standards subcommittee of the American academy of neurology, the child neurology society, and the American epilepsy society. Neurology. 2000;55:616–623. doi: 10.1212/wnl.55.5.616. [DOI] [PubMed] [Google Scholar]
- 16.Gaillard WD, Chiron C, Cross JH, et al. Guidelines for imaging infants and children ith recent-onset epilepsy. Epilepsia. 2009;50:2147–2153. doi: 10.1111/j.1528-1167.2009.02075.x. [DOI] [PubMed] [Google Scholar]
- 17.Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 18.Berg AT, Levy SR, Testa FM, D’Souza R. Remission of epilepsy after 2 drug failures in children: a prospective study. Ann Neurol. 2009;65:510–519. doi: 10.1002/ana.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg AT, Vickrey BG, Testa FM, et al. How long does it take epilepsy to become intractable? A prospective investigation. Ann Neurol. 2006;60:73–79. doi: 10.1002/ana.20852. [DOI] [PubMed] [Google Scholar]
- 20.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–1077. doi: 10.1111/j.1528-1167.2009.02397.x. [DOI] [PubMed] [Google Scholar]
- 21.Sillanpaa M, Jalava M, Kaleva O, Shinnar S. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715–1722. doi: 10.1056/NEJM199806113382402. [DOI] [PubMed] [Google Scholar]
- 22.Berg AT, Langfitt J, Shinnar S, et al. How long does it take for partial epilepsy to become intractable? Neurology. 2003;60:186–190. doi: 10.1212/01.wnl.0000031792.89992.ec. [DOI] [PubMed] [Google Scholar]
- 23.Berg AT, Lin J, Ebrahimi N, et al. Modeling remission and relapse in pediatric epilepsy: application of a Markov process. Epilepsy Research. 2004;60:31–40. doi: 10.1016/j.eplepsyres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Berg AT. Defining intractable epilepsy. In: Blume WT, Carlen PL, Starrveld E, et al., editors. Advances in Neurology: Intractable Epilepsies. Vol. 97. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 5–10. [PubMed] [Google Scholar]
- 25.Dlugos D, Sammel M, Strom B, Farrar J. Response to first drug trial predicts outcome in childhood temporal lobe epilepsy. Neurology. 2001;57:2259–2264. doi: 10.1212/wnl.57.12.2259. [DOI] [PubMed] [Google Scholar]
- 26.Spooner CG, Berkovic SF, Mitchell LA, et al. New onset temporal lobe epilepsy in children: lesion on MRI predicts poor seizure outcome. Neurology. 2006;67:2147–2153. doi: 10.1212/01.wnl.0000248189.93630.4f. [DOI] [PubMed] [Google Scholar]
- 27.Sillanpaa M, Schmidt D. Natural history of treated childhood-onset epilepsy: prospective, long-term population-based study. Brain. 2006;129:617–624. doi: 10.1093/brain/awh726. [DOI] [PubMed] [Google Scholar]
- 28.Camfield C, Camfield P, Gordon K, et al. Outcome of childhood epilepsy: a population-based study with a simple predictive scoring system for those treated with medication. J Pediatr. 1993;122:861–868. doi: 10.1016/s0022-3476(09)90008-7. [DOI] [PubMed] [Google Scholar]
- 29.Berg AT, Langfitt JT, Testa FM, et al. Global Cognitive Function in Children with Epilepsy: A community-based study. Epilepsia. 2008;49:608–614. doi: 10.1111/j.1528-1167.2007.01461.x. [DOI] [PubMed] [Google Scholar]