

Abstract
TARP syndrome, comprising Talipes equinovarus, Atrial septal defect, Robin sequence (micrognathia, glossoptosis, and cleft palate), and Persistence of the left superior vena cava, is an X-linked condition with pre- or postnatal lethality in affected males. Based on linkage studies and massively parallel sequencing of X-chromosome exons in two families, the disease causing gene was identified as RBM10. We identified a maternally inherited frameshift mutation in an unrelated patient, confirming RBM10 as the disease gene. This is the first reported individual with TARP syndrome who survived past early infancy, thus expanding the phenotypic spectrum of this disorder. In addition to the characteristic cleft palate, atrial septal defect and persistent superior vena cava, he had low-set and posteriorly angulated ears, upslanting palpebral fissures, cryptorchidism and structural brain abnormalities including partial agenesis of the corpus callosum, dysplastic enlarged caudate, and cerebellar hypoplasia with megacisterna magna. Preterm delivery, suspected pulmonary hypoplasia and pulmonary hypertension resulted in chronic lung disease. At the age of 3 7/12 years, he remained ventilator-dependent at night, and he was fed exclusively through a gastro-jejunal tube. Sensorineural hearing loss required a hearing aid. Optic atrophy and cortical visual impairment were noted. He was unable to sit independently, was non-communicative and he had severe intellectual disability. Atrial flutter required recurrent ablation of intra-atrial re-entry pathways. The mother's heterozygosity for the RBM10 mutation underscored the importance of accurate diagnosis and counseling for TARP syndrome.
Keywords: ASD, cryptorchidism, persistent left superior vena cava, Pierre-Robin sequence, RBM10, talipes equinovarus, X-linked
Introduction
A syndrome comprising Pierre-Robin sequence (micrognathia, glossoptosis, and cleft palate), talipes equinovarus, atrial septal defect (ASD) and persistence of the left superior vena cava was described by Gorlin et al. [1970] in a large family with seven affected males. The proband died suddenly at age 3 months, and no other affected male survived past early infancy, strongly suggestive of an X-linked inheritance pattern with infantile male lethality [Gorlin et al., 1970]. This disorder was designated “TARP syndrome” for Talipes equinovarus, ASD, Robin sequence and Persistence of the left superior vena cava, and molecularly mapped to the X-chromosome by Kurpinski et al. [2003]. Applying massively parallel exon sequencing to DNA samples from the original family and from a second family segregating TARP syndrome in three affected individuals, Johnston et al. [2010] identified mutations in RBM10. Here we report the third family with TARP syndrome with a novel RBM10 mutation and thus confirm RBM10 as the disease associated gene. The longer survival of this patient, compared to previously reported patients, allows further delineation of the phenotype and the data suggest that cardiac conduction defects may be a cause of early mortality and that CNS defects are part of the syndrome.
Materials and Methods
Clinical Report
The patient was born to a 19-year-old gravida 2 para 0 mother of Mexican ancestry, whose first pregnancy resulted in a spontaneous first trimester loss. Delivery occurred by cesarean at 33 weeks gestation due to breech position and a premature rupture of membranes resulting in oligohydramnios. His birth weight was 1.5 kg (∼25th centile for gestational age), length 44.5 cm (∼50th centile) and OFC 28 cm (<10th centile; 50th centile for ∼29.5 weeks gestation). Apgar scores were 1, 5, and 6 at 1, 5, and 10 minutes, respectively. Multiple congenital anomalies included micrognathia and U-shaped cleft palate, low set and posteriorly angulated ears, and cryptorchidism. “Extreme dorsiflexion of the left foot” was seen at birth but resolved subsequently. Respiratory distress resulted in intubation and placement of a tracheostomy at age seven days. The patient required high frequency oscillatory ventilation and the use of nitric oxide, and he remained hospitalized for his first eight months. A combination of suspected pulmonary hypoplasia, pulmonary hypertension, and broncho-pulmonary dysplasia caused chronic lung disease. Oropharyngeal dysfunction and aspiration led to placement of a gastrostomy tube in combination with a Nissen fundoplication. He had clinically diagnosed cortical visual impairment with bilateral optic atrophy and mild myopia. Bilateral severe to profound sensorineural hearing loss was identified through auditory brain stem response testing and treated with hearing aides. The kidneys appeared echogenic on ultrasound studies and chronic kidney disease with renal tubular acidosis was treated medically. While dysplastic kidneys have not been excluded, these renal issues were considered secondary to postnatal hypoxic damage. Echocardiogram showed persistent left superior vena cava draining into the coronary sinus, and absence of the right superior vena cava. A sinus venosus type ASD allowed for inter-atrial communication and pulmonary hypertension was present. The ASD was closed surgically at age 3 months. Subsequently, he developed atrial flutter due to intra-atrial re-entry tachycardia, which required ablation at age 3 2/12 years, and in a different location at age 3 4/12 years.
At age 3 7/12 years, his length was 91 cm (10-25th centile), weight 14.7 kg (∼50th centile), and OFC 48.4 cm (3rd-10th centile). His head was brachycephalic, with right posterior plagiocephaly. The face was round, with flat supraorbital ridges, upslanting palpebral fissure and mild telecanthus with an inner canthal distance of 4 cm (>2 SD above mean for age) and outer canthal distance of 8.4 cm (75-90th centile) (Fig. 1). His ears were low set, posteriorly angulated and measured 4.8 and 4.9 cm in length (∼1 SD below mean for age) (Fig. 1). A tracheostomy and a gastro-jejunal tube were in place, and he had numerous well-healed scars after surgical procedures. Hands showed mild brachyclinodactyly of the fifth digit bilaterally, and the distal flexion crease of the right fourth finger was missing (Fig. 2). Toes showed very mild syndactyly of the second and third digits, bilaterally. The patient had limited head control and was unable to sit without support. His muscle tone varied with low tone in his upper extremities and trunk, and increased tone in his legs. He remained exclusively fed through the gastro-jejunal tube, required ventilator support during most hours of sleep and had no consistent evidence of expressive or receptive language capabilities. Hearing aids were used in school only.
Figure 1.
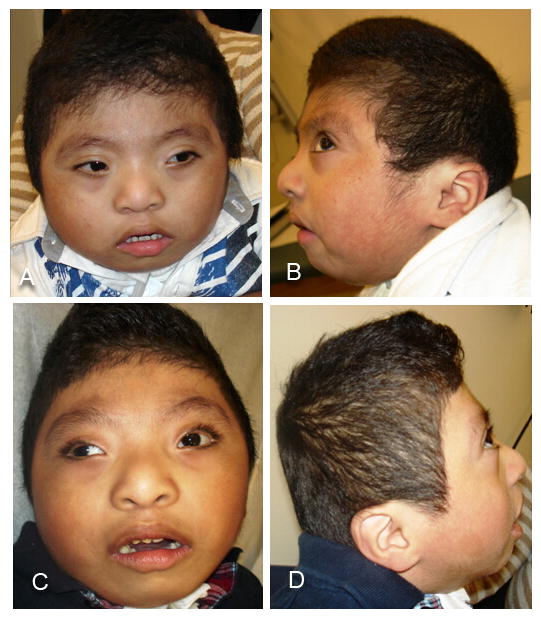
Photographs of the proband at ages 2 2/12 (A, B) and 3 7/12 years (C, D); note the round face, flat supraorbital ridges, upslanting palpebral fissures and mild telecanthus, highly arched eye brows, and low-set and posteriorly angulated ears.
Figure 2.
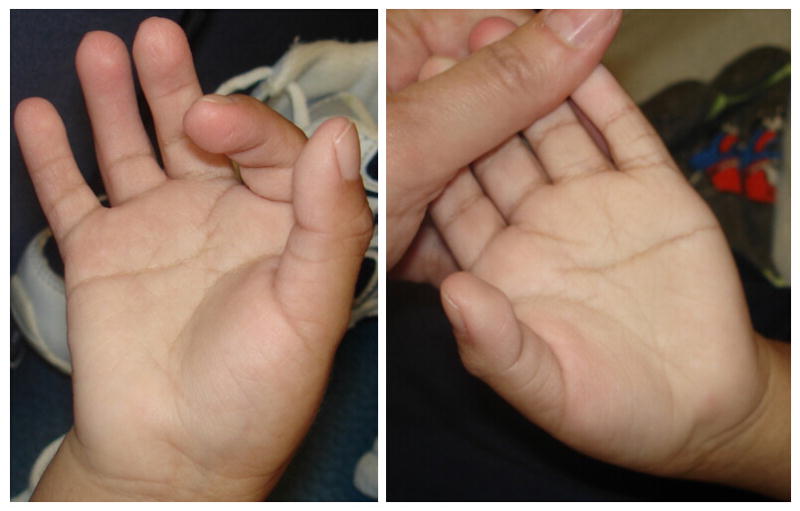
Palms and fingers at age 3 7/12 years, note the absent distal flexion crease in the right fourth finger, mild bilateral brachyclinodactyly of the fifth digit, persistent fetal pads and unusual palmar flexion creases.
Cranial MRI was first performed at term adjusted age, and heterotopia along the lateral ventricles, cerebellar vermis hypoplasia and a megacisterna magna were noted. A subsequent cranial MRI at age 1 year showed relatively small frontal lobes (Fig. 3), a small cerebellum and a megacisterna magna. Large cavum septum et vergae and an enlarged third ventricle were seen. The corpus callosum and the hippocampi were small. Enlarged dysplastic caudate nuclei were hard to differentiate from adjacent heterotopia.
Figure 3.

Cranial MRI at age 1 year showed a mildly sloping forehead and relatively small frontal lobes (A), markedly short and thin corpus callosum (large square in A), and megacisterna magna (round arrowhead in A). The lower posterior vermis (just above round arrowhead) is borderline small. The caudate nuclei appear large, with the left caudate very large and compressing the frontal horn of the left lateral ventricle (round arrowhead in B, and long arrows labeled “c” in D). Small heterotopias are seen in the subependymal region next to the anterior bodies of the lateral ventricles (all arrows in C, and arrows labeled “het” in D and E). They are adjacent to and difficult to distinguish from the caudate nuclei. The thin corpus callosum, large cavum septum et vergae, and large third ventricle are seen in the midline in B and D-F. The hippocampi are small and dysplastic (long arrow labeled “hip” in D; also seen in E and F). Abbreviations: c, caudate nuclei; het, heterotopia; hip, hippocampus).
Numerous clinical diagnostic studies, including SignatureChipWG™ CGH and sequencing of CHD7 and selected exons of MED12, did not yield a diagnostic result.
A younger brother was born at term after an uncomplicated pregnancy, he is in good health.
Molecular Analyses
The patient and his mother enrolled in a research study that was reviewed and approved by the NHGRI IRB. DNA was isolated by standard procedures from peripheral blood leucocytes. The exons and flanking introns of the RBM10 gene were amplified as described in Johnston et al. [2010]. Nucleotide numbering was determined in accordance with Human Genome Variation Society guidelines using reference sequence NM_005676.4.
Results
The sequencing data showed that the patient was hemizygous for a c.del159C variant in RBM10, which predicts p.Lys54SerfsX80. His mother was heterozygous for this variant.
Discussion
Recently, massively parallel exon sequencing of DNA samples from two families demonstrated that mutations in RBM10 cause the rare, X-linked TARP syndrome [Johnston et al., 2010]. Here we report an individual with TARP syndrome who is unrelated to the two families included in the exome study. A novel RBM10 mutation was identified in this patient, confirming RBM10 as the disease associated gene. This report shows the rapid translation of knowledge obtained through the research use of exome re-sequencing technology into clinical care.
In contrast to all previously reported individuals with TARP syndrome who died in early infancy, the patient reported here survived to his current age 3 7/12 years with intensive medical care. Long-term survival is thus consistent with a diagnosis of TARP syndrome, and additional patients may be identified amongst older individuals. An accurate diagnosis of TARP syndrome, even postmortem, is important for recurrence risk counseling because, at least so far, all heterozygous females are phenotypically normal. In the two previously reported families and in the family reported here, the proband's mothers were found to carry the pathogenic change, and thus had a 50% recurrence risk for each son.
The long term survival of the patient reported here allows further phenotype delineation, including the characteristic facial features of a round face with upslanting palpebral fissures and low-set, posteriorly angulated ears (Fig. 1). The single previously published facial photograph of a patient with TARP syndrome also shows a round face and upslanting palpebral fissures [Gorlin et al., 1970]. The hallmark findings of talipes, ASD, Robin sequence and persistence of the left superior vena cava are common, but not universally present (Table I), and may be underreported due the inclusion of deceased individuals with limited available information in previous publications. While low-set ears and cryptorchidism appear to be frequent in patients with TARP syndrome, these are neither specific nor life-threatening abnormalities. Sensorineural hearing loss and optic atrophy with cortical visual impairment are present in the patient reported here, but were not previously noted in TARP syndrome. Similar to these functional neurologic abnormalities, this patient's cardiac rhythm disturbance may have been present in other individuals, but remained unidentified due to their short life span and the severity of their other medical problems. The “periodic tachycardia” [Gorlin et al., 1970] may have reflected similar dysrhythmia. Pulmonary abnormalities reported on autopsies [Gorlin et al., 1970; Johnston et al., 2010] included “marked underdevelopment of alveoli”, and the patient reported here has chronic lung disease. Lung development and function are apparently affected by the apparent loss of function of RBM10.
Table I.
Findings in the Present Patient, Compared to the Two Prior Reported Families.
| Present Patient | Gorlin et al. [1970] | Johnston et al. [2010] | Combined N=11 | |
|---|---|---|---|---|
| Talipes equinovarus | - | 6/7 | 2/3 | 8/11 (73%) |
| Atrial septal defect | + | 3/7 | 1/1 | 5/9 (56%) |
| Cleft palate and micrognathia | + | 4/6 | 3/3 | 8/10 (80%) |
| Persistent left superior vena cava | + | 4/7 | 0/0 | 5/8 (63%) |
| Low-set ears | + | + in proband | + in proband | 3/3 (100%) |
| Cryptorchidism | + | + in proband | + in proband | 3/3 (100%) |
| Structural brain anomaly | Short and thin corpus callosum, dysplastic caudate and hippocampi; cavum septum et vergae; megacisterna magna | N/A | N/A | |
| Hearing | Sensorineural hearing loss | N/A | N/A | |
| Eye and vision | Myopia, optic atrophy, cortical visual impairment | “possible early… cataracts” | N/A | |
| Pulmonary issues | Chronic lung disease | “Lung … congestion, fibrosis, and some bronchiectasis” on autopsy | “marked under-development of alveoli” on autopsy; “hyaline membrane disease” | |
| Cardiac rhythm | Atrial flutter, requiring ablation twice | “periodic tachycardia … on several occasions” | N/A | |
| Death in infancy | - | 7/7 | 3/3 | 10/11 (91%) |
The structural brain abnormalities reported here are notable, and may prove to be distinctive for TARP syndrome. While the small frontal lobes, the short and thin corpus callosum, and the megacisterna magna are non-specific, the combination of large cavum septum et vergae, dysplastic hippocampi, enlarged and dysplastic caudate nuclei, and heterotopia is unusual. In light of these structural brain abnormalities, the patients' neurological abnormalities and his apparently severe intellectual disability are not surprising. Cortical visual impairment and sensorineural hearing loss were also present.
We conclude that long-term survival can occur in individuals with TARP syndrome. In addition to the previously noted hallmark findings, its presentation encompasses characteristic facial features, cryptorchidism, structural brain abnormalities, severe intellectual disability, sensorineural hearing loss, and pulmonary disease. A suspected clinical diagnosis may be confirmed by identification of an RBM10 mutation. If the proband is deceased and no DNA is available, testing the mother may be considered to establish the diagnosis and recurrence risk.
Acknowledgments
We appreciate the family's generosity in allowing us to share this information. This work was supported in part by funding from the Intramural Program of the National Human Genome Research Institute of the National Institutes of Health.
References
- Gorlin RJ, Cervenka J, Anderson RC, Sauk JJ, Bevis WD. Robin's syndrome. A probably X-linked recessive subvariety exhibiting persistence of left superior vena cava and atrial septal defect. Am J Dis Child. 1970;119:176–178. [PubMed] [Google Scholar]
- Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, NIH Intramural Sequencing Center. Chong K, Mullikin JC, Biesecker LG. Massively Parallel Sequencing of Exons on the X Chromosome Identifies RBM10 as the Gene that Causes a Syndromic Form of Cleft Palate. Am J Hum Genet. 2010;86:743–748. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurpinski KT, Magyari PA, Gorlin RJ, Ng D, Biesecker LG. Designation of the TARP syndrome and linkage to Xp11.23-q13.3 without samples from affected patients. Am J Med Genet Part A. 2003;120A:1–4. doi: 10.1002/ajmg.a.10201. [DOI] [PubMed] [Google Scholar]