

Abstract
The performance of several two-step scoring approaches for molecular docking were assessed for their ability to predict binding geometries and free energies. Two new scoring functions designed for “step 2 discrimination” were proposed and compared to our CHARMM implementation of the linear interaction energy (LIE) approach using the Generalized-Born with Molecular Volume (GBMV) implicit solvation model. A scoring function S1 was proposed by considering only “interacting” ligand atoms as the “effective size” of the ligand, and extended to an empirical regression-based pair potential S2. The S1 and S2 scoring schemes were trained and five-fold cross validated on a diverse set of 259 protein-ligand complexes from the Ligand Protein Database (LPDB). The regression-based parameters for S1 and S2 also demonstrated reasonable transferability in the CSARdock 2010 benchmark using a new dataset (NRC HiQ) of diverse protein-ligand complexes. The ability of the scoring functions to accurately predict ligand geometry was evaluated by calculating the discriminative power (DP) of the scoring functions to identify native poses. The parameters for the LIE scoring function with the optimal discriminative power (DP) for geometry (step 1 discrimination) were found to be very similar to the best-fit parameters for binding free energy over a large number of protein-ligand complexes (step 2 discrimination). Reasonable performance of the scoring functions in enrichment of active compounds in four different protein target classes established that the parameters for S1 and S2 provided reasonable accuracy and transferability. Additional analysis was performed to definitively separate scoring function performance from molecular weight effects. This analysis included the prediction of ligand binding efficiencies for a subset of the CSARdock NRC HiQ dataset where the number of ligand heavy atoms ranged from 17 to 35. This range of ligand heavy atoms is where improved accuracy of predicted ligand efficiencies is most relevant to real-world drug design efforts.
Keywords: CDOCKER, CHARMM, Protein-Ligand Interactions, Docking, Scoring Functions, Distance Dependent Pair Potential, Decoys, Molecular Weight, Fragment, Kinase, p38alpha, p38MAP, Fragment-Based-Design
1. Introduction
The application of computational methods in drug discovery has been ever increasing with the growth of computational power and accumulation of new experimental data. During the past decade, various new methodologies have continued to improve in addressing the two fundamental steps of computational drug design: docking1–3 and scoring.4, 5 Docking generates and identifies native or native-like configurations of the receptor-ligand complexes. Then an appropriate scoring function approximates the binding affinities using their geometries. In a virtual screening process, these two steps are used to distinguish the hits from a large library of small molecules. While numerous docking methods are capable of producing near native docking poses,1–3 the development of a high-accuracy scoring function still remains a challenge. A good scoring function has several requirements. 1) It should be able to distinguish the near native poses from the non-native poses of a particular ligand molecule. 2) The predicted protein-ligand binding affinities should correlate with the experimentally derived values. 3) The scoring function should have a low computational cost, making it applicable to a large set of ligand molecules. Ferrara et al. evaluated the performance of 9 different scoring functions using 189 protein-ligand complexes from the Ligand-Protein DataBase (LPDB).4 Several of the scoring functions, including CHARMm6, DOCK-energy1, DrugScore7, ChemScore8, and AutoDock9, performed well in distinguishing near-native poses from mis-docked poses. However, the binding affinities predicted by most of the scoring functions correlated poorly with the experimental binding energies. Among them, ChemScore performed the best with R2 = 0.51. Using 800 protein-ligand complexes, Wang et al. evaluated 14 different scoring functions for their ability to predict experimental binding affinities.5 They also concluded that the predicted and experimental binding affinities correlated only moderately. Among the scoring functions, X-Score10, DrugScore, Sybyl::ChemScore11, and Cerius2::PLP12 produced better correlations. Even in an ideal case where all of the binding geometries are predicted with perfect accuracy, an inaccurate scoring function for ranking compounds will still produce a large number of false positives in the screening process. This impairs the utility of virtual screening as a tool for drug discovery. Thus the development of a reliable and accurate scoring function has been the focus of many ongoing studies.
Force-field based linear interaction energy (LIE) methods have been widely employed to predict protein-ligand binding free energies with reasonable accuracy.13–18 Typical implementation of a LIE estimates the binding free energy by averaging the interaction energies of protein-ligand complexes extracted from molecular dynamics simulations. More recently this method has been successfully applied using only single-point energy minimizations along with high-accuracy continuum electrostatic solvent approaches (either Poisson-Boltzmann or Generalized-Born implicit solvation). Different interaction energy terms for van der Waals, electrostatic and solvation are scaled by empirical parameters that are optimized using a set of protein-ligand complexes. The greatest advantage of the LIE method compared to alchemical transformation methods (free energy perturbation and thermodynamic integration) is that it may be applied over thousands of ligands with dramatic differences in size and topology. In spite of this advantage, the application of the LIE method still suffers from the fact that the parameters may not be perfectly transferable across significant variations in ligand classes and functional groups, and over significant changes in binding sites.
Empirical scoring functions have been very useful for their simplicity and low computational cost. The derivation of most standard empirical scoring functions is based on the non-rigorous Kirkwood superposition approximation, namely that the binding affinity can be estimated by the addition of individual interaction terms. In many of these implementations, regression-based methods are applied to estimate the weights of the individual terms using experimentally derived configurations and binding affinities of a set of receptor-ligand complexes. ChemScore8, GOLD19 and AutoDock9 have been among the most popular empirical scoring functions. In spite of their success, poor transferability of parameters has been one of the major disadvantages of empirical scoring functions. As a general rule, the parameters that regulate the relative contributions of the individual terms are dependent on the training set used for the regression analysis. Several factors, including molecular surface area, hydrogen bonds, ligand rotational entropy, ionic interactions, hydrophobic interactions, and desolvation energies, were taken into account while constructing empirical scoring functions.8, 9, 19 The exploration and incorporation of other novel factors can also improve performance of empirical scoring approaches as experimental datasets grow larger and more diverse.20
For ligands that bind with reasonable affinity, the size of the ligand correlates directly with the protein-ligand binding affinity and thus it can be considered as a factor in the empirical scoring scheme. Kuntz et al. surveyed the correlation between the sizes and the binding free energies of a large number of ligands.21 They demonstrated that each non-hydrogen ligand atom contributed a maximum of ~ −1.5 kcal/mol to the protein-ligand binding free energy. The total binding free energy reached a limit of ~ −15 kcal/mol for larger number of heavy atoms per molecule. They suggested that the decrease in the average contribution per atom with increasing number of ligand atoms was due to the shielding of van der Waals and hydrophobic interactions. Hajduk et al. deconstructed 18 highly optimized drug leads into fragments and analyzed the consequent potency change.22 They observed a linear relationship between the binding affinity and the ligand molecular weight. Both of these studies suggest that as long as a given ligand is known to bind with a reasonable affinity, the binding free energy scales as a function of ligand size. In the spirit of these observations, we have developed a scoring function S1 based on a direct quantitative relationship between the binding free energy and the ligand size. For large ligands that bind strongly to a receptor, the contribution to ΔGbind from ligand atoms that are not strongly interacting with receptor atoms are significantly reduced due to shielding effects.21 Therefore ligand atoms that form close contacts are the major contributors to the binding free energy. Thus, in our derivation of S1, we considered only these “interacting” ligand atoms in determining the “effective size” of the ligand. We have demonstrated that this method is successful in removing the non-linearity in the binding-affinity ligand-size relationship. We have further extended S1 in order to construct an empirical pair potential-based scoring function S2, which was initially trained and validated on a diverse set of 259 protein-ligand complexes from the Ligand Protein Database (LPDB),23 and then additionally assessed in the CSARdock 2010 benchmark test (NCR HiQ set) (reference). In our pair interaction energy scheme S2, we classified and counted only the directly interacting protein-ligand atomic pairs and used them as variables in our derivation of binding free energy. S2 is entirely empirical in nature, which is different from the knowledge-based pair potentials. Our primary objective of this manuscript is to assess the performance of S1 and S2. In order to do this, we also compared S1 and S2 results to other variations of these schemes (S3, S2h and S3w) in order to further understand the origin of good or bad performance. For example, as a variation on S2, we have also constructed S2h, which considers the interactions among only the heavy atoms.
Many knowledge-based scoring functions are developed by systematic analyses of atomic interactions between adjacent receptor and ligand surfaces. Using the crystal structures of 38 protein-ligand complexes, Wallqvist et al. determined the statistical preferences of the atomic interactions between amino acids.24, 25 They constructed a model to estimate the binding free energy based on the classifications of the atomic pairs buried in the interfacial surface. Using the model, they could predict the binding free energies of 10 HIV protease complexes with an accuracy of ±1.5 kcal/mol. Verkhivker et al. derived a knowledge-based distance-dependent pair potential using 30 HIV-1, HIV-2 and SIV protein-ligand complexes and were able to achieve a good correlation between the experimental and predicted ΔGbind.26 DeWitte et al. developed an inter-atomic interaction-based scoring function using a large set of protein-ligand crystal structures.27 They incorporated this knowledge-based potential into a Metropolis Monte Carlo molecular growth algorithm, SMoG, for de novo ligand design. DrugScore and DrugScore(CSD)28 are the most widely used and validated distance dependent pair potential based scoring function for protein-ligand interactions. DrugScore has been successful in identifying native poses of ligands7 as well as in predicting their binding affinities28. As a control, we have compared our S1 and S2 scoring schemes to a distance-dependent pair potential S3 that was derived from the same protein-ligand dataset that was used for regression analysis to develop S1 and S2.
We have compared the performance of these scoring functions against our implementation of LIE for several steps of the virtual drug screening process. The parameters of the LIE, S1, S2, and S2h scoring functions demonstrate reasonable transferability in a five-fold cross-validation for both the prediction of binding geometries and free energies. The performance of S2 is significantly better than the performance of LIE for predicting the binding free energies of a diverse set of 259 protein-ligand complexes from LPDB. We have evaluated the ability of the scoring functions to discriminate the well-docked native-like poses from the mis-docked poses and calculated the discriminative power (DP) of the scoring functions to identify native poses. Finally, we have tested the scoring functions for their ability to distinguish active compounds from the non-active decoy compounds in a virtual screening process. We combined active and decoy ligands obtained from the directory of useful decoys (DUD)29 that are specific for representative proteins from four target classes. The results of this study demonstrate the usefulness of the S1, S2, and S2h scoring functions in various stages of virtual screening process, especially compared to the S3 and S3w variations.
2. Methods
2.1 Description of the data sets
259 protein-ligand complexes from the LPDB (LPDB259) are used for training and validation of the scoring functions for their ability to predict binding free energies. The scoring functions S1 and S2 are also examined using two new sets from the CSARdock 2010 benchmark test sets: NRC HiQ set1 consisting of 176 protein-ligand complexes and NRC HiQ set2 consisting of 167 protein-ligand complexes. LPDB160, a subset of LPDB259 consisting of 160 complexes, is used for the calculations of discriminative power. Both of the LPDB259 and LPDB160 datasets were randomly split into five separate groups for a five-fold cross-validation study (see Supplementary Materials).
2.2 CHARMM-based molecular docking method
Our approach to molecular docking uses the program CHARMM3, 30, 31 for an all-atom force field potential energy description of the protein-ligand complexes. CHARMM has been used extensively over the past two decades to model the structure and dynamics of biological macromolecules (proteins, DNA, RNA, lipids, small molecules, and solvent molecules), as well as for multi-scale modeling in structural biology32, and has been widely validated for numerous systems. A significant advantage of using CHARMM as an engine for molecular docking is that many different conformational searching strategies have been implemented, such as MD and Monte Carlo (MC) simulation techniques, which can be applied to the docking problem. The docking and the scoring methods are summarized in Figure 1. Docking consists of a sequence of independent trials that are composed of a large number of individual docking attempts (where a single conformation of a ligand is docked to the protein). An independent docking trial proceeds by generating a series of random initial ligand conformations, and then generating a series of random orientations that are docked to the protein binding site. In our docking method, a 3-D grid is used to describe the static conformation of the protein binding site, in which the interaction energy among 20 types of probe atoms is calculated for every point on the grid. The granularity of this grid is 1.0 Å. The flexible ligand is modeled with an all-atom representation including all hydrogen atoms. The CHARMm force field originally parameterized by Momany and Rone has been extended to describe ligands in the Ligand-Protein Database (LPDB) and was used to build potential energy functions for all ligands.6, 23 During a docking trial, random configurations of the flexible ligand are generated by running one thousand 2-fs time steps of MD at 1,000 K in vacuo. During this generation of alternative starting conformations, all electrostatic interactions are turned off to avoid favorable intermolecular electrostatic interactions, which lead to significant errors in docking. For each random conformation generated, random rigid-body rotations about the center of mass are used as the initial orientations in the grid representation of the binding site. The all-atom model of the ligand interacts with the potential energy of the grid using a soft-core repulsion term for both van der Waals (VDW) and electrostatic interactions. The soft-core repulsion term allows the ligand to penetrate into the interior of the protein with a relatively small energetic penalty. This lowers the energy of conformational transition barriers and facilitates a more efficient conformational search within the 3-D grid. MD simulated annealing is used, starting from the generated ligand conformations, to search for low energy conformations of the all-atom ligand on the grid. The heating phase consists of four thousand 2-fs MD steps heating from a temperature of 300–700 K. The cooling phase consists of ten thousand 2-fs MD steps from 700 K back down to 300 K. The potential energy is then minimized with steepest descent minimization of 1,000 steps. Then, the 3-D grid potential of the protein is removed and the all-atom representation of the rigid protein is restored. The potential energy in the all-atom protein-ligand representation is then minimized, fixing the coordinates of the protein, using the standard hard-core repulsion for both VDW and electrostatics with a distance-dependent dielectric function (Rdie). Other extensive details of the setup and protocol have been published previously.3, 33–35 Various components of the final minimized potential energy are employed to construct CHARMm-based scores for ranking the final ligand pose. Several of these CHARMm-based scoring functions have been previously assessed by Ferrara et al. and compared to other widely used potential functions.4 In this manuscript, we assess six CHARMm-based scores for geometry discrimination: Cdie(Tot) [total interaction energy with a constant dielectric], Rdie(Tot) [total interaction energy with a distance dependent dielectric], Rdie(Tot)+LigInt [Total Rdie interaction energy and the ligand internal energy (angles, bonds, torsions, impropers)], GBMV(Tot) [Total interaction energy including gbmv solvation term], LIE(Rdie) [α*(ΔVDW))+β(Δelec)], LIE(GBMV) [α(ΔVDW)+β(Δ(elec+gbmv)+γ(Nrot)]. The initial use and implementation of LIE(Rdie) and LIE(GBMV) has been described previously.36, 37
Figure 1.

Overview of CHARMM-based molecular docking protocol and scoring functions used for step 1 (geometry) and step 2 (binding free energy) discrimination.
2.3 Binding free energy calculation using scoring schemes (step 2 discrimination)
At the end of docking, the lowest energy poses of the protein-ligand complexes were scored by five scoring functions:
2.31. Linear Interaction Energy: LIE(GBMV)
Energy minimization of the protein-ligand complex was performed to construct a LIE score starting from each of the lowest energy poses. A Generalized Born using Molecular Volume (GBMV)38, 39 method was used to account for the solvent effect implicitly, which is the most accurate of the CHARMM Generalized Born implicit solvent methods.38, 39 Following the minimization with the GBMV implicit solvent, van der Waals (VDW), electrostatic (ELEC) and solvation (SOLV) energy components were calculated for the “free” and “bound” states from the single minimized configuration of the protein-ligand complex. A three-parameter form of the LIE13–18 scheme was used to calculate the binding free energy (ΔGbind) from these components:
| (1) |
The empirical scaling parameters α, β and γ were optimized to match the calculated ΔGbind with the experimental ΔGbind of the LPDB complexes. Nrot represents the number of rotatable bonds in the ligand.
2.3.2 S1 scoring function: based on effective ligand size
We developed S1 following the work of Kuntz et al.21 However, S1 is different from the function used by Kuntz et al. in several respects. They considered the total number of heavy atoms in the ligand. Instead, we counted the number of interacting ligand atoms (Nint) for each of the lowest energy protein-ligand complexes. We also included the light (hydrogen) atoms in the counting. A ligand atom was counted as “interacting” if any protein atom was found within a sphere of a constant radius (cutoff) from it. Then the binding free energy was estimated using the equation:
| (2) |
The empirical scaling parameter δ was optimized to match the calculated ΔGbind with the experimental ΔGbind of LPDB complexes. We note that S1 has a logarithmic form which was not used by Kuntz et al.21
2.3.3 S2 scoring function: an empirical regression-based pair potential
Interacting atom-pairs were counted for each of the lowest energy protein-ligand complexes. In an atom-pair, one atom belongs to the protein and the other atom belongs to the ligand. All possible atom-pairs were considered, using a very “naïve” atom typing strategy where each atomic atom only has one atom type. For example, there is only one atom type for hydrogen and there is no difference between a polar hydrogen and a non-polar hydrogen. This is in contrast with many other strategies that use complex atom typing. An atom-pair was counted as “interacting” if the distance between the two atoms was less than a cutoff value. The interacting atom-pairs were then classified in several groups. A scoring function S2 was constructed to derive ΔGbind by scaling the number of a particular atom-pair type with a weight and adding their contributions:
| (3) |
The empirical scaling parameters σ and λi (i = OH, CH, HH, NH, CC, ON, OO, OC, NC, other) were optimized to match the calculated ΔGbind with the experimental ΔGbind of the LPDB complexes. For example, NOH is the number of atom-pairs containing one O and one H and λOH is its scaling factor. Atom-pairs that did not match with the above-mentioned 9 types were naively counted as “other” pairs. “Other” pairs include interactions involving halogens (F, Cl, Br), phosphorus (P), and metals (Zn, Ca, Na). We selected these 9 types of atom-pairs considering their high frequencies of occurrence. The next most frequent pair types in order of frequency were: HF, NN, PC, CF, PN, and PO.
In previous approaches to empirical scoring functions, it has been very common to perform regression analysis to determine the weights for a series of protein-ligand complex descriptor terms20 (for example, VDW energy, electrostatic energy, hydrogen bonds, SASA descriptors, hydrophobicity, polar ligand surface, N rotatable bonds, etc), but less common to perform regression analysis to determine the weights of contact pairs. Some examples of widely used scoring functions that were developed using these common regression approaches20 include ChemScore8, GOLD19 and AutoDock9. However, in most previous contact pair potentials, it has been much more common to use a knowledge-based approach rather than a regression approach to weight the terms. To the best of our knowledge, the functional form of S2 is most similar to the previous work of Wallqvist et al. and DeWitte et al., except that both of these approaches utilized statistical preferences of the atomic pair interactions in their contact pair potential scores.24, 25, 27 S2 uses linear regression against experimental binding affinities to determine the weights, λpair, that should be applied to each contact pair, rather than statistical preferences. In the work of Wallqvist et al., the equivalent of the S2 weight λpair is determined by both knowledge-based statistical probability for a pair, Ppair, and two regression parameters (γ and δ), where λpair = γ + δ*ln(Ppair) where, the regression parameters γ and δ are also optimized to a set of protein-ligand complex structures to minimize the difference between predicted and experimental ΔGbind (see Wallqvist equation 6).24 In the work of Dewitte et al., there are no regression terms and the contact pairs are weighted only by a statistical probability for contact pairs.
2.3.4 S2h scoring function: an empirical regression-based pair potential using heavy atoms
Following S2, another scoring function S2h was constructed that involved the interactions between only the heavy atoms.
| (4) |
The constants φ and θ have similar meanings as in S2. The less-frequently occurring NN interactions are also included in this scheme to account for all the heavy atom interactions.
2.3.5 Determination of cutoff value
The determination of the cutoff value is a crucial step in developing the scoring functions S1, S2 and S2h. In the past, arbitrary values like 6 Å7 or 12 Å40 were used as cutoff for protein-ligand atomic interactions. Instead of adopting a particular cutoff value, we assessed the performance of S1, S2 and S2h at different cutoff values in order to identify the cutoff values that correspond to the best performance. For each cutoff, the parameters of the scoring functions were optimized and the performances were determined by the average unsigned errors (AUE) between the calculated and experimental ΔGbind for LPDB complexes. AUE was calculated by taking an average of the absolute values of the differences between calculated and experimental values (errors). AUE is a measure of the average deviation of a predicted value from the experimentally observed value. Thus, a low AUE reflects a high accuracy of the prediction. The lowest AUE was observed at a cutoff of 3.75 Å for S1, and this cutoff value was applied for S1 in the rest of this work. We note that the value of Nint in S1 steadily increases with the increment of the cutoff value until it reaches the limit of the “total number of ligand atoms”. The performance of S1 for predicting ΔGbind is optimal at shorter cutoff values. Obviously, using a cutoff value in S1 improves the fitting of ΔGbind as compared to counting the total number of ligand atoms, as done by Kuntz et al.21 Short contact distances (2.4 Å hydrophilic, 2.6–2.8 Å hydrophobic) have been employed in the past to count protein-ligand contacts in various implementations of the DOCK-contact score.41
For S2, the lowest AUE was observed at a longer cutoff of 5.25 Å. This cutoff value was applied for S2 in the rest of the work reported here. The lowest AUE was observed for S2h at a slightly higher cutoff value of 6.5 Å. The performance of scoring functions S1, S2, and S2h in terms of AUE are plotted as a function of cutoff distance in Figure S1 (Supplementary Material). However, we applied the same cutoff value of 5.25 Å for S2h as well. This should have an insignificant effect on the results due to the low variability of the performance of S2h with different cutoff values. It is interesting to note that our optimal cutoff of 5.25 Å for S2 is similar to the cutoff of 5.0 Å used by DeWitte et al. in their knowledge-based contact potential in SMoG.27 DeWitte et al. proposed that by using an interaction radius of 5.0 Å (which is similar to the correlation length of solvent ordering) the statistical probabilities of specific contacts will include the effect of an average over the contribution of solvation entropy to ΔGbind.27 In the spirit of this argument, it is also possible that our regression weighted terms λpair for S2 may implicitly incorporate some important contributions from solvation effects.
2.3.6 S3 scoring function: a distance dependent pair potential
The primary objective of considering the scoring function S3, was to compare the regression-based approach of S2 against the performance of a distance-dependent pair potential using the exact same “naïve” atom typing with the exact same set of pairs, derived from the same dataset of protein-ligand complexes. Interacting atom pairs were counted as a function of distance for each of the lowest energy protein-ligand complexes in LPDB259, to construct radial pair distribution functions gpair(r) using a bin size of 0.25 Å. Atom pairs that were used to construct S2 were considered out to a distance of 6 Å. The radial pair distribution functions are converted to a distance dependent pair potential, Wpair(r), by taking the natural log of the ratio of the normalized pair distribution function gpair(r) for a given pair to the normalized pair distribution function for all pairs gtotal(r),
| (5) |
This formulation for Wpair(r) is similar to the one that was used to construct the protein-ligand scoring function DrugScore.7 Many others have taken similar approaches to construct knowledge-based distance dependent pair potentials.26, 42, 43 A scoring function S3 was constructed to approximate ΔGbind by adding the distance dependent score of each pair type Σpair(Wpair(r)). An atom-pair was counted as “interacting” if the distance between the two atoms was less than a cutoff distance of 6 Å:
| (6) |
Atom pairs that did not match with the above mentioned 9 types were naively counted as “other” pairs. Unlike S2, the “other” atom pairs were ignored, as it was assumed that the average gpair(r) of multiple atom types averaged together may introduce error. Distance dependent pair potentials Wpair(r) were implemented as look-up tables using a bin size of 0.25 Å (Table S1 Supplementary Material). For unfavorable short-range contacts, each distance dependent pair potential Wpair(r) was truncated at different short-range cutoffs to have a maximum penalty of 2.0 kcal/mol so energetic penalties for short-range contacts are not dominated by sparse data. All docking poses assessed by this scoring function are assumed to be energy minimized by a hard-core CHARMM-based molecular mechanics energy function, so significantly unfavorable short-range repulsions should be minimal. We carefully note that the good performance of other similar approaches to construct knowledge-based distance dependent pair potentials in the literature is due to the use of complex atom typing rather than our naïve atom typing, and it is expected that the naïve atom typing may lead to a reduction in performance.26, 42, 43
2.3.7 S3w scoring function: a distance dependent pair potential including regression weighting terms
Interacting atom-pairs were counted as a function of distance for each of the lowest energy protein-ligand complexes in LPDB259, to construct radial pair distribution functions gpair(r), and corresponding distance dependent pair potentials Wpair(r) as above for S3. A very similar scoring function S3w was constructed to approximate ΔGbind by adding the distance dependent score of each pair type, but also including a regression-based weight λ for each distance dependent pair potential Wpair(r), that is optimized by fitting the binding free energies, similar to S2:
| (7) |
The empirical scaling parameters λi (i = OH, CH, HH, NH, CC, ON, OO, OC, NC) were optimized to match the calculated ΔGbind with the experimental ΔGbind of the LPDB complexes (see Table S2 Supplementary Material).
2.4 Calculation of discriminative power (step 1 discrimination for binding geometry)
The scoring functions were evaluated for their abilities to discriminate between the near native ligand poses (≤ 2 Å) and the misdocked decoy poses, following the previous work by Ferrara et al.4 160 protein-ligand complexes were obtained from LPDB (LPDB160) along with corresponding low-energy cluster representatives for misdocked and docked ligand poses which were energy minimized using CHARMm and Rdie(Tot). Root Mean Square Deviation (RMSD) was calculated for each pose to quantify its resemblance to the native crystal structure, and the RMSD values for these docked and misdocked decoys range from 0.1 to greater than 40 Å RMSD from the native pose. The poses with RMSD ≤ 2 Å were categorized as well-docked poses and poses with RMSD ≥ 4 Å were categorized as misdocked poses. The Z score4, 34 was calculated for each pose using
| (8) |
where E is the binding energy of the protein-ligand complex, Ē is the mean and σ is the standard deviation of the binding energy distribution of either the well-docked or misdocked conformations. The Discriminative Power (DP)4 is defined as:
| (9) |
where i represents a particular protein-ligand complex and N is the total number of complexes (=160). and are the Z scores of the lowest energy conformer among the well-docked and misdocked conformations respectively. fi is the fraction of the well-docked conformations with Z scores lower than those of the misdocked conformations. The scoring functions were applied to calculate the DP using the poses generated by CHARMm. A more negative value of the DP implies better discriminative power of the scoring function.
2.5 Calculation of enrichment factors and ROC curves (step 2 discrimination)
Several scoring functions were evaluated for their abilities to separate actives from a large set of decoys in a virtual screening process. Target specific sets of active and decoy ligands were obtained from the “Directory of Useful Decoys” (DUD)29 for for eight protein targets: androgen receptor, glutocorticoid receptor, thrombin, trypsin, cox1, cox2, p38α MAP kinase, and vegfr2 kinase. The DUD target specific decoy sets contain decoys that are very challenging for enrichment as decoys were selected based on similarity to actives in both physical properties (molecular weight, number of hydrogen bond acceptors, number of hydrogen bond donors, number of rotatable bonds and logP), as well as similarity of important functional groups. Therefore, these target specific DUD decoy sets are more challenging for enrichment than other random large databases. The p38a complex 1ouk was recently shown to be able to provide a high docking accuracy for p38α ligands compared to other p38a receptor conformations,36 and most results presented in this manuscript for docking to the p38a 1ouk crystal structure conformation. Results are also presented for vgfr2 kinase ligands docking to the 2oh4 conformation, as the DUD receptor conformation was outdated and did not include adequate electron density for the activation loop.
During the docking process, the ligand was flexible but the protein was kept rigid. For each ligand’s top-ranked pose, ΔGbind was calculated using scoring functions with parameters that were optimized using the LPDB complexes. Results for step 2 discrimination were compared by calculating the scores [LIE(GBMV), S1, S2, S2h, S3, S3w ] for the top-scoring pose identified by LIE(GBMV), as this latter scoring function had the optimal performance in step 1 discrimination. The ligands were sorted according to their ranks (predicted ΔGbind). The Enrichment Factors (EF) at different % of database was reported and the Receiver Operating Characteristic (ROC) curves were constructed following44, 45:
| (10) |
| (11) |
where Actn is the number of true high affinity actives and Decn is the number of decoys in the top n ranked compounds. Acttotal (=230) is the total number of high affinity actives and Dectotal (=8942) is the total number of decoys in the dataset. The ROC curve is a two-dimensional rendering of the performance of a scoring function in separating actives from decoys. A scoring function that randomly assigns a score to a ligand without discriminating between actives and decoys should coincide with the diagonal line. On the other hand, a scoring function that favors the actives over decoys should have higher true positive rate than false positive rate near the beginning of the database, producing a curve above the diagonal line. For the sake of comparison, the performance of a scoring function in ROC curve can be condensed in one scalar value, the area under the ROC curve (AUC). The value of AUC can be anything between 0 and 1.0 because it measures an area under a unit square. AUC of a perfectly random scoring function should be 0.5. A higher value of AUC represents better enrichment of actives over decoys, such that excellent performance is anything greater than 0.9 and modest performance is anything between 0.6 and 0.5.
2.6 Cross validation
The robustness of the parameters of the scoring functions is examined by a K-fold cross validation scheme. Considering the small size of the dataset, it is randomly divided into 5 subsets: group 1, 2, 3, 4 and 5. In trial 1, the scoring function parameters are optimized with a training set consisting of the complexes in groups 2, 3, 4, and 5. Then these optimized parameters are examined with set 1, which was not included in their training set. This process was repeated 5 times in a similar fashion to validate all 5 sets. This analysis was performed for both ΔGbind predictability as well as discriminative power. This cross-validation procedure was performed for LIE(GBMV), S1, S2, and S2h, but was not performed for S3 and S3w due to poor performance.
3. Results and Discussion
3.1 Binding free energy prediction
Table 1 shows the performance of the optimized scoring functions in predicting the experimental ΔGbind for the LPDB259 dataset. The average unsigned error (AUE) in kcal/mol and the best fit linear correlation coefficient (R2) were calculated for each fit (Table 1). Table 2 shows the optimal parameters from least-squares fitting, and the predicted and experimental values of ΔGbind are compared in Figure 2. Clearly, both in terms of AUE and R2, the best overall performance is obtained with S2, the empirical regression-based pair potential. The worst overall performance was for S3 and S3w, the two distance dependent pair potentials. In terms of AUE, the scoring functions S1, S2, and S2h all outperformed LIE(GBMV). LIE(GBMV) did have a slightly better linear correlation with the experimental data than S1, but S1 had a lower AUE. The fact that the 1 parameter scheme S1 performs better than one of the most accurate scoring functions, LIE, is quite surprising. This strongly suggests that the functional form of S1 (Equation 2) correctly describes the relationship between ΔGbind and the effective size of the ligand. The ligand size in the training set ranges from 17 to 116 atoms. However, it is interesting that the scatter plot of experimental vs. S1-predicted ΔGbind does not demonstrate a non-linearity for larger ligands as observed by Kuntz et al.21 While Kuntz et al. compared experimental ΔGbind with the number of heavy atoms in the ligand, S1 differs from their scheme in three respects: i) S1 considers only the number of interacting ligand atoms (Nint) instead of all ligand atoms; ii) S1 includes the light hydrogen atoms in addition to the heavy atoms; iii) S1 has a logarithmic form.
Table 1.
Comparison of predicted and experimental ΔGbind for different scoring functions optimized to the LPDB259 dataset.
| Scoring Function | AUE (kcal/mol) | R2 |
|---|---|---|
| LIE (GBMV) | 2.86 | 0.32 |
| S1 | 2.26 | 0.29 |
| S2 | 2.02 | 0.44 |
| S2h | 2.17 | 0.35 |
| S3 | 4.16 | 0.25 |
| S3w | 3.64 | 0.27 |
Table 2.
Scoring function parameters optimized to match experimental ΔGbind for LPDB259, LPDB160 and the five cross-validation subgroups of LPDB259.
| LPDB259 | NRC HiQ set1 | NRC HiQ set2 | LPDB160 | LPDB259 | LPDB259 | LPDB259 | LPDB259 | LPDB259 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| all | all | all | all | group1 | group2 | group3 | group4 | group5 | ||
| LIE | α | 0.2058 | 0.2444 | 0.2092 | 0.2157 | 0.2038 | 0.1995 | 0.1974 | ||
| β | 0.0517 | 0.0504 | 0.0501 | 0.0564 | 0.0505 | 0.0478 | 0.0533 | |||
| γ | 0.0531 | 0.1701 | 0.0564 | 0.0794 | 0.0470 | 0.0440 | 0.0274 | |||
|
| ||||||||||
| S1 | δ | −2.3585 | −2.3991 | −2.4137 | −2.3891 | −2.3713 | −2.3371 | −2.3667 | −2.3587 | −2.3594 |
|
| ||||||||||
| S2 | σ | −4.0731 | −4.2933 | −3.6214 | −4.7736 | −3.7644 | −3.9780 | −4.3851 | −4.0368 | −4.1141 |
| λOH | 0.0182 | 0.0210 | 0.0417 | 0.0219 | 0.0157 | 0.0199 | 0.0213 | 0.0143 | 0.0189 | |
| λCH | −0.0140 | −0.0124 | −0.0273 | −0.0031 | −0.0092 | −0.0233 | −0.0161 | −0.0131 | −0.0087 | |
| λHH | −0.0013 | 0.0022 | 0.0153 | −0.0051 | −0.0035 | 0.0023 | 0.0003 | −0.0024 | −0.0032 | |
| λNH | 0.0320 | 0.0299 | −0.0348 | 0.0188 | 0.0336 | 0.0401 | 0.0297 | 0.0321 | 0.0251 | |
| λCC | 0.0150 | 0.0042 | 0.0218 | −0.0196 | 0.0053 | 0.0340 | 0.0164 | 0.0156 | 0.0057 | |
| λON | −0.1067 | −0.0217 | −0.0867 | −0.1037 | −0.1021 | −0.1090 | −0.0940 | −0.0979 | −0.1264 | |
| λOO | −0.1130 | −0.0075 | −0.0668 | −0.1777 | −0.1414 | −0.1204 | −0.1241 | −0.0871 | −0.0908 | |
| λOC | 0.0145 | −0.0212 | −0.0252 | 0.0371 | 0.0208 | 0.0164 | 0.0113 | 0.0184 | 0.0063 | |
| λNC | −0.0173 | −0.0339 | 0.0338 | 0.0023 | −0.0249 | −0.0254 | −0.0084 | −0.0285 | −0.0024 | |
| λother | −0.0219 | −0.0283 | −0.0069 | −0.0107 | −0.0198 | −0.0294 | −0.0246 | −0.0190 | −0.0192 | |
|
| ||||||||||
| S2h | φ | −4.5527 | −4.7290 | −4.3205 | −4.8203 | −4.8011 | −4.3219 | −4.4595 | ||
| λCC | −0.0320 | −0.0414 | −0.0331 | −0.0342 | −0.0307 | −0.0300 | −0.0327 | |||
| λON | −0.0920 | −0.0761 | −0.0907 | −0.1040 | −0.0679 | −0.0812 | −0.1192 | |||
| λOO | −0.1217 | −0.1755 | −0.1510 | −0.1116 | −0.1335 | −0.1098 | −0.1012 | |||
| λOC | 0.0368 | 0.0587 | 0.0424 | 0.0392 | 0.0346 | 0.0356 | 0.0319 | |||
| λNC | 0.0412 | 0.0417 | 0.0359 | 0.0493 | 0.0356 | 0.0271 | 0.0626 | |||
| λNN | −0.0995 | −0.0959 | −0.0818 | −0.0914 | −0.0788 | −0.1047 | −0.1594 | |||
Figure 2.
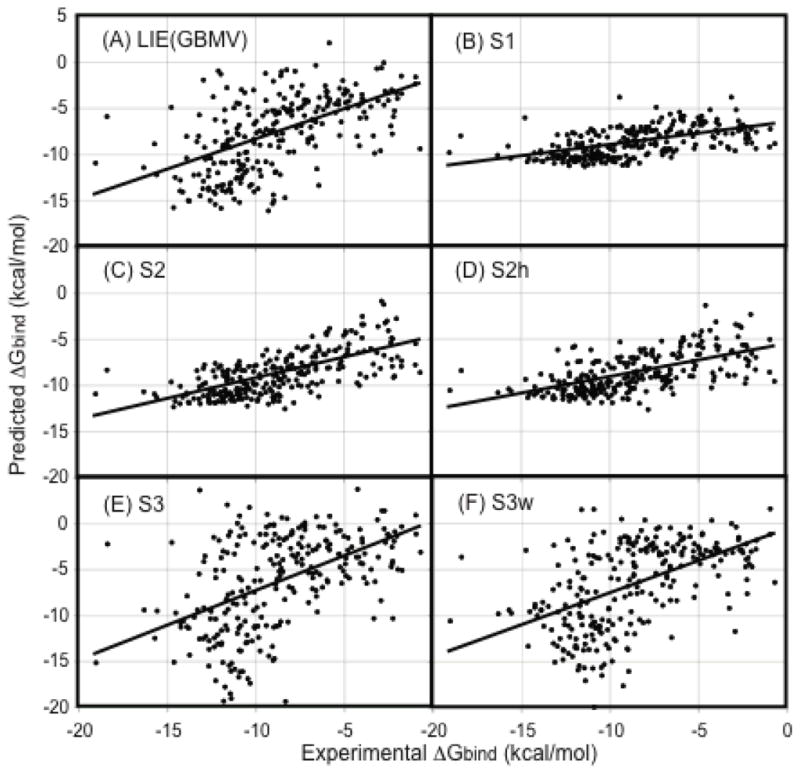
Comparison of predicted and experimental ΔGbind (kcal/mol) for six scoring functions optimized for LPDB259: (A) LIE(GBMV), (B) S1, (C) S2, (D) S2h, (E) S3, (F) S3w.
S2 performs the best among the scoring functions, with an AUE of 2.02 kcal/mol and R2 of 0.44. This performance is comparable with results obtained by Ferrara et al. who estimated the ability of 9 scoring functions to predict experimental ΔGbind.4 They used a set of 189 LPDB complexes (set 1a) and concluded that the performance of ChemScore8 was the best, with an R2 value of 0.51. However, the R2 value was 0.43 when 69 complexes used to calibrate ChemScore and Autodock were removed from the set (set 2). The success of S2 strongly suggests its ability to predict the experimental ΔGbind of protein-ligand complexes of wide diversity. In Figure 2, two high-affinity outlier complexes can be seen for all of the scoring functions. These two complexes are 7cpa (carboxypeptidase A) and 1stp (streptavidin), both of which are very high affinity complexes that are known to have charge transfer polarizability. When these two complexes are removed from the fitting and comparison, S2 has an AUE of 1.95 kcal/mol and an R2 of 0.46 for the remaining 257 diverse LPDB complexes.
The performance of S1 for predicting binding affinity is impressive considering the fact that it has only 1 adjustable parameter compared to 11 in S2 and 7 in S2h. A comparison among the λi and θi for S2 and S2h respectively also suggests the relative importance of the atom-pair types that reflect the strength of physical interactions (Table 2). Most importantly, the OO, ON and NN atom-pair types were the dominant attractive terms. These reflect the strong electrostatic interactions between the polarized oxygen and nitrogen atoms. These interactions are known to provide strong enthalpic contributions to the protein-ligand interactions. For S2, λNH (= 0.0320) had the highest positive value and λOO (=−0.1130) had the highest negative value, indicating the most repulsive and most attractive terms contributing to the estimation of ΔGbind. These weights λNH and λOO for S2 can also be rationalized from features of the S3 distance dependent pair potentials: WNH(r) did not exhibit any strong energetically favorable minima, while WOO(r) exhibited a strong −1.6 kcal/mol minima at 3.0 Å, and a second −0.4 kcal/mol minima at 5.0 Å. For S2h, θNC (=0.0412) had the highest positive value and θOO (= −0.1217) had the highest negative value. Again, the S2h weight θNC can also be rationalized from the S3 distance dependent pair potential: WNC(r) did not exhibit any strong energetically favorable minima, but is just slightly more favorable at distances from 5.25 to 5.75 Å However, in S2 it is noteworthy that the total contribution of each type of atom-pair interaction also depends on the number of occurrences (NOO, etc.). Understanding the relative importance of the different atom-pair types is further complicated by the large cutoff value of 5.25 Å. Currently S2 and S2h treat the short-ranged and long-ranged atom-pair interactions equally, and do not incorporate distance dependence.
The scoring functions S1 and S2 are applied to two new datasets NRC HiQ set1 and NRC HiQ set2 to examine their abilities to predict ΔGbind. One of the desirable criteria in a training set is a poor correlation between the experimental ΔGbind and the MW of the ligand because it ensures that the good performance of a scoring function is not a direct consequence of the fact that the scoring function assigns a higher score for a ligand with high MW. The R2 values of experimental ΔGbind vs the MW of the ligand were 0.17 and 0.36 for set1 and set2, respectively. These reasonably low R2 values should make tests on set 1 more difficult than set 2. The scoring function parameters were reoptimized for each dataset using least square fits. Table 3 shows the performances of the scoring functions in terms of AUE and R2 and Table 2 shows the optimized parameters. Figure 3A and B show the correlation between the predicted and experimental ΔGbind for NRC HiQ set1 and set2, respectively. Although, the performance of S1 was only average, S2 performed quite well with R2 of 0.44 and 0.54 for set1 and set2, respectively. This illustrates the fact that S2 can be trained using a completely new, diverse and difficult dataset to predict ΔGbind with good accuracy. Later in the manuscript, we demonstrate that these parameter fits also exhibit reasonable transferability.
Table 3.
Comparison of predicted and experimental ΔGbind for S1 and S2 optimized to NRC HiQ set1 and set2 dataset, with parameters transferred from LPDB259 and parameters transferred between NRC HiQ set1 and set2. All statistical metrics for the CSARdock 2010 benchmark are shown, Pearson R2, Spearman ρ, Kendal τ, average unsigned error (AUE), median absolute error (MAE), and root mean squared error (RMSE).
| Scoring Function | Dataset Used for Parameter Optimization | Calc. NRC HiQ Dataset | Parameter Description | Pearson R2 | Spearman ρ | Kendall τ | Average Unsigned Error (AUE) kcal/mol | Median Absolute Error (MAE) kcal/mol | Root Mean Squared Error (RMSE) kcal/mol |
|---|---|---|---|---|---|---|---|---|---|
| S1 | LPDB dataset | set1 | transferred | 0.24 | 0.48 | 0.34 | 2.15 | 1.77 | 2.72 |
| S1 | NRC HiQ set1 | set1 | optimized | 0.24 | 0.48 | 0.34 | 2.15 | 1.83 | 2.71 |
| S1 | NRC HiQ set2 | set1 | transferred | 0.24 | 0.48 | 0.34 | 2.15 | 1.79 | 2.71 |
|
| |||||||||
| S1 | LPDB dataset | set2 | transferred | 0.39 | 0.61 | 0.43 | 1.87 | 1.48 | 2.38 |
| S1 | NRC HiQ set2 | set2 | optimized | 0.39 | 0.61 | 0.43 | 1.86 | 1.47 | 2.39 |
| S1 | NRC HiQ set1 | set2 | transferred | 0.39 | 0.61 | 0.43 | 1.86 | 1.42 | 2.39 |
|
| |||||||||
| S2 | LPDB dataset | set1 | transferred | 0.17 | 0.43 | 0.30 | 2.30 | 1.78 | 2.91 |
| S2 | NRC HiQ set1 | set1 | optimized | 0.44 | 0.69 | 0.50 | 1.72 | 1.33 | 2.33 |
| S2 | NRC HiQ set2 | set1 | transferred | 0.30 | 0.58 | 0.41 | 2.18 | 1.90 | 2.70 |
|
| |||||||||
| S2 | LPDB dataset | set2 | transferred | 0.38 | 0.61 | 0.43 | 1.93 | 1.69 | 2.38 |
| S2 | NRC HiQ set2 | set2 | optimized | 0.54 | 0.73 | 0.53 | 1.57 | 1.32 | 2.02 |
| S2 | NRC HiQ set1 | set2 | transferred | 0.47 | 0.71 | 0.51 | 1.78 | 1.66 | 2.20 |
Figure 3.
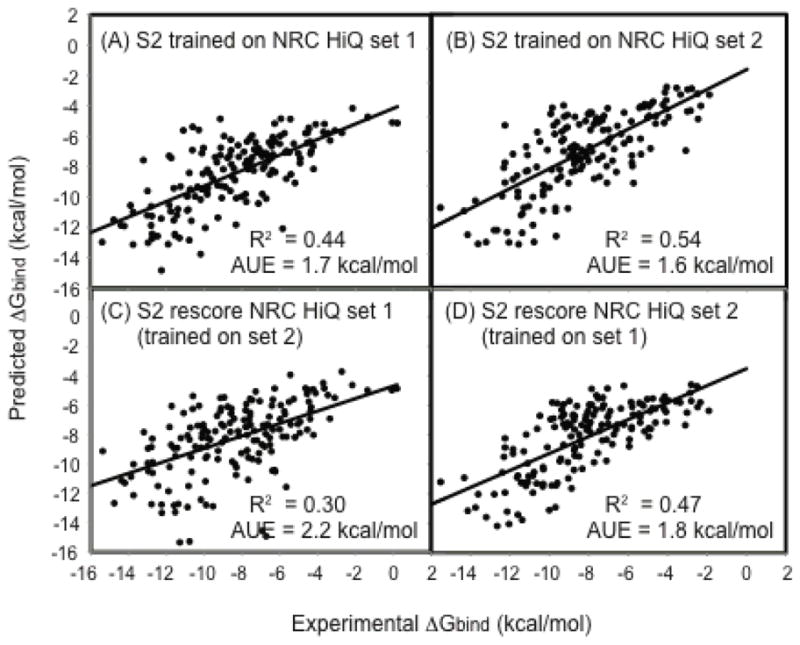
Comparison of predicted and experimental ΔGbind (kcal/mol) for the S2 scoring function for NRC HiQ data sets. (A) S2 parameters optimized on HiQ set 1 (B) S2 parameters optimized on NRC HiQ set 2 (C) S2 rescore set 1 (with parameters optimized from set 2) (D) S2 rescore set 2 (with parameters optimized from set 1).
3.2 Discrimination of well-docked and misdocked poses
In this section we analyze the abilities of the scoring functions in discriminating native-like poses from decoy poses. The discriminative power (DP) of each scoring function is calculated using LPDB160, a subset of LPDB259. Table 4 shows the calculated DP over the LPDB160 set using the parameters optimized from LPDB259. For the CHARMm-based scores it was found that Rdie(Tot)+LigInt < Rdie(Tot) < Cdie(Tot), which is in qualitative and near quantitative agreement with the Ferrara et al. study.4 Compared to these three CHARMm-based scores, both LIE variations were found to have improved discriminative power, with LIE(GBMV) having the best DP (most negative). In comparison, applying the equally weighted GBMV term to the total interaction energy [GBMV(Tot)] has significantly worse discriminative power than using GBMV with the LIE weights. Figure 4A shows the DP of the various CHARMM-based scores plotted as a function of ligand molecular weight (MW). This comparison shows that the improved performace of Rdie(Tot) over Cdie(Tot) or GBMV(Tot) holds up over the entire MW range. In comparing Rdie(Tot) to Rdie(Tot)+LigInt, the largest improvement in DP for including the internal ligand internal energy (angles, bonds, torsions, impropers) is in the higher molecular weight range, MW 400–1000, where the larger number of degrees of freedom can result in poses that have favorable interaction energies but with more strained internal geometry. In general, LIE(GBMV) has an improved DP over the entire MW range compared to LIE(Rdie), but the most significant feature of this difference is over the range MW 200–600, where there is a significantly improved average DP. Figure 4B compares LIE(GBMV) with three different values for the electrostatic parameter β while the van der Waals parameter was fixed at α=0.20. This shows that the LIE(GBMV) fit to the LPDB259 has an improved DP over the entire MW range compared to using equally weighted VDW and electrostatics (β=0.20) or using only the VDW interaction energy (b=0.00). Detailed examination of the DP of individual complexes showed that the differences in average DP over the dataset are not from individual complexes that dominate the average (e.g., as might occur if for some reason DP of an individual complex could improve from −6 to −12).
Table 4.
Discriminative power (DP) for the LPDB160 test set of scoring functions S1, S2, S2h, S3, S3w, trained on LPDB259, compared to various CHARMm based scoring functions: Cdie(Tot) [total interaction energy with a constant dielectric], Rdie(Tot) [total interaction energy with a distance dependent dielectric], Rdie(Tot)+LigInt [Total Rdie interaction energy and the ligand internal energy (angles, bonds, torsions, impropers)], GBMV(Tot) [Total interaction energy including gbmv solvation term], LIE(Rdie) [α*(ΔVDW))+β(Δelec)], LIE(GBMV) [α(ΔVDW)+β(Δ(elec+gbmv)+γ(Nrot)].
| Scoring Function | DP |
|---|---|
| Cdie(Tot) | −0.57 |
| Rdie(Tot) | −1.21 |
| Rdie(Tot)+LigInt | −1.40 |
| GBMV(Tot) | −0.53 |
|
| |
| LIE (Rdie) | −1.53 |
| LIE (GBMV) | −1.62 |
|
| |
| S1 | −0.12 |
| S2 | −1.64 |
| S2h | −1.52 |
| S3 | −0.19 |
| S3w | −0.18 |
Figure 4.
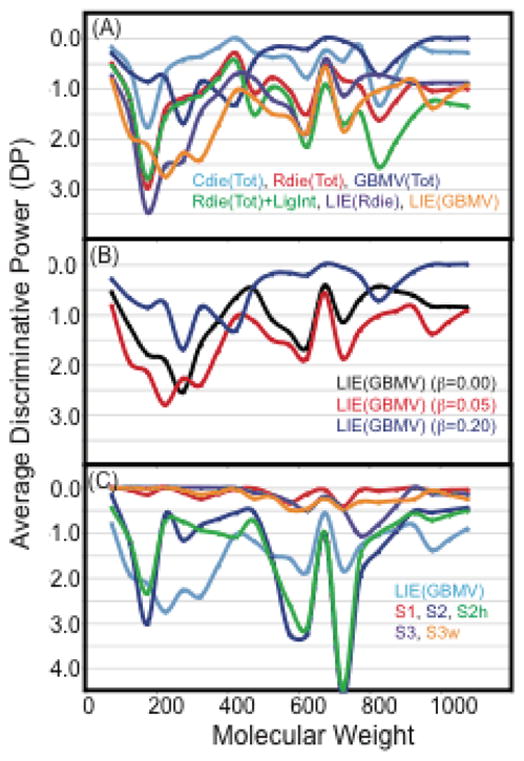
Average discriminative power (DP) of various scoring functions as a function of molecular weight. DP has been averaged over LPDB entries using a 50 dalton window of ligand size, and interpolated for windows with less than three LPDB entries to produce a smooth plot. (A) DP of several CHARMM-based scoring functions compared to LIE(Rdie) and LIE(GBMV) fit to the LPDB259. (B) DP of LIE(GBMV) with three different values of the electrostatic parameter β, while the van der Waals parameter is fixed at α=0.20. (C) DP of LIE(GBMV), S1, S2, S2h, S3, S3w all fit to LPDB259.
In comparison to the CHARMm-based scores, S2 and S2h have very good average discriminative power (Table 4), but S1, S3 and S3w all had very poor ability to predict native-like binding poses. In comparing the performance of LIE(GBMV) with S2 and S2h over the entire molecular weight range (Figure 4C), it is clear that LIE(GBMV) performs better in the lower MW range, 200–500, while S2 and S2h perform better in the higher MW range, 500–800. This may partially be rationalized by the fact that the greater number of total protein-ligand interactions in the higher molecular weight range may be easier for the S2 and S2h schemes to discriminate native from non-native, because strongly weighed atom pairs may occur numerous times (multiple hydrogen bonds in for example peptidomimetic inhibitors). In contrast, using the LIE(GBMV) it is possible for a decoy conformation with a greater VDW interaction energy and fewer hydrogen bonds to score better. In examining several peptidomimetic ligand complexes in the MW range of 500–800, there are several complexes where the the two scoring functions have huge differences in DP.. For example, [1ppk, 1epp, 1hih, 4phv, 1hps, 9hvp] have LIE(GBMV) DP values of [0.0, −0.7, −1.8, −1.9, −3.0, −1.5] while corresponding values from S2 are [−5.3, −5.6, −15.2 −8.2, −16.1, and −12.1]. These large improvements in DP seem to result from strong weights on pairs involving numerous peptidomimetic ligand hydrogen bonds. Another factor that may contribute to the improved performance of S2 and S2h over LIE(GBMV) is the neglect of internal ligand energy (angles, bonds, torsions, impropers) in the LIE(GBMV) score.
The performance of S1 was poor for prediction of binding pose geometry, which can be partially rationalized by the fact that the number of close contacts (distance < 3.75 Å) for low energy, non-native ligand poses were found to be similar to the number for native poses. However, we have shown that this metric of close contacts has a greater power to discriminate between different ligands in an enrichment study (see below). We attribute the poor performance of our S3 and S3w scoring functions to the fact that our dataset is very small. Previous work on the performance of DrugScore has shown that sparse datasets result in lower predictive power using this formalism, and the developers have demonstrated impressive performance improvements with newer and very large datasets of protein-ligand complexes.28 However, we have also demonstrated that the S2 functional form was able to retrieve more predictive power with the same sparse dataset. The good performance of S2 compared to LIE(Rdie) and LIE(GBMV) was not expected, especially since there is no direct incorporation of any implicit solvation information. However, it is likely that some empirical information regarding solvation is contained in the fit for the parameters.
The calculation of DP using the parameters optimized with LPDB259 as well as with LPDB160 are shown in Table 5. It is interesting that the DP of S2 and S2h were slightly better with the parameters optimized with LPDB259 than with LPDB160. This suggests that, for these schemes, the parameters that are optimized to reproduce the ΔGbind might not be the best parameters for DP calculation. In this particular case, the parameters optimized to reproduce the ΔGbind of a larger data set (LPDB259) might produce better DP for a subset (LPDB160). This was also true for LIE, but there was not much difference between the two sets of optimized LIE α and β parameters. However, the best parameters for LPDB259 (α=0.2058, β=0.0517) give a slightly better DP than the best parameters for LPDB160 (α=0.2444, β=0.0504). The largest difference in the LIE fit to LPDB259 and LPDB160 was in the LIE parameter γ, which does not affect the calculation of DP, as this parameter only contributes to a step 2 discrimination between two ligands with different numbers of rotatable bonds.
Table 5.
Calculated discriminative power (DP) of scoring functions for LPDB160 after being trained on LPDB259 and LPDB160.
| Scoring Functions | LPDB259
|
LPDB160
|
||||
|---|---|---|---|---|---|---|
| Training | Training | Validation | Training | Training | Validation | |
| AUE (kcal/mol) | R2 | DP | AUE (kcal/mol) | R2 | DP | |
| LIE (GBMV) | 2.86 | 0.32 | −1.62 | 2.45 | 0.44 | −1.58 |
| S1 | 2.26 | 0.29 | −0.12 | 1.96 | 0.41 | −0.12 |
| S2 | 2.02 | 0.44 | −1.64 | 1.77 | 0.52 | −1.52 |
| S2h | 2.17 | 0.35 | −1.52 | 1.81 | 0.49 | −1.38 |
3.3 Cross-validation and parameter transferability
LPDB259 is used to cross validate the ability of the scoring functions to predict ΔGbind. Table 6 summarizes the results of this analysis. In general, all the scoring functions performed well when applied to the validation set using the parameters optimized using the training set, demonstrating the robustness of the parameters. The average performance of the 5 trials was only slightly worse than the performance when optimizing the parameters with the full dataset. During cross-validation the optimized LIE parameters for the 5 trials remained quite close to (α=0.20, β=0.05), and the parameter γ had the widest variation, from 0.056 to 0.027 (Table 2). Of the 11 parameters for S2, weights λHH and λCH and λCC are the least robust (λHH even changes sign), but they also have some of the smallest values. The lack of robustness for λHH may also be partially rationalized from features of the S3 distance dependent pair potentials: WHH(r) does exhibit a moderately strong 1.0 kcal/mol minimum at 2.5 Å, but this pair potential trails off to zero by 4.0 Å. Thus, the observed variation in λHH across training sets may reflect noise due to the long S2 cutoff distance (5.25 Å), and a shorter cutoff distance for this pair may improve performance. In comparison, the S2h parameters are more robust presumably because the heavy atom contacts typically have minima in the range of 4.0 to 5.25 Å (Figure S2 Supplementary materials).
Table 6.
Binding free energy prediction from cross validation using LPDB259 after training on five random cross-validation subgroups of LPDB259.*
| Trial | LIE(GBMV)
|
S1
|
S2
|
S2h
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Training | Validation | Training | Validation | Training | Validation | Training | Validation | |||||||||
| AUE | R2 | AUE | R2 | AUE | R2 | AUE | R2 | AUE | R2 | AUE | R2 | AUE | R2 | AUE | R2 | |
| all | 2.86 | 0.32 | - | - | 2.26 | 0.29 | - | - | 2.02 | 0.44 | - | - | 2.17 | 0.35 | - | - |
|
| ||||||||||||||||
| 1 | 2.85 | 0.33 | 3.01 | 0.31 | 2.26 | 0.29 | 2.24 | 0.29 | 1.97 | 0.46 | 2.19 | 0.35 | 2.11 | 0.39 | 2.42 | 0.22 |
| 2 | 2.89 | 0.30 | 2.74 | 0.43 | 2.32 | 0.27 | 2.10 | 0.45 | 2.02 | 0.47 | 2.24 | 0.30 | 2.20 | 0.34 | 2.16 | 0.42 |
| 3 | 2.91 | 0.33 | 2.70 | 0.30 | 2.28 | 0.29 | 2.17 | 0.31 | 2.07 | 0.43 | 2.02 | 0.44 | 2.24 | 0.33 | 1.98 | 0.42 |
| 4 | 2.83 | 0.36 | 3.06 | 0.17 | 2.30 | 0.32 | 2.11 | 0.18 | 2.09 | 0.43 | 1.78 | 0.45 | 2.24 | 0.34 | 1.87 | 0.35 |
| 5 | 2.84 | 0.31 | 2.99 | 0.37 | 2.15 | 0.29 | 2.69 | 0.29 | 1.90 | 0.45 | 2.56 | 0.38 | 2.02 | 0.39 | 2.85 | 0.23 |
|
| ||||||||||||||||
| Avg | 2.86 | 0.33 | 2.90 | 0.32 | 2.26 | 0.29 | 2.26 | 0.30 | 2.01 | 0.45 | 2.16 | 0.38 | 2.16 | 0.36 | 2.26 | 0.33 |
AUE is in units of kcal/mol.
LPDB160 is used for the cross validation of the scoring functions for discriminative power. Table 7 summarizes the results of this analysis. The scoring functions retain their discriminative power when the parameters optimized with the training set are applied to the validation set. The average DP of the 5 trials is comparable to the DP obtained with the parameters optimized with the full set. This demonstrates that the scoring function parameters are robust for DP calculations. Of these four scoring functions, LIE(GBMV) was found to have the most robust discriminative power (DP) during cross validation, and we conclude that out of all of the scoring functions assessed in our study, LIE(GBMV) has the best overall DP. The DP of S2 and S2h during cross validation are still quite good and are comparable to the CHARMM-based scoring function Rdie(Tot)+LigInt, which was found to have the best DP in the Ferrara et al study.4
Table 7.
Calculated discriminative power (DP) from cross validation using LPDB160 after training on the five random cross-validation subgroups of LPDB160.*
| Trial | LIE(GBMV)
|
S1
|
S2
|
S2h
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Training | Validation | Training | Validation | Training | Validation | Training | Validation | |||||
| AUE | R2 | DP | AUE | R2 | DP | AUE | R2 | DP | AUE | R2 | DP | |
| all | 2.45 | 0.44 | −1.58 | 1.96 | 0.41 | −0.12 | 1.77 | 0.52 | −1.44 | 1.81 | 0.49 | −1.38 |
|
| ||||||||||||
| 1 | 2.38 | 0.46 | −1.61 | 2.05 | 0.38 | −0.14 | 1.76 | 0.53 | −1.58 | 1.81 | 0.50 | −1.38 |
| 2 | 2.51 | 0.41 | −1.19 | 1.87 | 0.43 | −0.12 | 1.76 | 0.51 | −1.15 | 1.78 | 0.50 | −1.15 |
| 3 | 2.36 | 0.48 | −2.44 | 1.95 | 0.46 | −0.21 | 1.78 | 0.53 | −2.32 | 1.85 | 0.50 | −2.35 |
| 4 | 2.47 | 0.43 | −1.29 | 1.92 | 0.39 | −0.09 | 1.71 | 0.53 | −0.93 | 1.73 | 0.51 | −1.01 |
| 5 | 2.46 | 0.46 | −1.37 | 2.01 | 0.41 | −0.09 | 1.74 | 0.54 | −0.97 | 1.84 | 0.49 | −1.03 |
|
| ||||||||||||
| Avg | 2.44 | 0.45 | −1.58 | 1.96 | 0.41 | −0.13 | 1.75 | 0.53 | −1.39 | 1.80 | 0.50 | −1.39 |
AUE is in units of kcal/mol.
As the parameters for LIE(GBMV) seemed to be quite similar over the cross-validation for prediction of ΔGbind, and given the unexpectedly poor DP of the GBMV(Tot) scoring function, we calculated the DP for the LPDB160 dataset as a function of the LIE electrostatic parameter β (Figure 5), while keeping the van der Waals parameter α fixed at 0.20 (which is the average value of α from cross validation). For LIE(Rdie) the DP improved (became more negative) linearly as the electrostatic contribution was reduced from β=0.20 down to the minimum at β=0.02 (Figure 5A). Compared to all the other scoring functions in this work, LIE(Ride) still retains a very good DP over this entire range of β. For LIE(GBMV) the DP was quite poor with the full electrostatic contribution (β=0.20), but improved substantially as the electrostatic contribution was reduced down to the minimum at β=0.05. It was quite striking that the LIE parameters (α=0.20, and β=0.05), which provide the maximum discriminative power (most negative DP), also are very similar to the optimized LIE parameters for ΔGbind over 259 LPDB complexes (α=0.2058, and β=0.0517). Basically the same optimal parameters were arrived at from two completely independent free energy assessments, one being for geometry discrimination, and one for binding free energy discrimination. We also calculated the DP of LIE(GBMV) for the five random cross validation groups from the LPDB160 dataset as a function of the LIE electrostatic parameter β (Figure 5B), and demonstrate that for each of these cross-validation groups, the most negative DP value always occurred for β in the range from 0.03 to 0.07. The most negative DP was found at β=0.06, 0.04, 0.05, 0.03 and 0.07 for groups 1 through 5, respectively. These values are also in reasonable agreement with other recent CHARMM-based LIE implementations that have been reported by Caflisch and coworkers (kinases: α=0.2898, and β=0.0442)15 and Karplus and coworkers (α=0.36, and β=0.16).46 They are also close to the values from another LIE(GBMV) implementation using molecular dynamics rather than energy minimizations by Armen et al. (α=0.165, and β=0.037).36 The LIE scheme parameters are known to have small differences when optimized using different proteins. For kinases the reported values are α=0.2898, and β=0.044215 while for aspartic proteases they are α=0.274, and β=0.180.47 We observe the same characteristics here, with slight differences of the values for α, β and γ when optimized using HIV protease, trypsin, or p38α complexes (data not shown).
Figure 5.
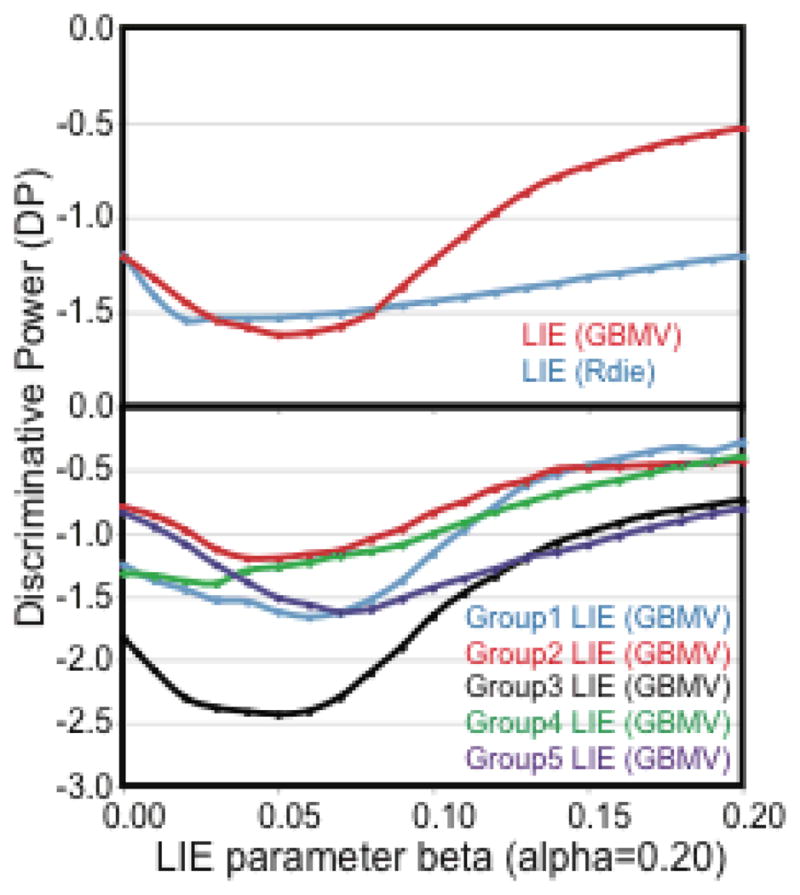
Discriminative power (DP) of LIE scoring functions as a function of the LIE electrostatic parameter β, keeping the van der Waals parameter α fixed at 0.20. (A) DP for LIE(GBMV) and LIE(Rdie) over the LPDB160 dataset. (B) DP for LIE(GBMV) calculated across 5 cross validation subgroups of LPDB160.
The results of the cross validation as shown in Table 6 and Table 7 suggest the robustness of LIE(GBMV), S1, S2 and S2h. However, it is evident that the LIE(GBMV) parameters change when optimized for specific receptors, likely due to the dominance of different types of protein-ligand interactions in different types of complexes. In order to explore comparable behavior in the other schemes (S1, S2 and S2h), we optimized them to reproduce the experimental ΔGbind of 28 HIV PR, 25 trypsin and 12 p38α MAP kinase complexes as separate groups as well as together. The S1 parameter δ, optimized using the full data set was comparable to its values optimized using the individual complex type. Similar to LIE(GBMV) parameters, the parameters of S2 and S2h changed moderately when optimized for a specific receptor (data not shown).
The transferability of the S1 and S2 parameters were further investigated using the NRC HiQ datasets. The parameters optimized using LPDB259 were transferred to the NRC HiQ set1 and set2 datasets and the performance of S1 and S2 were examined (Table 3). The performance of S1 remained the same, both in terms of AUE and R2; the performance of S2 deteriorated. This indicates that the parameters of S2 might not be transferable between two completely different sets of protein-ligand complexes (i.e. LPDB and NRC HiQ here). However, reoptimization of the parameters using a particular dataset produced good performance of S2. The NRC HiQ set1 and set2 were constructed in part as a way to benchmark the transferability of parameters, particularly for a regression based scoring function like S1 and S2 (reference). Thus, we transferred the parameters optimized using HiQset1 and applied to HiQset2 and vice versa (Figure 3C and D). Table 3 shows that the performance of S1 remains the same if the parameters are exchanged. The performance of S2 deteriorates both in terms of AUE and R2 values. This suggests that the S1 parameter is absolutely transferable and S2 parameters are reasonably transferable between these two datasets. Because the NRC HiQ set1 and set2 are proposed as benchmarks for the test of scoring function parameter transferability, the transferability of S1 and S2 parameters can be compared directly with those of other standard scoring functions.
We have further analyzed the size dependence of S1 and S2 by comparing the predicted and experimental ligand binding efficiencies using a narrow dataset consisting of ligands with sizes relevant to drug discovery. The range of 17–35 heavy atoms is where it is most important to calculate ligand efficiencies with high accuracy in order to have an impact on real world drug design optimization and fragment based design efforts. Protein-ligand complexes with ligands containing 17–35 heavy atoms (HA) were separated from NRC HiQ set1 and set2 to construct two smaller datasets. In these two new subsets of set1 and set2 there was a very low correlation between the experimental ΔGbind and the MW of the ligands (R2 = 0.03 and 0.05 for set1 and set2, respectively). The experimental ligand binding efficiencies were calculated by dividing experimental ΔGbind by HA and the predicted ligand binding efficiencies were calculated by dividing S1 or S2 scores by HA. For the new subset of NRC HiQ set1, the R2 between S1 ligand efficiency and experimental ligand efficiency was 0.22 and the R2 between S2 ligand efficiency and experimental ligand efficiency was 0.62. These values for the new subset of NRC HiQ set2 were 0.23 and 0.44, respectively. These results clearly show that the good performance of these scoring functions, especially S2 are not due to their dependencies on the ligand size but due to their abilities to measure the strength of specific protein-ligand interactions. This analysis has been extended to address the issue of parameter transferability as well. The parameter optimized using the two datasets were interchanged and the same procedure as before was followed. This deteriorated the R2 values. For NRC HiQ set1, the R2 for S1 and S2 were 0.22 and 0.39, respectively. For set2, they were 0.23 and 0.33, respectively. These suggest that the scoring functions, especially S2, have reasonable accuracy in predicting ligand binding efficiencies even with the transferred parameters (Figure 6). These tests confirm the robustness of these scoring functions, which are ligand size independent and transferable between different datasets.
Figure 6.
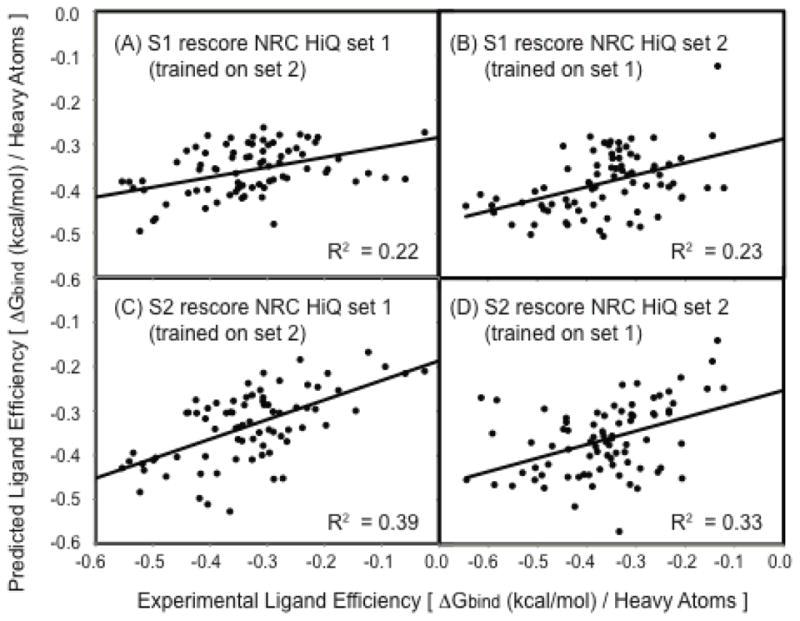
Comparison of predicted and experimental ligand efficiencies for subsets of the NRC HiQ data sets containing 17–35 heavy atoms. (A) S1 rescore NRC HiQ set 1 (parameters optimized on set 2) (B) S1 rescore NRC HiQ set 2 (parameters optimized on set 1 (C) S2 rescore set 1 (with parameters optimized from set 2) (D) S2 rescore set 2 (with parameters optimized from set 1). For these data subsets there is a very low correlation between binding affinity and molecular weight.
3.4 Enrichment of actives over decoys
Finally, we investigated the performance of the scoring functions in a virtual screening process (see Methods section for details). In this virtual screening experiment, we used the scoring function with the best discriminative power, LIE(GBMV), to identify the lowest energy binding poses. Then for each ligand, the single lowest energy pose was used to calculate the S1, S2, and S3 scores to rank the compounds. The parameters trained with LPDB259 were used, and in addition, the S2 parameters trained from NRC HiQ set 1 and set 2. In this way, we assessed the transferability of the S2 parameters in the context of enrichment performance over 8 protein targets representing 4 different target classes. We calculated Receiver Operator Characteristic (ROC) curves and calculated the area under the curve (AUC) and enrichment properties for each target shown in Table 8. The results of the enrichment studies summarized in Table 8 represent a single consistent two-step scoring strategy applied to all target classes. It is clear that specific parameter sets have improved performance for individual targets, and it would be possible to optimize performance for any specific target. As in the “Directory of Useful Decoys” (DUD)29 manuscript the best enrichment results were obtained in the nuclear hormone receptor and cyclooxygenase target classes, likely because these targets are characterized by more buried and lower dielectric binding sites.
Table 8.
The performance of the scoring functions in enriching actives over decoys using several DUD data sets. The scoring functions parameters were transferred from LPDB259, HiQset1 and HiQset2. The AUC of ROC, EF at 1% of the database and EF at 20% of the database are shown.
| Target Class | Target Name | Dataset Details | Parameters from | Parameters from | |||||
|---|---|---|---|---|---|---|---|---|---|
| Actives | decoys | LPDB | LPDB | LPDB | LPDB | NRC HiQ set1 | NRC HiQ set2 | ||
| LIE | S1 | S2 | S3 | S2 | S2 | ||||
| N | N | AUC | AUC | AUC | AUC | AUC | AUC | ||
| nuclear hormone rec. | androgen rec. | 78 | 2610 | 0.74 | 0.68 | 0.97 | 0.67 | 0.93 | 0.57 |
| nuclear hormone rec. | glutocort. rec. | 70 | 2739 | 0.71 | 0.74 | 0.79 | 0.48 | 0.67 | 0.54 |
| serine protease | thrombin | 66 | 2070 | 0.54 | 0.60 | 0.45 | 0.60 | 0.49 | 0.52 |
| serine protease | trypsin | 48 | 1402 | 0.53 | 0.65 | 0.46 | 0.66 | 0.58 | 0.53 |
| cyclooxygenase | cox1 | 25 | 744 | 0.62 | 0.43 | 0.61 | 0.56 | 0.56 | 0.43 |
| cyclooxygenase | cox2 | 330 | 5599 | 0.78 | 0.62 | 0.90 | 0.81 | 0.82 | 0.58 |
| kinase | p38a | 153 | 8944 | 0.55 | 0.63 | 0.69 | 0.41 | 0.61 | 0.74 |
| kinase | vegfr2 | 84 | 2574 | 0.46 | 0.52 | 0.55 | 0.56 | 0.49 | 0.72 |
|
| |||||||||
| EF1 | EF1 | EF1 | EF1 | EF1 | EF1 | ||||
|
|
|
|
|||||||
| nuclear hormone rec. | androgen rec. | 78 | 2610 | 7.4 | 11.2 | 8.7 | 9.9 | 11.2 | 3.7 |
| nuclear hormone rec. | glutocort. rec. | 70 | 2739 | 13.5 | 12.1 | 10.8 | 0.0 | 8.1 | 6.7 |
| serine protease | thrombin | 66 | 2070 | 0.0 | 8.6 | 0.0 | 0.0 | 0.0 | 0.0 |
| serine protease | trypsin | 48 | 1402 | 3.8 | 5.8 | 0.0 | 1.9 | 0.0 | 0.0 |
| cyclooxygenase | cox1 | 25 | 744 | 7.4 | 3.7 | 11.2 | 3.7 | 11.2 | 0.0 |
| cyclooxygenase | cox2 | 330 | 11633 | 6.8 | 7.6 | 14.7 | 3.8 | 17.0 | 2.1 |
| kinase | p38a | 153 | 8944 | 1.3 | 5.1 | 3.2 | 0.6 | 6.51 | 0.87 |
| kinase | vegfr2 | 84 | 2574 | 4.5 | 11.3 | 2.3 | 1.1 | 2.2 | 9.8 |
|
| |||||||||
| EF20 | EF20 | EF20 | EF20 | EF20 | EF20 | ||||
|
|
|
|
|||||||
| nuclear hormone rec. | androgen rec. | 78 | 2610 | 2.1 | 2.6 | 4.7 | 2.4 | 3.7 | 1.4 |
| nuclear hormone rec. | glutocort. rec. | 70 | 2739 | 2.4 | 3.3 | 2.6 | 1.0 | 2.2 | 2.0 |
| serine protease | thrombin | 66 | 2070 | 1.5 | 1.8 | 0.7 | 1.3 | 0.9 | 1.4 |
| serine protease | trypsin | 48 | 1402 | 1.9 | 1.5 | 0.3 | 1.5 | 1.3 | 0.9 |
| cyclooxygenase | cox1 | 25 | 744 | 1.4 | 1.5 | 1.7 | 1.2 | 1.4 | 0.6 |
| cyclooxygenase | cox2 | 330 | 11633 | 2.6 | 1.7 | 3.6 | 2.7 | 3.3 | 1.5 |
| kinase | p38a | 153 | 8944 | 0.8 | 1.5 | 2.3 | 0.5 | 2.09 | 2.7 |
| kinase | vegfr2 | 84 | 2574 | 1.0 | 1.8 | 1.1 | 1.4 | 0.8 | 2.5 |
Using the best-fit parameters to the LPDB database, overall the best performance was found with S2, although S2 did not perform well for serine proteases. For the other six targets, S2 performed as well or outperformed LIE(GBMV). For serine proteases and p38α, S1 was able to outperform LIE(GBMV). S3 showed the worst overall performance, although still showing a reasonable performance for the less challenging targets. Good performance was also observed for the S2 scoring function with parameters optimized for the NRC HiQ set 1 and set 2 datasets. A noteworthy example is for trypsin where the parameters optimized for NRC HiQ set 1 and 2 performed better than for S2 optimized to the LPDB. Other than this example, the S2 scoring function showed a reasonable transferability of performance among the target classes. It is interesting to note that the S2 parameters optimized to the NRC HiQ set 1 exhibited better performance in the nuclear hormone receptor and cyclooxygenase targets (more buried and lower dielectric) while the S2 parameters optimized to set 2 exhibited better performance in the kinase targets.
It is likely that the poor enrichment in serine proteases was a result of non-optimal LIE parameters in prediction of binding geometry. As mentioned previously, the LIE scheme parameters are known to have small differences when optimized using different proteins. Other recent CHARMM-based LIE literature values reported for kinases were α=0.2898, and β=0.044215 while for aspartic proteases they were α=0.274, and β=0.180.47 In this manuscript, it was determined that the LIE parameters with the optimal discriminative power over the LPDB160 datset were (α=0.20, and β=0.02) for LIE(Rdie) and (α=0.20, and β=0.05) for LIE(GBMV). Eleven trypsin crystal structures of the LPDB160 dataset were analyzed as a subset, and the LIE parameters with the optimal discriminative power for trypsin were found to be (α=0.20, and β=0.11) for LIE(Rdie) and (α=0.20, and β=0.08) for LIE(GBMV). These parameters both reflect that a stronger electrostatic component weight was required for correct determination of ligand geometry. The DUD dataset for trypsin was then redocked using these LIE(Rdie) and LIE(GBMV) parameters, and enrichment properties were shown to improve, due to more accurately protein-ligand geometries. Rescoring the new trypsin DUD dataset with S2 scoring function parameters optimized for the HiQset1 and HiQset2 datasets, the ROC (AUC) improved to 0.64 and 0.61 for HiQ set 1 and 2 parameters respectively. This demonstrates that the same S2 scoring functions with transferred parameters from HiQ were able to show improved enrichment performance when the protein-ligand geometries were determined with greater accuracy.
3.5 Scoring function performance over high and low molecular weight ranges
Specifically for the p38α dataset, it was important to consider the molecular weight range of both the actives and the decoys (Figure 7A). In our enrichment studies, the true enrichment factor and Receiver Operator Characteristic (ROC) consider only actives and decoys in the same MW range of 320–450, in order to avoid artificial enrichment that may occur from actives that are higher in MW than the decoys.48 It is well known in enrichment studies that the MW composition of the actives and decoys is crucial to the enrichment and ROC properties, mainly because high MW actives or decoys can easily have lower scores than both actives and decoys of lower MW.48 We have also split the dataset into two MW groups (Figure 7B) to consider each scoring function’s ability to enrich actives in the low MW range (320–375) and in the high MW range (375–450). The DUD decoys selected for p38a have similar physical properties but different topology to p38a actives, so this represents a challenging dataset for enrichment.
Figure 7.
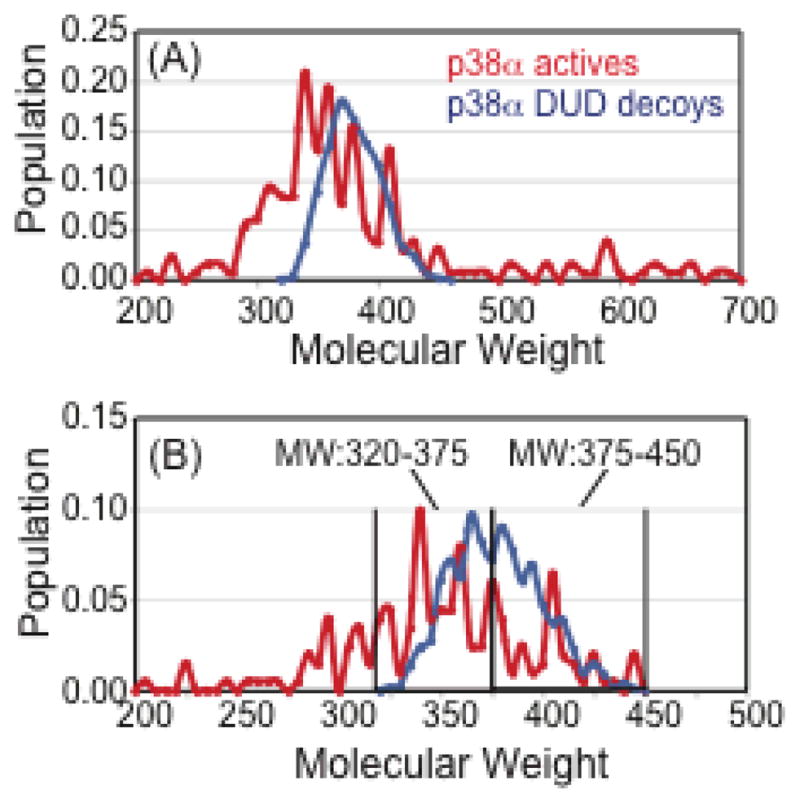
Distribution of the molecular weight (MW) for p38a MAP kinase active ligands and decoy ligands taken from the DUD. (A) Distribution of the entire MW range of actives from 200–700 using 10 dalton bins. DUD decoys only cover the range MW 320–450. (B) Distribution of the MW range from 200 to 450 using 5 dalton bins. The molecular weight range of 320–450 is shown broken into two groups for Enrichment Factor and ROC curve analysis: low MW range (320–375) and high MW range (375–450).
Early enrichment of the actives was assessed by plotting the enrichment factor (EF) at different % of the database (Figure 8). For the entire MW range of 320–450 (Figure 8A), the S1 and the S2 scoring function had by far the best enrichment over the initial 10% of the database. For both S1 and S2 the EFs ranged from the maximum theoretical enrichment of 59, where S1 had an average EF of 19.8 over the initial 1% of the database, and S2 had an average EF of 12.1. The next best scoring function for early enrichment was S3, but it had a maximum EF of only 6.6 and an average EF of 1.6 over the initial 1% of the database. The next best scoring function for early enrichment was LIE(GBMV), but it had a maximum EF of 4.2 and an average EF of 1.9 over the initial 1% of the database. For the low MW range of 320–375 (Figure 8B), the S2 scoring function had by far the best enrichment over the initial 10% of the database, with EF ranging up to the maximum theoretical value of 46 and an average EF of 14.8 over the initial 1% of the database. In this low MW range, over the initial 1% of the database S1, LIE(GBMV) and S2h were the next best with average EF over the top 1% of 5.8, 3.5, and 2.5 respectively. For the high MW range of 375–450 (Figure 8B), the S1 scoring function had by far the best enrichment over the initial 10% of the database, where the EFs ranged up to the maximum theoretical enrichment of 88. In this higher MW range, S1 had an average EF of 40.3 over the initial 1% of the database, and S2 had an average EF of 7.1. The average EF over the top 5% of the database is reported in Table 9, which also demonstrates that S1 and S2 have the best overall performance for early enrichment.
Figure 8.
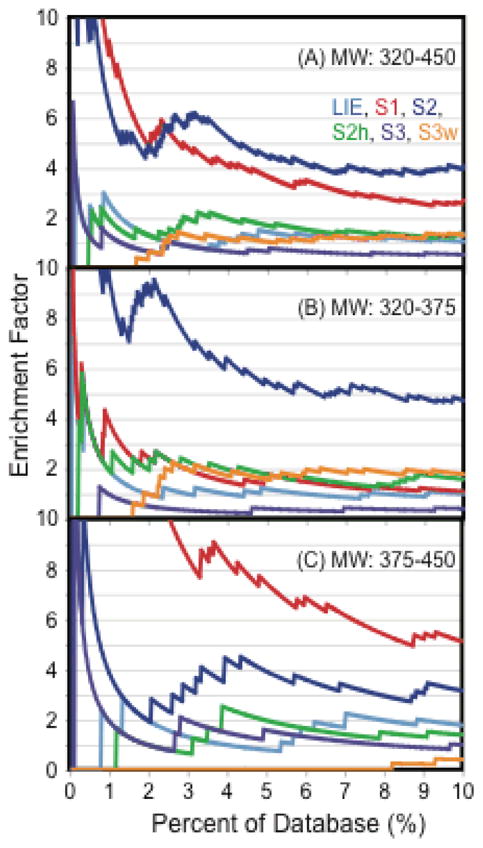
Early enrichment for the top 10 percent of the database. EF factors are shown for the six scoring functions over three molecular weight (MW) ranges: (A) MW: 320–450 which reflects the true total enrichment over the database (B) low MW range from 320–375 (C) high MW range from 375–450.
Table 9.
Performance of the scoring functions in enrichment of active compounds compared to DUD decoys that are specific for p38a. The average enrichment factor <EF> is reported over the first 5% of the database. The area under the curve (AUC) is reported for the Receiver Operating Characteristic (ROC) curves. These properties are calculated for the entire molecular weight (MW) range (MW 320–450), as well as for the low molecular weight range (MW 320–375) and the high molecular weight range (MW 375–450).
| MW Range | Overall | Low MW | High MW | Overall | Low MW | High MW |
|---|---|---|---|---|---|---|
| 320–450 | 320–375 | 375–450 | 320–450 | 320–375 | 375–450 | |
| Scoring Function | <EF> | <EF> | <EF> | AUC | AUC | AUC |
| LIE(GBMV) | 1.4 | 1.6 | 1.2 | 0.55 | 0.57 | 0.60 |
| S1 | 8.0 | 2.8 | 16.7 | 0.59 | 0.53 | 0.80 |
| S2 | 6.6 | 8.8 | 4.1 | 0.75 | 0.77 | 0.73 |
| S2h | 1.6 | 2.2 | 1.1 | 0.65 | 0.66 | 0.64 |
| S3 | 1.0 | 0.4 | 1.9 | 0.43 | 0.39 | 0.54 |
| S3w | 0.7 | 1.1 | 0.0 | 0.57 | 0.62 | 0.50 |
ROC curves are also constructed to assess the performance of these scoring functions (Figure 9). S2 retained the best enrichment of the actives throughout the database (Figure 9A). S1, LIE(GBMV) and S3w are all closer to the diagonal (random) line, indicating performance that is not much better than random at various ranks in the database. The ROC curve of S3 was below the diagonal (random) line, indicating poor enrichment where the sign of the scoring function is actually incorrect, scoring decoys better than actives in a systematic way. The area under the ROC curve (AUC) is calculated from the ROC curves of the scoring functions and compared with each other (Table 9). For p38α over the entire MW range 320–450, the performance of S2 was the best (AUC=0.75) followed by S2h (AUC=0.65), and then S1 (AUC=0.59). In the low MW range S2 is the best by far (AUC=0.77), followed by S2h (AUC=0.66). In the high MW range S1 is the best (AUC=0.80), followed by S2 (AUC=0.73). The LIE(GBMV) scoring function had improved performance in the high MW range (AUC=0.60), compared to the low MW range (0.57). For the two distance dependent pair potentials, the overall performance was quite poor, but S3w exhibited improved performance in the low MW range (AUC=0.62). Although data is only presented for docking the actives against the DUD dataset to one p38a crystal structure conformation for clarity, our results docking to several other p38a crystal structure conformations (1a9u, 1ouk, 1oz1, 1w84, 1kv2, and 1w83) also verify a similar assessment of the performance of S1 and S2.
Figure 9.
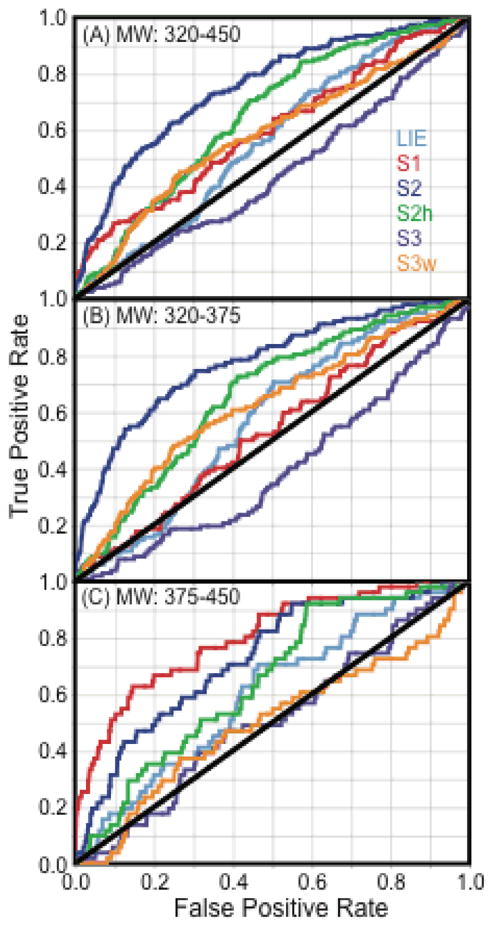
Receiver Operating Characteristic (ROC) curve for database. ROC curves are shown for the six scoring functions over three molecular weight (MW) ranges: (A) MW: 320–450 which reflects the true total enrichment over the database (B) low MW range from 320–375 (C) high MW range from 375–450. ROC of random is shown as a black line on the diagonal.
These observations were also found to hold true if the entire p38α dataset was re-ranked with different step 1 scoring functions to identify the single lowest energy scoring poses, and then the step 2 scores were recalculated and re-ranked. This procedure was performed for the p38α dataset with the three best performing CHARMM-based scoring for step 1 geometry discrimination [Rdie(Tot), LIE(Rdie), and LIE(GBMV) ], and the overall conclusions regarding step 2 performance did not change (Supplementary Table S3). In this analysis, we also assessed enrichment using the entire low MW range of the actives including MW 200–320, and were able to show that S2 also had the best performance in separating low MW actives (MW 200–375) against the range of low MW decoys (MW 320–375) (Supplementary Table S3). The overall conclusion remains that S2 has an improved ability to separate actives from decoys over a large MW range, including the low MW range, while S1 exhibits optimal ability to separate actives from decoys in the higher MW range. This observation was also similar for the other target classes. One reason for improved performance of S1 in the higher MW range may be that for higher MW ligands, it is likely that decoys have a larger percentage of non-interacting atoms than actives, and thus the shorter distance cutoff of S1 is more discriminating than the larger cutoff for S2. On the other hand, in the lower MW region more of the total ligand surface may participate in favorable and specific contacts, and thus the short distance contact is not sufficient to discriminate as actives are now more similar to decoys in the number of interactions that they contribute.48
3.6 Separation of scoring function performance from molecular weight effects
It is very important to separate the performance of a scoring function from its correlation with MW effects. When comparing the predicted ΔGBind to experimental ΔGBind from protein-ligand crystal structures, it is important that a scoring function perform well not only when there is a strong correlation between experimental ΔGBind and MW, but also when there is very low correlation. We demonstrated a reasonable performance for S1 and S2 in subsets of NRC HiQ datasets that has very low correlation of ΔGBind and MW in section 3.3, where it was straightforward that the scoring function outperformed MW effects. As mentioned previously, in critical analysis of enrichment studies, the MW composition of a data set is very important. Artificial enrichment may result from a large number of high MW actives competing against a much larger number of low MW decoys.48 On the other hand, in a different dataset lower values of ROC (AUC) may result from the scores of many high molecular weight decoys competing against a large number of low molecular weight actives. In an ideal world, these low MW actives should be able to be separated from high MW actives, but this does not always happen even using the best scoring functions. One strategy that can definitively separate the effects of MW composition of a dataset from the true performance of a scoring function is to compare the enrichment of actives against only decoys of identical MW.
Ideally, it would be possible to create a set of decoys that have the same valence of an active, but with a different topology and identical MW. In analyzing our DUD decoy sets, we found that there were an insufficient number of decoys with the identical valence as the actives. However, we performed a test that was very similar to this. In each target specific DUD sets, we identified groups of 5 or more decoys that had the same valence (identical MW and molecular formula). These groups of similar decoys varied in size from 5 to up to 40 decoys. The enrichment of actives against these sets of similar decoys was calculated within very narrow molecular weight ranges (bins of 5 daltons). In this way, the enrichment of active ligands was compared against groups of similar decoys with nearly identical MW. The ROC (AUC) for each of these bins for the various scoring functions were then compared to merely sorting the bins by MW. This analysis is presented for p38a (Table 10) and cox2 (Table 11) as these datasets were the largest and had the most DUD decoys. The results demonstrate that S1 and S2 both clearly are able to separate actives out of these groups of decoys independent of MW effects. This result is robust for both p38a and cox2 over a large MW range: 330–425 (where it was possible to perform this analysis). Overall, it is clear that S2 had the best performance in this test. However, these results are also a convincing demonstration that the performance of S1 to separate actives from decoys is independent of MW effects.
Table 10.
The performance of the scoring functions in enriching p38α actives over groups of DUD decoys that have the identical MW and molecular formula. The dataset has been divided into subcategories based on MW and the AUCs were calculated in each narrow MW range.
| Sort by MW | LIE | S1 | S2 | S3 | |||
|---|---|---|---|---|---|---|---|
| MW range | N actives | N decoys | ROC (AUC) | ROC (AUC) | ROC (AUC) | ROC (AUC) | ROC (AUC) |
| 330–335 | 7 | 100 | 0.30 | 0.75 | 0.86 | 0.72 | 0.38 |
| 335–340 | 20 | 135 | 0.44 | 0.85 | 0.89 | 0.99 | 0.29 |
| 340–345 | 8 | 146 | 0.20 | 0.56 | 0.52 | 0.61 | 0.59 |
| 345–350 | 9 | 427 | 0.30 | 0.72 | 0.93 | 0.63 | 0.52 |
| 350–355 | 9 | 498 | 0.47 | 0.71 | 0.68 | 0.82 | 0.32 |
| 355–360 | 16 | 335 | 0.36 | 0.45 | 0.40 | 0.61 | 0.31 |
| 360–365 | 5 | 688 | 0.47 | 0.54 | 0.89 | 0.77 | 0.46 |
| 365–370 | 5 | 518 | 0.35 | 0.64 | 0.71 | 0.58 | 0.44 |
| 370–375 | 12 | 424 | 0.36 | 0.48 | 0.31 | 0.52 | 0.34 |
| 375–380 | 8 | 577 | 0.41 | 0.44 | 0.61 | 0.85 | 0.29 |
| 380–385 | 2 | 455 | 0.86 | 0.60 | 0.76 | 0.60 | 0.27 |
| 385–390 | 5 | 331 | 0.29 | 0.34 | 0.47 | 0.75 | 0.63 |
| 390–395 | 2 | 381 | 0.48 | 0.29 | 0.73 | 0.91 | 0.20 |
| 395–400 | 3 | 199 | 0.37 | 0.55 | 0.79 | 0.58 | 0.41 |
| 400–405 | 13 | 171 | 0.51 | 0.50 | 0.84 | 0.78 | 0.56 |
| 405–410 | 4 | 180 | 0.82 | 0.41 | 0.97 | 0.74 | 0.52 |
| 410–415 | 3 | 72 | 0.64 | 0.53 | 0.85 | 0.68 | 0.66 |
| 415–420 | 1 | 18 | 0.76 | 0.59 | 0.65 | 0.41 | 0.76 |
| 420–425 | 4 | 62 | 0.20 | 0.27 | 0.90 | 0.75 | 0.61 |
Table 11.
The performance of the scoring functions in enriching cox2 actives over groups of DUD decoys that have the identical MW and molecular formula. The dataset has been divided into subcategories based on MW and the AUCs were calculated in each narrow MW range.
| MW range | N actives | N decoys | Sort by MW | LIE | S1 | S2 | S3 |
|---|---|---|---|---|---|---|---|
| ROC (AUC) | ROC (AUC) | ROC (AUC) | ROC (AUC) | ROC (AUC) | |||
| 335–340 | 4 | 42 | 0.24 | 0.57 | 0.74 | 0.94 | 0.72 |
| 340–345 | 15 | 231 | 0.37 | 0.70 | 0.79 | 0.77 | 0.78 |
| 345–350 | 14 | 174 | 0.57 | 0.84 | 0.68 | 0.79 | 0.77 |
| 350–355 | 10 | 48 | 0.34 | 0.69 | 0.47 | 1.00 | 0.98 |
| 355–360 | 15 | 294 | 0.51 | 0.77 | 0.50 | 0.80 | 0.77 |
| 360–365 | 15 | 157 | 0.26 | 0.76 | 0.65 | 0.92 | 0.86 |
| 365–370 | 14 | 159 | 0.33 | 0.60 | 0.38 | 0.90 | 0.74 |
| 370–375 | 15 | 280 | 0.38 | 0.56 | 0.45 | 0.85 | 0.68 |
| 375–380 | 9 | 203 | 0.52 | 0.86 | 0.69 | 0.86 | 0.81 |
| 380–385 | 21 | 194 | 0.38 | 0.80 | 0.34 | 0.94 | 0.86 |
| 385–390 | 19 | 153 | 0.52 | 0.67 | 0.47 | 1.00 | 0.81 |
| 390–395 | 11 | 87 | 0.79 | 0.90 | 0.78 | 0.94 | 0.87 |
| 395–400 | 15 | 87 | 0.55 | 0.94 | 0.40 | 0.84 | 0.83 |
| 400–405 | 14 | 104 | 0.49 | 0.88 | 0.52 | 0.97 | 0.85 |
4. Conclusions
The performance of several two-step scoring approaches for molecular docking were assessed for their ability to predict binding geometries and free energies. Two new scoring functions, S1 and S2, designed for “step 2 discrimination” were compared to CHARMM-based scoring functions including two LIE variations. The LIE(GBMV), S1, S2, and S2h scoring functions were trained and five-fold cross-validated on a diverse set of 259 protein-ligand complexes from the Ligand Protein Database (LPDB). The parameters for the LIE scoring function with the optimal (DP) for geometry were also very similar to the best-fit parameters for binding free energy over a large number of protein-ligand complexes. Although it is well-known that LIE fits to individual receptor-ligand series can dramatically improve LIE model performance, the convergence of LIE parameters in independent assessments for step 1 and step 2 discrimination indicate that these are the optimally transferable parameters for virtual screening with widely diverse ligands and receptors. However despite this convergence of parameters for LIE(GBMV), both the S1 and S2 scoring functions were also shown to demonstrate improved prediction of binding free energy and separation of actives from decoys compared to LIE(GBMV). The transferability of the S1 and S2 parameters was established using several assessment strategies: prediction of binding affinity (using cross-validation and transfer of optimized parameters), prediction of ligand efficiency, and separation of active compounds from decoys.
The performance of these scoring functions was also assessed over various MW ranges, and analysis was performed to separate scoring function performance from MW effects. Of all of the scoring functions, LIE(GBMV) had the best DP in the lower MW range, 200–500, while S2 and S2h show improved performance in the higher MW range, 500–800. The S1 scoring function was shown to have improved separation of actives in the higher molecular weight range, and S2 has superior performance over the entire molecular weight range and in the low molecular weight range (MW 200–375). This study shows how a two-step scoring function scheme can be trained and optimized for performance in specific MW ranges of interest. This approach would be applicable for improved accuracy in fragment docking in the low MW range or for efficient virtual screening for high MW hits from large databases.
Supplementary Material
Acknowledgments
This work has been supported by the National Institutes of Health to CLB (GM037554) and to RSA (GM076836), in the form of an NRSA Postdoctoral Fellowship. This work has also been supported by the National Science Foundation to MT (0941318 and 0922657), by the Army to MT (54723-CS) and by the CONCyT fellowship to MT. OR and DJD were also supported by the National Institutes of Health (P42ES010344). RSA coordinated and planned this work, which was executed by RSA and OR. OR developed the S1, S2, and S2h scoring function in collaboration with RSA, MT, DJD and CLB. RSA did all of the work regarding the CHARMM-based scoring functions, the LIE variations, and the S3 and the S3w scoring functions.
Abbreviations
- fs
femtosecond
- ps
picosecond
- MD
molecular dynamics
- LPDB
Ligand-Protein Database
- Rdie
Distance Dependent Dielectric Function
- LIE
Linear Interaction Energy
Footnotes
Supporting Information Available: Additional information is available regarding the scoring functions derived from LPDB data, as noted in the text. Cross-validation groups are provided for the LPDB259 and LPDB160 datasets. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Ewing TJA, Kuntz ID. Critical evaluation of search algorithms for automated molecular docking and database screening. J Comput Chem. 1997;18 (9):1175–1189. [Google Scholar]
- 2.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19 (14):1639–1662. [Google Scholar]
- 3.Wu GS, Robertson DH, Brooks CL, Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER - A CHARMm-based MD docking algorithm. J Comput Chem. 2003;24 (13):1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL. Assessing scoring functions for protein-ligand interactions. J Med Chem. 2004;47 (12):3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 5.Wang R, Lu Y, Fang X, Wang S. An Extensive Test of 14 Scoring Functions Using the PDBbind Refined Set of 800 Protein-Ligand Complexes. J Chem Inf Comput Sci. 2004;44:2114–2125. doi: 10.1021/ci049733j. [DOI] [PubMed] [Google Scholar]
- 6.Momany FA, Rone R. Validation of the General-Purpose Quanta(R)3.2/Charmm(R) Force-Field. J Comput Chem. 1992;13 (7):888–900. [Google Scholar]
- 7.Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol. 2000;295 (2):337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 8.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions .1. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput-Aided Mol Des. 1997;11 (5):425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 9.Goodsell DS, Olson AJ. Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins. 1990;8 (3):195–202. doi: 10.1002/prot.340080302. [DOI] [PubMed] [Google Scholar]
- 10.Wang RX, Lai LH, Wang SM. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput-Aided Mol Des. 2002;16 (1):11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 11.SYBYL, version 6.9. Tripos; St. Louis, MO: 2002. [Google Scholar]
- 12.Cerius2, version 4.6. Accelrys; San Diego, CA: 2001. [Google Scholar]
- 13.Aqvist J, Medina C, Samuelsson JE. New Method for Predicting Binding-Affinity in Computer-Aided Drug Design. Protein Eng. 1994;7 (3):385–391. doi: 10.1093/protein/7.3.385. [DOI] [PubMed] [Google Scholar]
- 14.Huang D, Caflisch A. Efficient evaluation of binding free energy using continuum electrostatics solvation. J Med Chem. 2004;47 (23):5791–5797. doi: 10.1021/jm049726m. [DOI] [PubMed] [Google Scholar]
- 15.Kolb P, Huang D, Dey F, Caflisch A. Discovery of kinase inhibitors by high-throughput docking and scoring based on a transferable linear interaction energy model. J Med Chem. 2008;51 (5):1179–1188. doi: 10.1021/jm070654j. [DOI] [PubMed] [Google Scholar]
- 16.Zhou T, Huang D, Caflisch A. Is quantum mechanics necessary for predicting binding free energy? J Med Chem. 2008;51 (14):4280–4288. doi: 10.1021/jm800242q. [DOI] [PubMed] [Google Scholar]
- 17.Carlsson J, Boukharta L, Aqvist J. Combining docking, molecular dynamics and the linear interaction energy method to predict binding modes and affinities for non-nucleoside inhibitors to HIV-1 reverse transcriptase. J Med Chem. 2008;51 (9):2648–2656. doi: 10.1021/jm7012198. [DOI] [PubMed] [Google Scholar]
- 18.Huang DZ, Luthi U, Kolb P, Cecchini M, Barberis A, Caflisch A. In silico discovery of beta-secretase inhibitors. J Am Chem Soc. 2006;128 (16):5436–5443. doi: 10.1021/ja0573108. [DOI] [PubMed] [Google Scholar]
- 19.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267 (3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 20.Sotriffer CA, Sanschagrin P, Matter H, Klebe G. SFCscore: scoring functions for affinity prediction of protein-ligand complexes. Proteins. 2008;73 (2):395–419. doi: 10.1002/prot.22058. [DOI] [PubMed] [Google Scholar]
- 21.Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proc Natl Acad Sci US A. 1999;96 (18):9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajduk PJ. Fragment-based drug design: How big is too big? J Med Chem. 2006;49 (24):6972–6976. doi: 10.1021/jm060511h. [DOI] [PubMed] [Google Scholar]
- 23.Roche O, Kiyama R, Brooks CL. Ligand-Protein DataBase: Linking protein-ligand complex structures to binding data. J Med Chem. 2001;44 (22):3592–3598. doi: 10.1021/jm000467k. [DOI] [PubMed] [Google Scholar]
- 24.Wallqvist A, Jernigan RL, Covell DG. A Preference-Based Free-Energy Parameterization of Enzyme-Inhibitor Binding - Applications to Hiv-1-Protease Inhibitor Design. Protein Sci. 1995;4 (9):1881–1903. doi: 10.1002/pro.5560040923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallqvist A, Covell DG. Docking enzyme-inhibitor complexes using a preference-based free-energy surface. Proteins. 1996;25 (4):403–419. doi: 10.1002/prot.1. [DOI] [PubMed] [Google Scholar]
- 26.Verkhivker G, Appelt K, Freer ST, Villafranca JE. Empirical Free-Energy Calculations of Ligand-Protein Crystallographic Complexes .1. Knowledge-Based Ligand-Protein Interaction Potentials Applied to the Prediction of Human-Immunodeficiency-Virus-1 Protease Binding-Affinity. Protein Eng. 1995;8 (7):677–691. doi: 10.1093/protein/8.7.677. [DOI] [PubMed] [Google Scholar]
- 27.DeWitte RS, Shakhnovich EI. SMoG: de Novo design method based on simple, fast, and accurate free energy estimates. 1. Methodology and supporting evidence. J Am Chem Soc. 1996;118 (47):11733–11744. [Google Scholar]
- 28.Velec HFG, Gohlke H, Klebe G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J Med Chem. 2005;48 (20):6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- 29.Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49 (23):6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Charmm - a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem. 1983;4 (2):187–217. [Google Scholar]
- 31.MacKerell AD, Jr, Brooks B, Brooks CL, III, Nilsson L, Roux B, Won Y, Karplus M. CHARMM: The Energy Function and Its Parameterization with an Overview of the Program. In: Schleyer P, et al., editors. The Encyclopedia of Computational Chemistry. Vol. 1. John Wiley & Sons; Chichester, UK: 1998. pp. 271–277. [Google Scholar]
- 32.Feig M, Karanicolas J, Brooks CL. MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graphics Modell. 2004;22 (5):377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Vieth M, Hirst JD, Dominy BN, Daigler H, Brooks CL. Assessing search strategies for flexible docking. J Comput Chem. 1998;19 (14):1623–1631. [Google Scholar]
- 34.Vieth M, Hirst JD, Kolinski A, Brooks CL. Assessing energy functions for flexible docking. J Comput Chem. 1998;19 (14):1612–1622. [Google Scholar]
- 35.Taufer M, Crowley M, Price DJ, Chien AA, Brooks CL. Study of a highly accurate and fast protein-ligand docking method based on molecular dynamics. Concurr Comp-Pract E. 2005;17 (14):1627–1641. [Google Scholar]
- 36.Armen RS, Chen J, Brooks CL. An Evaluation of Explicit Receptor Flexibility in Molecular Docking Using Molecular Dynamics and Torsion Angle Molecular Dynamics. J Chem Theory Comput. 2009;5 (10):2909–2923. doi: 10.1021/ct900262t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taufer M, Armen RS, Chen JH, Teller PJ, Brooks CL. Computational Multiscale Modeling in Protein-Ligand Docking. IEEE Eng Med Biol Mag. 2009;28 (2):58–69. doi: 10.1109/MEMB.2009.931789. [DOI] [PubMed] [Google Scholar]
- 38.Lee MS, Feig M, Salsbury FR, Brooks CL. New analytic approximation to the standard molecular volume definition and its application to generalized born calculations. J Comput Chem. 2003;24 (11):1348–1356. doi: 10.1002/jcc.10272. [DOI] [PubMed] [Google Scholar]
- 39.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem. 2004;25 (2):265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 40.Muegge I, Martin YC. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J Med Chem. 1999;42 (5):791–804. doi: 10.1021/jm980536j. [DOI] [PubMed] [Google Scholar]
- 41.Shoichet BK, Bodian DL, Kuntz ID. Molecular Docking Using Shape Descriptors. J Comput Chem. 1992;13 (3):380–397. [Google Scholar]
- 42.Mitchell JBO, Laskowski RA, Alex A, Forster MJ, Thornton JM. BLEEP - Potential of mean force describing protein-ligand interactions: II. Calculation of binding energies and comparison with experimental data. J Comput Chem. 1999;20 (11):1177–1185. [Google Scholar]
- 43.Mitchell JBO, Laskowski RA, Alex A, Thornton JM. BLEEP - Potential of mean force describing protein-ligand interactions: I. Generating potential. J Comput Chem. 1999;20 (11):1165–1176. [Google Scholar]
- 44.Hawkins PCD, Warren GL, Skillman AG, Nicholls A. How to do an evaluation: pitfalls and traps. J Comput-Aided Mol Des. 2008;22 (3–4):179–190. doi: 10.1007/s10822-007-9166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain AN. Bias, reporting, and sharing: computational evaluations of docking methods. J Comput-Aided Mol Des. 2008;22 (3–4):201–212. doi: 10.1007/s10822-007-9151-x. [DOI] [PubMed] [Google Scholar]
- 46.Spichty M, Taly A, Hagn F, Kessler H, Barluenga S, Winssinger N, Karplus M. The HSP90 binding mode of a radicicol-like E-oxime determined by docking, binding free energy estimations, and NMR 15N chemical shifts. Biophys Chem. 2009;143 (3):111–23. doi: 10.1016/j.bpc.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang D, Luthi U, Kolb P, Cecchini M, Barberis A, Caflisch A. In silico discovery of beta-secretase inhibitors. J Am Chem Soc. 2006;128 (16):5436–43. doi: 10.1021/ja0573108. [DOI] [PubMed] [Google Scholar]
- 48.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, Taylor RD, Watson P. Virtual screening using protein-ligand docking: avoiding artificial enrichment. J Chem Inf Comput Sci. 2004;44 (3):793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.