

Abstract
Changes within the glucocorticoid receptor (GR) cellular signaling pathway were evaluated in adolescent mice exposed to 50 ppb arsenic during gestation. Previously, we reported increased basal plasma corticosterone levels, decreased hippocampal GR levels and deficits in learning and memory performance in perinatal arsenic-exposed mice. The biosynthesis of members of the mitogen-activated protein kinase (MAPK) signaling pathway, known to be involved in learning and memory, is modulated by the binding of GR to glucocorticoid response elements (GREs) in the gene promoters. Two genes of the MAPK pathway, Ras and Raf, contain GREs which are activated upon binding of GRs. We evaluated the activity of GRs at Ras and Raf promoters using chromatin immunoprecipitation and real-time PCR and report decreased binding of the GR at these promoters. An ELISA-based GR binding assay was used to explore whether this decreased binding was restricted to in vivo promoters and revealed no differences in binding of native GR to synthetic GREs. The decreased in vivo GR binding coincides with significantly decreased mRNA levels and slight reductions of protein of both H-Ras and Raf-1 in perinatally arsenic-exposed mice. Nuclear activated extracellular-signal regulated kinase (ERK), a downstream target of Ras and Raf, whose transcriptional targets also play an important role in learning and memory, was decreased in the hippocampus of arsenic-exposed animals when compared to controls. GR-mediated transcriptional deficits in the MAPK/ERK pathway could be an underlying cause of previously reported learning deficits and provide the link to arsenic-induced deficiencies in cognitive development.
Keywords: Arsenic(As), Glucocorticoid Receptor (GR), Glucocorticoid Response Element (GRE), Extracellular-signal regulated kinase (ERK), Hippocampal formation, Mitogen-activated protein kinase (MAPK) Signaling Pathway
1. INTRODUCTION
The persistence of arsenic in the environment calls for continued research on arsenic and its health effects. Drinking water contamination is of particular concern as arsenic has been shown to be permissive to a variety of cancers, vascular diseases and cellular disruption (Agency for Toxic Substances and Disease Registry Arsenic, 2007). In 2001, the EPA lowered the acceptable arsenic limit from 50 ppb to 10 ppb (United States Environmental Protection Agency, 2001). However, millions of people in the US are continuously exposed to levels exceeding 10 ppb. Common sources of contaminated drinking water still above the EPA limit are untreated wells in areas with naturally occurring high levels, areas near mining and smelting sites, and areas affected by agricultural runoff (United States Environmental Protection Agency, 2010). Several studies have identified arsenic as a potent neurotoxin posing a threat to cognitive development in both rodents (Martinez et al., 2008; Martinez-Finley et al., 2009; Rodriguez et al., 2001, 2002, 2003) and humans (Calderón et al., 2001; Von Ehrenstein et al., 2007; Wasserman et al., 2004).
Activation of glucocorticoid receptors (GRs), through the binding of glucocorticoids, mediates a variety of cellular and physiological events. In mammals, the hypothalamic pituitary adrenal (HPA) axis is modulated by the hippocampus which helps to integrate neuroendocrine, cognitive, emotional and autonomic inputs (Joels et al., 2004; McEwen, 2000). Some of the strongest GR expression is found within the CA1 and CA2 regions of the hippocampus (Sousa et al., 1998). Developmental modifications in hippocampal GR function, can lead to dysregulation of the HPA feedback system, which, in turn, can impact the functioning of the hippocampus (McEwen, 2002). Moderate doses of arsenic have been shown to perturb glucocorticoid receptor (GR) mediated gene transcription in cell culture models (Bodwell et al., 2006; Kaltreider et al., 2001). Both increases and decreases in transcription of genes under the control of GRs have been described as one consequence of altered GR levels (Bodwell et al., 2006). At concentrations between 1–3 M, the effects of arsenic on GR-mediated gene activation are inhibitory (Bodwell et al., 2006). Hamilton and colleagues showed that a single, low, non-overtly toxic dose of arsenite significantly altered basal and inducible mRNA expression of the GRE-controlled phosphoenolpyruvate carboxykinase gene in cell culture and in a whole-animal model (1998). In addition, arsenite had no effect on expression of GRE-non-inducible or constitutively expressed genes (Hamilton et al., 1998). In support of these observations, Kaltreider and colleagues have shown that nontoxic doses of arsenite can selectively inhibit GR-mediated transcription of the phosphoenolpyruvate carboxykinase gene in rat hepatoma (H4IIE) cells (2001) without altering GR nuclear translocation.
Events that disrupt maturation of the HPA axis, particularly during gestation, have been shown to permanently alter GR expression in the adult (Challis et al., 2000; O’Regan et al., 2001). Recently, we have shown that GR and mineralocorticoid receptor (MR) protein levels in the hippocampus of brains of offspring perinatally exposed to arsenic are significantly decreased compared to age-matched controls at postnatal day (PND) 35 (Martinez-Finley et al., 2009). Using our perinatal exposure model, we have also reported significant deficits in spatial learning and object recognition tasks in the arsenic exposed adolescent offspring (Martinez-Finely et al., 2009). In addition to our own findings of arsenic-induced learning and memory deficits, a recent study using a combined pre- and postnatal arsenic exposure found deficits in fear conditioned learning (Martínez et al., 2011).
Given that the GR controls the transcription of a number of genes, the present work investigated the effects of decreased levels of the GR in the hippocampus from perinatal arsenic-exposed mice on two genes shown to be transcriptionally activated by the GR and critically involved in learning and memory. Ras and Raf-1, two important members of the mitogen-activated protein kinase (MAPK) signaling pathway, are transcriptional targets of the GR (Revest et al., 2005). H-Ras and Raf-1 are core elements of the MAPK signaling pathway and their activation ultimately leads to phosphorylation, activation and nuclear localization of extracellular signal-regulated kinase (ERK1/2). The MAPK/ERK pathway plays a crucial role in learning and memory (Sweatt, 2001, 2004). At the biochemical level, the MAPK pathway is linked to synaptic plasticity, which contributes to memory formation and is important in information processing (reviewed in Thomas and Huganir, 2004). Induction of long-term potentiation (LTP), the best studied form of cellular learning and memory, relies on the regulation of protein phosphorylation (reviewed in Thomas and Huganir, 2004) and many forms of LTP have been shown to be ERK-dependent (Atkins et al., 1998; English and Sweatt, 1996; Ohno et al., 2001).
The present study focused on the connection between perinatal arsenic exposure, decreased hippocampal GR, and genes of the MAPK signaling pathway in brains of exposed offspring as alterations in these cascades could underlie the learning and memory deficits observed in these animals.
2. MATERIALS AND METHODS
Note: Arsenic is classified as a probable human carcinogen and all arsenicals were handled with caution (ATSDR, 2007).
2.1. Perinatal Arsenic Exposure Paradigm
The arsenic exposure paradigm employed in these studies was approved by the UNM Health Sciences Center Institutional Animal Care and Use Committee. All mice (C57BL/6) were bred and maintained on a reversed 12-h light/dark cycle (lights off from 0800 to 2000 h) with food and water ad libitum in a temperature controlled (22°C) room in the Animal Resource Facility. All dosing suspensions were prepared fresh weekly. Arsenic water at 50 ppb concentration (sodium arsenate; Sigma, St. Louis, MO; #A-6756) was prepared using standard tap water. Control mice were administered untreated tap water from the University of New Mexico which contains approximately 5 ppb arsenic. Mice were bred according to our established protocol (Martinez et al., 2008; Martinez-Finley et al., 2009). Dams were started on the arsenic treated water two weeks prior to breeding and continued on the treatment throughout gestation (fig. 1). Tap water replaced the 50 ppb arsenic water at weaning. Offspring were weaned at 23 days of age and maintained in litter-mate housed cages with ad libitum access to untreated tap water and standard mouse chow until they were used in experimental procedures at 35–40 days of age. This level and duration of exposure will produce inorganic arsenic concentrations in whole brain tissue 2.24 ± 0.02 ppb in the arsenic exposed offspring compared to 1.0 ± 0.24 ppb in controls at 35 days of age (Martinez-Finley et al., 2009). Male pups were used in all analyses except for the GR-GRE binding assay where the protein requirements necessitated the use of a few female tissue samples. Sample sizes represents the total number of litters with one pup per litter to preclude litter effect.
Figure 1.

Perinatal Arsenic Exposure Paradigm. Black bars represent standard tap water [5 ppb]. Grey bars represent arsenic dissolved in standard tap water [55ppb]. Female mice were started on the arsenic or control water 2 weeks prior to pregnancy. Arsenic exposed pups were weaned at PND23 and transferred to standard tap water until experimentation at PND35. PND=postnatal day
2.2. Nuclear Extracts
Adolescent offspring, 35–40 days of age, were sacrificed by decapitation and the hippocampal formation was rapidly dissected on ice. Preparation of nuclear extracts and protein determinations were performed as described in Buckley and Caldwell (2004) and Weeber et al. (2001), respectively.
2.3. mRNA isolation
mRNA was isolated using the Oligotex Direct mRNA Mini Kit (Qiagen, Valencia, CA; #72012) per manufacturer’s protocol. The mRNA concentration was determined (OD 260 nm) using a NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). Purified mRNA was stored at −80°C.
2.4. cDNA synthesis
First-Strand cDNA Synthesis reactions were performed using M-MLV Reverse Transcriptase (Invitrogen; Carlsbad, CA; #28025013) following the manufacturer’s protocol. Our reaction mixtures contained 20 ng mRNA, 500 ng oligo dT, and 1 mM of each dNTP. Synthesized cDNA was stored at −20°C. The suitability of the cDNA for PCR was determined using 2 μl of cDNA added to a 40 μl PCR reaction. A control reaction containing 2 μl of purified mRNA was run without reverse transcription to test for the presence of DNA contamination in the purified mRNA samples. The PCR products were run out on a 1.5% (w/v) agarose gel and visualized using Fluor-S MultiImager (Bio-Rad, Hercules, CA) and Quantity-One Analysis Software (Bio-Rad).
2.5. Primers
Oligonucleotide sequences for H-Ras and Raf-1 were based on sequences reported by Revest et al. (2005) and β-actin as reported by Caldwell et al. (2008). The specificity of each primer pair was confirmed by the identification of a single PCR product of predicted size on 1.5% (w/v) agarose gels.
2.6. Semi-quantitative determination of transcript levels by Real-time PCR
Real-time PCR was conducted in a Gene Amp 7300 sequence detection system (Applied Biosystems, Foster City, CA) using a MicroAmp® Fast 96-well plate (Applied Biosystems). The relative quantification of the H-Ras and Raf-1 genes in different tissue samples was determined using the 2−ΔΔCt method described by Livak and Schmittgen (2001). Initially, the ΔCt value, the difference between the average of the triplicate Ct values for the H-Ras and Raf-1 and the internal control β-actin, for each sample was calculated (ΔCtsample = ΔCtRas – ΔCtB-actin). Next, the average of the ΔCt values for the control animals (ΔCtCon av) was determined and subtracted from each sample (ΔCtsample – ΔCt Con Av), giving a ΔCt value for each. This was used to calculate the 2−ΔΔCt value for each. Because 2° is one, the mean 2−ΔΔCt value for each control sample was approximately one depending on the variability in the samples, thus the mean 2−ΔΔCt value for the arsenic samples was expressed relative to one.
2.7. ERK, phospho-ERK, H-Ras, and Raf-1 protein
Hippocampal nuclear fractions from arsenic-exposed and control animals were analyzed for phosphorylated ERK (nuclear fraction) and the ERK regulated gene products, H-Ras and Raf-1 via western blotting according to Buckley and Caldwell (2004). While ERK is also present in the cytosol, only nuclear localized ERK is involved in the regulation of gene expression. Extracts were thawed on ice, diluted in 4X NuPAGE® LDS Sample Buffer (Invitrogen; #NP0007), and heated at 70°C for 10 minutes. Samples were separated using NuPAGE® Novex 4–12% Bis-Tris Gels (Invitrogen; #NP0322BOX) and transferred to 0.45 μm nitrocellulose membranes (Invitrogen; #LC2001). The membranes were blocked with 0.25% I-Block™ Protein-Based Blocking Reagent (Applied Biosystems; #T2015) in TBS-T (25 mM Tris-HCl pH 7.2, 150 mM NaCl and 0.05% Tween-20). Blots were then incubated with either polyclonal primary antibody to Rabbit Anti-phospho-ERK1/2 (1:3,000; Cell Signaling Technology, Danvers, MA; #9101) for ERK, Mouse Anti-H-Ras (1:500; Santa Cruz Biotechnology, Santa Cruz, CA; F-235) for H-Ras, or Rabbit Anti-Raf-1 (1:500; Santa Cruz Biotechnology; C-12) for Raf-1. Either Goat Anti-rabbit IgG (H+L): HRP (1:50,000; Pierce, Rockford, IL; #31460) for ERK and Raf-1 or Goat Anti-mouse IgG (H+L): HRP (1:50,000, Pierce; #31430) for H-Ras was used for the secondary antibody in 0.25% I-Block™. Membranes were then incubated in either SuperSignal West Pico Chemiluminescent Solution (Pierce; #34080) or Western Lightning Plus ECL (PerkinElmer, Waltham, MA; NEL104001EA) and exposed to F-BX57 film (Phenix Research Products, Candler, NC). Film was developed in Kodak D-19 developer then washed and fixed in Kodak fixer. The blots were stripped with Restore Western Blot Stripping Buffer (Thermo Scientific, Rockford, IL; #21059), the activated ERK blots probed for total ERK (1:50,000; Cell Signaling Technology; #9102) and then stripped again, and all blots were probed for β-actin, a constitutively expressed protein, which serves as a loading control. β-Actin was probed for with Mouse Anti-β-Actin (1:2000; Sigma; A5441-.2ML) followed by the Goat Anti-mouse IgG (H+L): HRP (1:40,000). Blotting for ERK 1 and 2 follow the protocol described by Buckley and Caldwell (2004). For activated ERK, the linear range of the phosphorylated and total protein was calculated to ensure that the protein was not saturating the blot. Samples from control and perinatal arsenic-exposed animals were loaded on the same gel. The developed film was scanned (Hewlett Packard Scan Jet 5P) and immunoreactivities quantified by measurements of optical densities using Quantity-One Analysis Software. The amount of p-ERK was compared to the total ERK of the sample. The amounts of ERK, p-ERK, H-Ras, and Raf-1 were all compared to β-Actin.
2.8. Chromatin Immunoprecipitation (ChIP) Real-time PCR Assays
We followed a modified Upstate Biotechnology ChIP protocol, as described here. Hippocampal tissue was collected and homogenized on ice in 1% cross-linking solution (10% formaldehyde, 0.1M NaCl, 1 mM EDTA, 0.5 mM EGTA, 50 mM Hepes, and ddH 0, pH 8.0) for 15 minutes. Cells were centrifuged at 1,000 × g at 4ºC for 6 minutes and washed five times with 1.5 ml PBS containing 1 μl/ml protease inhibitor cocktail (Sigma; #P8340). Cell pellets were resuspended in cell lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% 2-[2-(4-nonylphenoxy)ethoxy]ethanol(IGEPAL-CA-630)) and allowed to sit on ice for 5 minutes. The cells were then pelleted (1,000 × g at 4°C for 6 minutes), resuspended in lysis buffer, and repelleted. Cells were then resuspended in nuclei lysis buffer (50 mM Tris pH 8.1, 10 mM EDTA, 1% SDS). At this point, samples were frozen and stored at −80°C overnight. Samples were thawed and cross-linked DNA was sheared via sonication (Kontes Micro Ultrasonic Cell Disrupter; setting = 25%, four times 10 seconds, waiting a couple minutes between rounds) to yield approximately 500–600 base pairs. To check the size of chromatin, 10 μl of sample was treated with 2 μl of 10 mg/ml proteinase K for 20 minutes at 50°C to reverse the cross-links. The samples were run on a 1.5% (w/v) agarose gel. After brief sonication to shear genomic DNA, the samples were centrifuged (minimum of 10,000 × g but not exceeding 15,000 × g at 4°C for 10 minutes). To prepare the beads, protein G agarose (Sigma; #P7700) was swelled in PBS (approximately 80 μl settled beads per sample) and spun (1,000 × g at 4°C for 1 minute) and resuspended in blocking buffer (25 μl 10 mg/ml sonicated salmon sperm DNA, 100 μl 1 mg/ml BSA, brought to 1 ml with H2O). Samples were blocked 15 minutes at 4°C while rotating. The mixture was then washed two times in 1.5 ml PBS and resuspended in 50/50 slurry with ChIP dilution buffer. To pre-clean the cell lysate, 150 μl of the supernatant of cell lysate was diluted with 900 μl ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8.1, 167 mM NaCl, 1 μl/ml protease inhibitor cocktail) and 80 μl protein G slurry to eliminate nonspecific binding. Samples were rotated at 4°C for 1 hour followed by pelleting of protein G (1,000 × g for 1 minute). The Protein G pellet was then discarded. A small fraction of the lysate (1%) was then saved as “input DNA” (positive control). Input DNA was treated in parallel with immunoprecipitated DNA during reverse crosslinking and purification steps. After discarding the protein G, cell lysates were incubated with 5 μg anti-glucocorticoid receptor antibody (GR, M-20; Santa Cruz Biotechnology; #sc-1004) or normal rabbit IgG (Santa Cruz Biotechnology; #sc-2027) as a negative control and rotated overnight at 4°C. The following day, 60 μl of the 50/50 protein G agarose slurry was added and samples were left to rotate for 2 hours at 4°C. Protein G was then pelleted (1,000 × g for 1 min.), supernatant was discarded and sample was washed with 1.5 ml of ChIP Dilution buffer and rotated for 5 minutes at 4°C. Samples were pelleted (1,000 × g for 1 min.). This wash step with ChIP dilution buffer was repeated. Samples were then washed with 1.5 ml TSE-500 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, and 500 mM NaCl) for 5 minutes and pelleted (1,000 × g for 1 min). Pellets were washed with 1.5 ml LiCl/Detergent (100 mM Tris pH 8.1, 500 mM LiCl, 1% IGEPAL, 1% deoxycholic acid) for 5 minutes and pelleted (1,000 × g for 1 min). Finally, the pellets were washed in 1.5 ml 1X TE for 5 minutes and pelleted (1,000 × g for 1 min). The DNA was eluted from the beads with 250 μl IP Elution Buffer (50 mM NaHCO3, 1% SDS). Samples were vortexed and left to rotate 15 minutes at room temperature. Protein G was pelleted (3,000 x g for 5 min.) and supernatant was saved. The elution step was repeated and the eluates were pooled. Crosslinks were reversed overnight at 65°C (20 μl 5 M NaCl). The saved input sample received 460 μl TE, 20 μl 5 M NaCl and was kept overnight at 65°C. The following day samples were incubated for 1 hour at 55–65°C in 32 μl (0.5 M EDTA, 1 M Tris pH 6.5, and 10 mg/mL proteinase K) solution. DNA was isolated using Qiaquick Spin Columns (Qiagen; #27104) according to the manufacturer’s protocol and quantified by NanoDrop® ND-1000 spectrophotometer. The ChampionCHIP One-Day Kit (SABiosciences, Frederick, MD; #GA-101) was also used according to the manufacturer’s instructions to confirm results. Purified DNA was used in PCR amplification for 40 cycles using the Power SYBR green master mix (Applied Biosystems; #4367659). The primers used for ChIP were based on sequences published in Revest et al. (2005) and purchased from Sigma. Non-immune rabbit IgG was used to determine non-specific antibody interactions and was subtracted from specific GR interactions. Results are presented as promoter binding relative to controls after subtracting for non-immune rabbit IgG interactions. Data represented are the result of four-five samples per condition, run in five independent experiments. PCR products were run out on 1.5% (w/v) agarose gel and results were visualized using a Fluor-S MultiImager and Quantity One Software.
2.9. GR-GRE Binding Assay
To test the ability of the native GR protein to bind to a synthethic GRE consensus sequence (5′-GGTACAnnnTGTTCT-3′) we used ELISA-based Trans-AM GR transcription factor kits (Active Motif, Carlsbad, CA; #45496). The GRE consensus sequence is immobilized on a 96-well ELISA plate and primary and secondary antibodies render the bound GR detectable. Perinatal arsenic-exposed and control protein samples were collected with the following method. Frontal cortex, hippocampus, and hypothalamus brain regions were dissected on ice and tissues combined into treatment and control groups. The samples were homogenized in 500 μl of homogenization buffer (20 mM Tris, 320 mM Sucrose, 1 mM EDTA, 20 mM β-glycerophsophate, 20 mM sodium pyrophosphate, 10 mM sodium fluoride, 200 μM sodium orthovanadate, 1 μl/ml Sigma protease inhibitor cocktail, 5 μM DTT, and ddH O, pH 7.4). Samples were centrifuged at 1000 × g for 8 minutes at 4°C. Supernatant was removed, discarded, and 300 μl of homogenization buffer containing 75 mM NaCl, 75 mM KCl, and 1% Triton X-100 was added. The samples were sonicated on ice twice for 15 seconds. The protein concentrations were determined by Bradford assay. We used the TransAM GR kit as described in the manufacturer’s protocol with the following changes: We loaded 200 μg of control or arsenic sample into each corresponding well and the wells were incubated then emptied. This was repeated two more times for a total of 600 μg of sample being loaded into each well. The method of three consecutive sample loadings was adapted from a protocol described by Robertson et al. (1987). HeLa nuclear extract, serving as a positive control, was loaded according to the protocol, incubated along with samples, emptied, and then filled with binding buffer and sealed for subsequent sample loadings. The development was run for 12 minutes. We ran a separate TransAM GR assay to test the dose-dependent linearity and threshold of the TransAM GR method described above. This assay followed the above method with the addition of control and arsenic samples being loaded at 25, 50, 100, 150, and 300 μg and all samples were run in triplicate. Absorbance readings for both assays were taken on an Infinite M200 (Tecan Group Ltd., Männedorf, Switzerland) with the absorbance wavelength set at 454 nm (after optimizing based on an absorbance scan) and the optical reference wavelength set at 655 nm.
2.10. Statistical analysis
All data were analyzed by t-test comparing the perinatal arsenic to the control. Significance was set at p ≤ 0.05. Data were analyzed and graphs were generated using GraphPad Prism software (v.4.03; GraphPad Software, San Diego, CA).
3. RESULTS
3.1. Arsenic Exposure Paradigm
The dose of arsenic used in the present study did not significantly alter maternal water consumption, maternal body weight, body weights of the newborn pups, brain wet weights or hippocampal wet weights (Martinez et al., 2008; Martinez-Finley et al., 2009).
3.2. Western Immunoblotting for ERK and phosphorylated-ERK
Due to the learning and memory deficits that we observed in our model (Martinez-Finley et al., 2009) we assessed ERK1/2 phosphorylation in the nucleus as ERK1/2 has been tied to learning and memory (figure 2). Treatment with arsenic resulted in decreased phosphorylation of ERK2 (p42/44), as quantified by densitometry and normalized to total levels (t(9) = 4.05, **p < 0.003). Data are presented as mean ± SEM of five to six litters. There was no difference in total ERK1/2.
Figure 2.

Activated ERK in hippocampal nuclear subcellular fraction. (a) Quantitation of p-ERK, total ERK, and β-actin bands in hippocampal nuclear subcellular fraction of arsenic exposed mice was based on densitometric analysis (**p = 0.003 vs. control). Results are expressed as a percentage of the p-ERK2 signal to total ERK2 signal, relative to control animals, and are presented as mean ± SEM of five-six litters. (b) Representative western blot of p-ERK1/2 and total ERK1/2 in the nuclear compartment of perinatal exposed and control mice.
3.3. H-Ras and Raf-1 mRNA levels
To determine if the decreased phosphorylation of ERK was due to decreases in upstream MAPK pathway components, we examined H-Ras and Raf-1 mRNA levels. In the perinatal arsenic-exposed offspring, H-Ras and Raf-1 mRNA expression was inhibited as determined by real-time reverse transcriptase-polymerase chain reaction. Figure 3a shows the H-Ras mRNA levels (t(16) = 4.35, ***p = 0.0005). Figure 3b shows the Raf-1 mRNA levels (t(15) = 3.55, **p = 0.003). The Cycle threshold (Ct) values were determined in triplicate for both groups. The average Ct values for β-actin, used as a housekeeping gene, were not different between the treatment and control groups. Data are presented as mean ± SEM of eight-ten litters.
Figure 3.

Reduced MAPK pathway mRNA levels in perinatal arsenic-exposed offspring compared to controls. (a) H-Ras mRNA levels in arsenic-exposed offspring and controls (***p = 0.0005 vs. control). (b) Raf-1 mRNA levels in arsenic-exposed offspring and controls (**p = 0.003 vs. control). Results are expressed as a percentage of the H-Ras or Raf-1 signal to the β-actin signal, relative to control animals, and are presented as mean ± SEM of eight-ten litters.
3.4. Western Immunoblotting for H-Ras and Raf-1
We used western blotting to semi-quantitatively assess the amounts of H-Ras and Raf-1 proteins in perinatal arsenic samples compared to controls (figure 4). Our blot densities were corrected to β-actin, a ubiquitous house-keeping protein, as a control for any sample loading discrepancies. The data were evaluated by t-test. We found there to be no statistically significant difference between H-Ras and Raf-1 protein levels in arsenic-exposed versus control (t(18) = 1.1747, p = 0.0986 and t(18) = 1.917, p = 0.0713, respectively). However, there was a noted trend toward a reduction in the arsenic exposed group. Data are presented as mean ± SEM, n = 10.
Figure 4.

MAPK pathway protein levels were only slightly reduced in perinatal arsenic-exposed offspring compared to controls. (a) H-Ras protein levels in arsenic-exposed offspring and controls (p = 0.098 vs. control). (b) Representative western blot of H-Ras and β-actin. (c) Raf-1 protein levels in arsenic-exposed offspring and controls (p = 0.07 vs. control). (d) Representative western blot of Raf-1 and β-actin. Results are expressed as a percentage of the H-Ras or Raf-1 signal to the β-actin signal, relative to control animals, and are presented as mean ± SEM of ten litters.
3.5. Chromatin Immunoprecipitation
To determine if the GR is able to bind to and regulate the GREs found in promoters of genes of the MAPK pathway we used ChIP to assess the in vivo association of the GR with H-Ras and Raf-1 promoters. Compared to controls, arsenic-exposed mice had lower GR binding to GREs in the promoter of the H-Ras gene (fig. 5a) (t(8) = 2.38; p = 0.04). Figures 5b and 5d are representative of the PCR products of each real-time assay run. Compared to controls, perinatal arsenic animals displayed lower GR binding to GREs in the Raf-1 promoter (fig. 5c) (t(7) = 2.15; p = 0.06) and although not statistically significant, there is an obvious reduction in binding. All experiments were run in duplicate. Data are presented as mean ± SEM of four to five litters.
Figure 5.

Perinatal arsenic exposure reduces glucocorticoid receptor (GR) binding to H-Ras and Raf-1 promoters. (a) Results of chromatin immunoprecipitation with anti-GR antibody in nuclear tissue extracts followed by real-time PCR for H-Ras (*p = 0.04 vs. control). (b) Gel showing real-time PCR product bands for Input (positive control), GR and IgG (negative control). (c) Results of chromatin immunoprecipitation with anti-GR antibody in nuclear tissue extracts followed by real-time PCR for Raf-1 (p = 0.06 vs. control). (d) Gel showing real-time PCR bands for Input (positive control), GR and IgG (negative control). Results are presented as relative promoter binding, relative to controls, after subtracting for non-immune rabbit IgG interactions. Results are representative of five separate experiments and are presented as mean ± SEM of four-five litters. GR: Glucocorticoid Receptor; IgG: normal rabbit IgG
3.6. GR-GRE Binding Assay
To ascertain if the reduced binding capability of the GR is due to a physical property change, we conducted ELISA-based, TransAM GR assays to detect ability of GR to bind to a synthetic sequence. GR-GRE binding was assessed based on the optical density (OD) readings of horseradish peroxidase (HRP) at 454 nm. The data are expressed as mean +/− SEM and were evaluated by t-test. The assay was conducted three separate times for a final n=8. Figure 6 shows the GR binding in control vs. arsenic treatment groups. Though the arsenic treatment group displayed lower OD readings, the reduction was not of statistical significance (t(14)=1.714, p=0.1086). To verify that we had not saturated the TransAM GR plate, resulting in the non-significant data stated above regardless of binding affinity, we ran a dose-dependent linearity and threshold test. We found that the 200 μg loading did not overwhelm the plate and that even with loading 300 μg the data still fit a dose-dependent linearity (data not shown).
Figure 6.
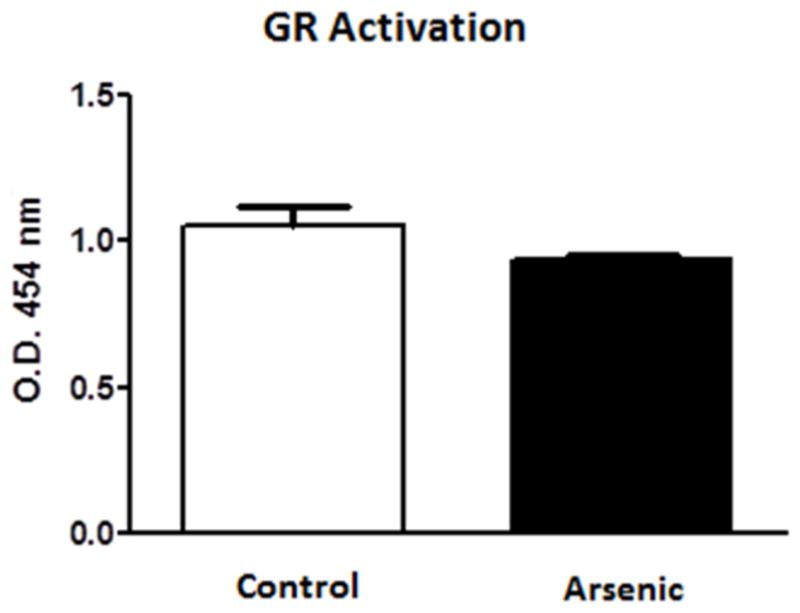
Perinatal arsenic does not interfere with the binding of native GR to a consensus GRE sequence. Assay was conducted three separate times. Data are mean optical density units (454 nm) ± SEM of eight litters.
4. DISCUSSION
Previously, we have reported learning and memory deficits along with decreases in GR and MR levels in the hippocampus of moderate (50 ppb) perinatal arsenic-exposed animals (Martinez-Finley et al., 2009). In an effort to assess a link between these two observations, the present study examined the levels of some of the genes that are associated with learning and memory and are also under the regulation of the GR. Studies have shown that the MAPK pathway is associated with learning and memory and depression (see Samuels et al., 2009 for review), two behavioral responses that were altered in perinatal arsenic-exposed mice (Martinez et al., 2008; Martinez-Finley et al., 2009), and many of its components are transcriptional targets of the glucocorticoid receptor (Revest et al., 2005). This work provides evidence for decreases in MAPK pathway transcripts, proteins (figs. 3 and 4), and activity (fig. 2) and binding activity of the GR to the GRE at in vivo (fig. 5) but not in vitro promoter sites (fig. 6) following moderate perinatal arsenic exposure.
Extracellular signal-regulated kinase 1 and 2 (ERK1/2), members of the mitogen-activated protein (MAP) kinase family, are widely expressed in post-mitotic neurons in the mammalian nervous system and play an important role in synaptic plasticity and learning and memory (English and Sweatt 1996; Sweatt 2001; Thomas and Huganir 2004). The ERK1/2 signaling cascade can be activated by a number of receptors and kinases within the hippocampus (Sweatt 2004), including through the Ras – Raf signaling pathway (Grewal et al. 1999; Sweatt 2001). Further, many of the mediators within the MAPK signaling cascade are upregulated by activation of the GR (Revest et al., 2010; Sarrazin et al., 2009). The decreased levels of phosphorylated ERK2 (fig. 2) in our prenatal arsenic exposed animals may provide the molecular basis for the perinatal arsenic-induced deficits in learning and memory (Martinez-Finley et al., 2009). Inhibition of this pathway has been shown to produce behavioral learning and memory deficits in other models. For example, intrahippocampal infusions of MAPK pathway inhibitors, prior to training, impair memory retention after training in the Morris Water Maze (Blum et al., 1999; Selcher et al., 1999).
Although, the decrease in pERK in the arsenic-exposed animals (fig 2) is in agreement with our earlier findings of learning and memory deficits (Martinez-Finley et al 2009), the lower pERK levels seen in the perinatal arsenic samples could be the result of differential loss of ERK-specific phosphatase activity. The method used to prepare the tissue in our study likely permitted continued phosphatase activity, thus preventing an accurate assessment of total pERK levels. Adequate postmortem preservation of phosphorylated proteins is a necessary measure in studies of this nature due to ongoing phosphatase activity. A study by O’Callaghan and Sriram evaluated three methods of sacrifice including decapitation, decapitation into liquid nitrogen and focused microwave irradiation, and analyzed the respective phosphorylation loss in multiple phosphoproteins including pERK (O’Callaghan and Sriram, 2004). They observed in the decapitated samples, levels of pERK were nearly absent as compared with the microwave irradiated samples, indicating that phosphatase activity is halted by microwave irradiation but continues following decapitation. While our preparation tried to reduce the impact of ongoing phosphatase activity by using a rapid dissection (< 60s) on ice followed by immediately transferring the tissue to a homogenization buffer containing, both are serine/threonine phosphatase inhibitors (beta-glycerophosphate and sodium pyrophosphate) and a tyrosine phosphatase inhibitor (sodium orthovanadate), an accurate evaluation of phosphorylated protein levels would require the microwave irradiation technique described by O’Callaghan and Sriram (2004). While an evaluation of other kinases within the MAP pathway would be interesting, these determinations will also suffer from a lack of validity because of the methods used for tissue preparations in the present study.
Revest and colleagues have shown that the GR-mediated activation of the MAPK pathway that culminates in the phosphorylation of ERK is preceded by an increase in the levels of proteins upstream from ERK (Revest et al., 2005). In their study, activation of the glucocorticoid receptors increased the expression of Ras, Raf-1, ERK 1 and 2 and Egr-1, all critical proteins within the MAPK signaling cascade. We report decreased levels of Ras and Raf-1 mRNA transcripts (fig. 3) in our perinatal arsenic exposed offspring. These lower mRNA levels are thought to be mediated by the GR, as transcription of Ras and Raf-1 are under the control of the GR (Revest et al., 2005). It is not always the case that mRNA levels correlate with protein levels nor that they always coincide with effects on cell function (Cox et al., 2005; Ghaemmaghami et al., 2003; Taniguchi et al., 2010; Tian et al., 2004). Despite lower H-Ras and Raf-1 mRNA levels, we were not able to find a corresponding significant decrease in protein levels. This could be the result of the limitations of quantification using western blotting or it could be the product of cellular regulatory mechanisms compensating for the reduced mRNA. The reduced expressions of Ras and Raf-1 along with the decrease in phosphorylated ERK 1/2 in the perinatal arsenic-exposed mice support the hypothesis that the MAPK pathway is involved in mediating the learning and memory deficits in response to the GR (Martinez-Finley et al., 2009).
The results of the ChIP-real-time PCR assay indicate decreased in vivo GR binding at H-Ras and Raf-1 promoter sites (fig. 5), thus the decrease in transcripts reported here is a result of the decrease in binding ability at these sites. The decrease in binding ability appears to be restricted to in vivo sites as GR isolated from arsenic-exposed pups was able to bind to the synthetic consensus GRE as well as that from control pups (fig. 6). Although there was a slight reduction in arsenic-exposed GR binding in this assay, it was not significant (fig. 6). The binding activity at the synthetic GRE sites was dose-dependent and linear for both control and arsenic (unpublished results). These data suggest that the reduced promoter activity we found in the ChIP assay was not due to a physical limitation of the GR itself.
There are a number of possible explanations for the decrease in binding ability of GR to GREs in arsenic exposed animals. The possibility of steric hindrance by arsenic at these sites has been ruled out albeit whole brain measures showed an arsenical level of 2 ppb, hippocampal arsenic amounts were below the level of detection (data not shown); thus it is unlikely that arsenic is sterically hindering these in vivo promoter sites. This conclusion is supported by a study by Bodwell et al. (2006), showing that arsenic (up to 10 M) had no effect on progesterone and GR interaction with the steroid response element in postnatal day 35 hippocampal samples.
An intriguing possibility is that of methylation changes at these promoter regions could be initiated by arsenic during the perinatal exposure period. Recent work by Martinez et al. (2011) has demonstrated that perinatal exposure to moderate levels of arsenic altered DNA methylation postnatally. Our own data shows decreased activity of the GR at in vivo promoters in perinatally arsenic-exposed offspring (fig. 5), yet native GR from these mice is able to bind to a synthetic GRE consensus sequence (fig. 6). These data are in agreement with an earlier in vitro arsenic study (Bodwell et al., 2006) which showed that arsenic did not interfere with the GR binding to the GRE. While there are other potential explanations for our findings, the hypothesis that perinatal arsenic exposure has altered the methylation status of certain promoter regions under GRE regulation is appealing and currently under investigation in our laboratory.
Arsenic has been shown to disrupt gene transcription in various models (Bodwell et al., 2006; Hamilton et al., 1998; Kaltreider et al., 2001). Our perinatal model is a unique view of arsenic disruption of gene regulation. In our perinatal exposure model arsenic is present only at a barely detectible level within the brain at the time these studies were conducted (PND 35). This suggests that the impact of arsenic on GR regulated gene expression is persistent and does not require significant levels of arsenic to be chemically present for the effect. In addition, it has been determined that chronic exposure to pharmacologically elevated corticosterone levels significantly reduces GR expression in the hippocampus (Lee et al., 2010). Although this effect did not persist long after exposure, it may give us insight into the mechanisms that disrupt normal function of the HPA axis. It is conceivable that arsenic inhibits the performance of the GR and, as a result, the GR is unable to re-set the HPA axis. This in turn creates the elevated corticosterone levels which then decrease GR expression, both of which are observed phenomena. Prolonged exposure to low concentrations of persistent neurotoxic environmental pollutants can lead to neuronal dysfunction, particularly during development when the brain is most sensitive. By analyzing changes in the gene expression profile of cells exposed to toxicants, it will be possible to find genes/gene clusters that are commonly or specifically challenged in response to damage associated with developmental neurotoxicants (Sunol, 2010). Overall, these data suggest that moderate perinatal arsenic has long-term effects on disruption of steroid hormone-regulated gene transcription well into adolescence, long after the arsenic has been metabolized. Down-regulation of GR-mediated gene transcription of key regulators within the MAP kinase pathway may be a mechanism leading to arsenic-induced cognitive dysfunction.
Highlights.
Reduced Expression of MAPK/ERK Genes in Perinatal Arsenic-Exposed Offspring Induced by Glucocorticoid Receptor Deficits - Martinez-Finley et al.
Changes within the glucocorticoid receptor (GR) cellular signaling pathway were evaluated in adolescent mice exposed to 50 ppb arsenic during gestation.
Levels of key MAP Kinase pathway components: ERK, Ras and Raf, were reduced in arsenic perinatal exposed mice.
A decrease level of GR activated Ras and Raf promoters was also seen in the perinatal arsenic mice relative to controls.
GR was able to bind to synthetic GRE sequences, suggesting that arsenic was not directly interfering with binding.
Findings suggest that GR-mediated transcriptional deficits in the MAPK/ERK pathway could be an underlying cause of previously reported learning deficits and provide the link to arsenic-induced deficiencies in cognitive development.
Acknowledgments
The authors thank David Leonard and for helpful discussions and technical assistance, as well as Dr. Kevin Caldwell and Dr. Xinyu Zhao for help with experimental design and data analysis. The project described was supported by 1RO1ES019583-01 (AMA) and by F31ES017196 (EJM-F) from the National Institute of Environmental Health Sciences, the University of New Mexico Graduate and Professional Society-Graduate Research Development Grant along with the legislature of the state of New Mexico; the Pfizer Global Research and Development Safety Scholars Fellowship and generous funds from the University of New Mexico College of Pharmacy. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences, the National Institutes of Health or Pfizer Global Research and Development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological profile for Arsenic. Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service; 2007. [Accessed 05 Jan 2008]. http://www.atsdr.cdc.gov/toxprofiles/tp2.html. [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–9. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Beato M, Chalepakis G, Schauer M, Slater EP. DNA regulatory elements for steroid hormones. J Steroid Biochem. 1989;32:737–47. doi: 10.1016/0022-4731(89)90521-9. [DOI] [PubMed] [Google Scholar]
- Blum S, Moore AN, Adams F, Dash PK. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–44. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodwell JE, Gosse JA, Nomikos AP, Hamilton JW. Arsenic Disruption of Steroid Receptor Gene Activation: Complex Dose-Response Effects Are Shared by Several Steroid Receptors. Chem Res Toxicol. 2006;19:1619–29. doi: 10.1021/tx060122q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CT, Caldwell KK. Fear conditioning is associated with altered integration of PLC and ERK signaling in the hippocampus. Pharmacol Biochem Behav. 2004;79:633–40. doi: 10.1016/j.pbb.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Calderón J, Navarro ME, Jimenez-Capdeville ME, Santos-Diaz MA, Golden A, Rodriguez-Leyva I, Borja-Aburto V, Díaz-Barriga F. Exposure to arsenic and lead and neuropsychological development in Mexican children. Environ Res. 2001;85(2):69–76. doi: 10.1006/enrs.2000.4106. [DOI] [PubMed] [Google Scholar]
- Caldwell KK, Sheema S, Paz RD, Samudio-Ruiz SL, Laughlin MH, Spence NE, Roehlk MJ, Alcon SN, Allan AM. Fetal alcohol spectrum disorder-associated depression: evidence for reductions in the levels of brain-derived neurotrophic factor in a mouse model. Pharmacology, Biochemistry and Behavior. 2008;90:614–24. doi: 10.1016/j.pbb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis J, Sloboda D, Matthews S, Holloway A, Alfaidy N, Howe D, Fraser M, Newnham J. Fetal hypothalamic-pituitary adrenal (HPA) development and activation as a determinant of the timing of birth, and of postnatal disease. Endocr Res. 2000;26(4):489–504. doi: 10.3109/07435800009048560. [DOI] [PubMed] [Google Scholar]
- Chalmers DT, Kwak SP, Mansour A, Akil H, Watson SJ. Corticosteroids regulate brain hippocampal 5-HT1A receptor mRNA expression. J Neurosci. 1993;13:914–23. doi: 10.1523/JNEUROSCI.13-03-00914.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B, Kislinger T, Emili A. Integrating gene and protein expression data: pattern analysis and profile mining. Methods. 2005;35(3):303–14. doi: 10.1016/j.ymeth.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Datson NA, Van der Perk J, de Kloet ER, Vreugdenhil E. Identification of corticosteroid-responsive genes in rat hippocampus using serial analysis of gene expression. Eur J Neurosci. 2001;14:675–89. doi: 10.1046/j.0953-816x.2001.01685.x. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Sybesma H, Reul HM. Selective control by corticosterone of serotonin1 receptor capacity in raphe-hippocampal system. Neuroendocrinology. 1986;42:513–21. doi: 10.1159/000124496. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–32. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- Geng CD, Pedersen KB, Nunez BS, Vedeckis WV. Human glucocorticoid receptor alpha transcript splice variants with exon 2 deletions: evidence for tissue- and cell type-specific functions. Biochemistry. 2005;44:7395–405. doi: 10.1021/bi047485e. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425(6959):737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Green S, Kumar V, Theulaz I, Wahli W, Chambon P. The N-terminal DNA-binding ’zinc finger’ of the oestrogen and glucocorticoid receptors determines target gene specificity. EMBO J. 1988;7:3037–44. doi: 10.1002/j.1460-2075.1988.tb03168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase singalling in neurons. Curr Opin Neurobiol. 1999;9:544–53. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Hamilton JW, Kaltreider RC, Bajenova OV, Ihnat MA, McCaffre J, Turpie BW, Rowell EE, Oh J, Nemeth MJ, Pesce CA. Molecular basis for effects of carcinogenic heavy metals on inducible gene expression. Environmental Health Perspectives. 1998;106 (Suppl 4):1005–15. doi: 10.1289/ehp.98106s41005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joels M, Karst H, Alfarez D, Heine V, Qin Y, van Riel E, Verkuyl M, Lucassen P, Krugers H. Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress. 2004;7:221–231. doi: 10.1080/10253890500070005. [DOI] [PubMed] [Google Scholar]
- Kaltreider RC, Davis AM, Lariviere JP, Hamilton JW. Arsenic Alters the Function of the Glucocorticoid Receptor as a Transcription Factor. Environmental Health Perspectives. 2001;109:245–51. doi: 10.1289/ehp.01109245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RS, Tamashiro KLK, Yang X, Purcell RH, Harvey A, Willour VL, Huo Y, Rongione M, Wand GS, Potash JB. Chonic Corticosterone Exposure Increases Expression and Decreases Deoxyribonucleic Acid Methylation of Fkbp5 in Mice. Endocrinology. 2010;151(9):4332–43. doi: 10.1210/en.2010-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martinez EJ, Kolb BL, Bell A, Savage DD, Allan AM. Moderate perinatal arsenic exposure alters neuroendocrine markers associated with depression and increases depressive-like behaviors in adult mouse offspring. Neurotoxicology. 2008;29(4):647–55. doi: 10.1016/j.neuro.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Finley EJ, Ali AM, Allan AM. Learning Deficits in C57Bl/6 mice following perinatal arsenic exposure: Consequence of lower corticosterone receptor levels? Pharmacology, Biochemistry and Behavior. 2009;94:271–7. doi: 10.1016/j.pbb.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez L, Jiménez V, García-Sepúlveda C, Ceballos F, Delgado JM, Niño-Moreno P, Doniz L, Saavedra-Alanís V, Castillo CG, Santoyo ME, González-Amaro R, Jiménez-Capdeville ME. Impact of early developmental arsenic exposure on promotor CpG-island methylation of genes involved in neuronal plasticity. Neurochem Int. 2011;58(5):574–81. doi: 10.1016/j.neuint.2011.01.020. [DOI] [PubMed] [Google Scholar]
- McEwen B. Allostasis, allostatic load, and the aging nervous system: role of excitatory amino acids and excitotoxicity. Neurochem Res. 2000;25:1219–1231. doi: 10.1023/a:1007687911139. [DOI] [PubMed] [Google Scholar]
- McEwen B. Sex, stress and the hippocampus: allostasis, allostatic load and the aging process. Neurobiol Aging. 2002;23:921–939. doi: 10.1016/s0197-4580(02)00027-1. [DOI] [PubMed] [Google Scholar]
- Meijer OC, Van Oosten RV, De Kloet ER. Elevated basal trough levels of corticosterone suppress hippocampla 5-hydroxytryptamine (1A) receptor expression in adrenally intact rats: implication for the pathogenesis of depression. Neuroscience. 1997;80:419–26. doi: 10.1016/s0306-4522(97)00008-0. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP, Sriram K. Focused microwave irradiation of the brain preserves in vivo protein phosphorylation: comparison with other methods of sacrifice and analysis of multiple phosphoproteins. J Neurosci Methods. 2004;135:159–68. doi: 10.1016/j.jneumeth.2003.12.006. [DOI] [PubMed] [Google Scholar]
- O’Regan D, Welberg L, Holmes MC, Seckl JR. Glucocorticoid programming of pituitary-adrenal function: mechanisms and physiological consequences. Semin Neonatol. 2001;6:319–29. doi: 10.1053/siny.2001.0067. [DOI] [PubMed] [Google Scholar]
- Ohno M, Frankland PW, Chen AP, Costa RM, Silva AJ. Inducible, pharmacogenetic approaches to the study of learning and memory. Nature Neuroscience. 2001;4:1238–43. doi: 10.1038/nn771. [DOI] [PubMed] [Google Scholar]
- Revest JM, Blasi FD, Kitchener P, Rouge-Pont F, Desmedí A, Turiault M, Tronche F, Piazza PV. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nature Neuroscience. 2005;85:664–72. doi: 10.1038/nn1441. [DOI] [PubMed] [Google Scholar]
- Revest JM, Kaouane N, Mondin M, Le Roux A, Rouge-Pont F, Valle’e M, Barik J, Tronche F, Desmedt A, Piazza PV. The enhancement of stress-related memory by lucocorticoids depends on synapsin-Ia/Ib. Mol Psychiatry. 2010;15:1140–51. doi: 10.1038/mp.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson NM, Kusmik WM, Grove BF, Miller-Diener A, Webb ML, Litwack G. Characterization of a monoclonal antibody that probes the functional domain of the glucocorticoid receptor. Biochem J. 1987;246:55–65. doi: 10.1042/bj2460055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez VM, Carrizales L, Jimenez-Capdeville ME, Dufour L, Giordano M. The effects of sodium arsenite exposure on behavioral parameters in the rat. Brain Res Bull. 2001;55(2):301–08. doi: 10.1016/s0361-9230(01)00477-4. [DOI] [PubMed] [Google Scholar]
- Rodriguez VM, Carrizales L, Mendoza MS, Fajardo OR, Giordano M. Effects of sodium arsenite exposure on development and behavior in the rat. Neurotoxicology Teratology. 2002;24:743–50. doi: 10.1016/s0892-0362(02)00313-6. [DOI] [PubMed] [Google Scholar]
- Rodriguez VM, Jimenez-Capdeville ME, Giordano M. The effects of arsenic exposure on the nervous system. Toxicol Lett. 2003;145:1–18. doi: 10.1016/s0378-4274(03)00262-5. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Saitta SC, Landreth GE. MAP’ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–7. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin N, Di Blasi F, Roullot-Lacarrière V, Rougé-Pont F, Le Roux A, Costet P, Revest JM, Piazza PV. Transcriptional effects of glucocorticoid receptors in the dentate gyrus increase anxiety-related behaviors. PLoS One. 2009;4(11):e7704. doi: 10.1371/journal.pone.0007704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoneveld OJ, Gaemers IC, Lamers WH. Mechanisms of glucocorticoid signaling. Biochim Biophys Acta. 2004;1680:114–28. doi: 10.1016/j.bbaexp.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–90. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa RJ, Tannery NH, Lafer EM. In situ hybridization mapping of glucocorticoid receptor messenger ribonucleic acid in rat brain. Mol Endocrinol. 1989;3:481–94. doi: 10.1210/mend-3-3-481. [DOI] [PubMed] [Google Scholar]
- Sunol C. Use of gene expression of neural markers in cultured neural cells to identify developmental neurotoxicants. Toxicol Sci. 2010;113:1–3. doi: 10.1093/toxsci/kfp240. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–7. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329(5991):533–8. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signaling and synaptic plasticity. Nat Rev Neuroscience. 2004;5:173–83. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, Doyle MJ, Yi EC, Dai H, Thorsson V, Eng J, Goodlett D, Berger JP, Gunter B, Linseley PS, Stoughton RB, Aebersold R, Collins SJ, Hanlon WA, Hood LE. Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol Cell Proteomics. 2004;3(10):960–9. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- Tsai SY, Carlstedt DG, Weigel NL, Dallman K, Gustafsson J, Tsai MJ, O’Malley BW. Molecular interactions of steroid hormone receptor with its enhancer element: evidence for receptor dimer formation. Cell. 1988;55:361–9. doi: 10.1016/0092-8674(88)90059-1. [DOI] [PubMed] [Google Scholar]
- United States Environmental Protection Agency (US EPA) National Primary Drinking Water Regulations; Arsenic and Clarifications to Compliance and New Source Contaminants Monitoring. Federal Register. 2001;66(14):6975–7066. [Google Scholar]
- United States Environmental Protection Agency (US EPA) Basic Information about Arsenic in Drinking Water. United States Environmental Protection Agency; 2010. [Accessed 06 Apr 2011]. http://water.epa.gov/drink/contaminants/basicinformation/arsenic.cfm. [Google Scholar]
- Von Ehrenstein OS, Poddar S, Yuan Y, Mazumder DG, Eskenazi B, Basu A, Hira-Smith M, Ghosh N, Lahiri S, Haque R, Ghosh A, Kalman D, Das S, Smith AH. Children’s intellectual function in relation to arsenic exposure. Epidemiology. 2007;18:444–51. doi: 10.1097/01.ede.0000248900.65613.a9. [DOI] [PubMed] [Google Scholar]
- Wasserman GA, Liu X, Parvez F, Ahsan H, Factor-Litvak P, van Geen A, Slavkovich V, Lolacono NG, Cheng Z, Hussain I, Momotaj H, Graziano JH. Water arsenic exposure and children’s intellectual function in Araihazar, Bangladesh. Environ Health Perspect. 2004;112:1329–33. doi: 10.1289/ehp.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber EJ, Savage DD, Sutherland RJ, Caldwell KK. Fear conditioning-induced alterations of phospholipase C-beta1a protein level and enzyme activity in rat hippocampal formation and medial frontal cortex. Neurobiol Learn Mem. 2001;76(2):151–82. doi: 10.1006/nlme.2000.3994. [DOI] [PubMed] [Google Scholar]